

Abstract
Nonalcoholic fatty liver disease (NAFLD) encompasses a range of liver pathology ranging from simple steatosis to varying degrees of inflammation, hepatocyte injury and fibrosis. Without intervention it can progress to end-stage liver disease and hepatocellular carcinoma. Given its close association with obesity, the prevalence of NAFLD has increased dramatically worldwide. Currently, there are no FDA-approved medications for the treatment of NAFLD and although lifestyle modifications with appropriate diet and exercise have been shown to be beneficial, this has been difficult to achieve and sustain for the majority of patients. As such, the search for effective therapeutic agents is an active area of research. Peroxisome proliferator-activated receptors (PPARs) belong to a class of nuclear receptors. Because of their key role in the transcriptional regulation of glucose and lipid metabolism, PPAR ligands have been investigated as possible therapeutic agents for NAFLD. Here we review the current evidence from preclinical and clinical studies investigating the therapeutic potential of PPAR ligands for the treatment of this spectrum of liver disorders.
Keywords: liver, NAFLD, NASH, PPAR, thiazolidinediones, fibrates
Introduction
Nonalcoholic fatty liver disease (NAFLD) is defined as hepatic steatosis in the absence of significant alcohol intake. Although there is clearly a genetic basis for susceptibility to NAFLD, development and progression of this disease is closely associated with obesity and insulin resistance. Consequently, with the obesity epidemic, NAFLD has now become the most common cause of liver disease in the United States. Prevalence estimates among the general population range from 20–50% in adults and 10–40% in children, with a much higher prevalence noted among obese individuals [1,2,3]. Worldwide prevalence of NAFLD is about 24% with estimates ranging from 6–35% [1,4]. The wide range in prevalence estimates reflects differences based on geographical location, ethnicity, definition, and mode of diagnosis. Additionally, such studies may also be underestimating the true prevalence as they fail to account for cases of cryptogenic cirrhosis, a significant percentage of which may be secondary to progressive NAFLD.
NAFLD encompasses a range of histological findings (Figure 1). A majority of patients have isolated steatosis, but a subset of patients can develop inflammation with varying degrees of fibrosis termed nonalcoholic steatohepatitis or NASH. Some patients can develop bridging fibrosis and cirrhosis and potentially hepatocellular carcinoma. Although isolated steatosis is generally considered benign, patients with biopsy findings consistent with NASH have a poorer prognosis and increased morbidity and all-cause mortality compared to the general population [5,6]. In a study involving 129 patients with biopsy confirmed NAFLD and elevated ALT, patients with NASH were more likely to die from cardiovascular disease and liver-related causes in a mean follow up time of 13 years [6]. Cirrhosis secondary to NASH accounted for 13.4% of liver transplants in 2015 (https://optn.transplant.hrsa.gov/data/view-data-reports/build-advanced/). This percentage has been steadily increasing over the past decade and with its rising prevalence and emergence of effective treatments for hepatitis C virus, NASH is predicted to become the leading cause of liver transplantation within the next decade [7].
Figure 1.
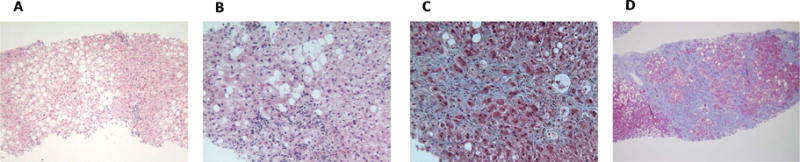
Histological spectrum of NAFLD. A. Steatosis without inflammation, hepatocyte ballooning, or fibrosis. B. Steatohepatitis with hepatocyte ballooning, Mallory-Denk bodies, lobular inflammation, and perisinusoidal fibrosis seen on haemotoxylin and eosin stain. C. Steatohepatitis with perisinusoidal fibrosis detected by trichome stain. D. Cirrhosis with remaining steatosis. Pictures were kindly provided by Dr. E. Brunt.
Studies estimate that 37–41% of NAFLD patients show fibrosis progression in follow up biopsies [6,8]. Although the progression of fibrosis is generally a slow process, the rate of change is highly variable among individual patients [5]. Prognostic factors have been investigated and studies suggest that diabetes and higher BMI are most highly associated with rapid progression of fibrosis [5,6]. Patton and colleagues retrospectively evaluated 245 children enrolled in the NASH clinical research network for features of metabolic syndrome. Not surprisingly, the study found metabolic syndrome to be common among children with NAFLD, and central obesity and insulin resistance were associated with increased severity of NAFLD including advanced fibrosis [9]. As such, therapies that target insulin resistance and metabolic abnormalities are believed to constitute an attractive target for treating the progression to NASH.
2. Pathophysiology of NAFLD
The pathophysiology of NAFLD is complex and multifactorial. There is a clear association with obesity, dysfunctional adipose tissue, and dysregulated de novo hepatic lipogenesis. Fatty acids in circulation and hepatic lipogenesis significantly contribute to accumulation of triglycerides in the liver while dietary lipids also account for a significant proportion of intrahepatic lipids [1,10].
A two hit model of injury has been proposed to explain the development and progression of NAFLD and NASH. The first hit is hepatic steatosis, which results in increased susceptibility to other forms of injury including exposure to gut-derived endotoxin, excessive inflammation, ER stress, oxidative stress, and mitochondrial dysfunction. This ultimately results in cell injury and death as well as activation of hepatic stellate cells, which produces extracellular matrix leading to fibrosis [11]. Studies have found that polymorphisms in PNPLA3 [12] and TM6SF2 [13] genes are associated with increased susceptibility to NAFLD and certain variants may be associated with more rapid progression to NASH [1,14] by mechanisms that are poorly understood.
Various animal models have been used to study NAFLD and NASH in tractable organisms. Each model has its advantages and disadvantages, and no one model perfectly recapitulates the disease in humans. Dietary models include the methionine and choline deficient (MCD) diet, high fat diets of varying compositions, fructose-containing diets, and high cholesterol diets [15,16]. That these diet compositions fail to accurately represent physiologic conditions and reproduce human dietary factors are major criticisms of all models. While the MCD diet consistently results in significant steatohepatitis, it may not model NAFLD in humans as mice fed a MCD diet tend to lose weight and have decreased plasma triglyceride and cholesterol levels. A high fat diet can induce obesity and insulin resistance with liver injury that tends to be less severe than that induced by the MCD diet [15,16]. Cholesterol-containing diets can reliably produce findings consistent with NASH, but the amount of cholesterol may be much higher than physiologic conditions in humans [17]. Frequently-used genetic models include ob/ob mice, db/db mice, as well as genetically-engineered mice often superimposed with dietary or toxin administration [15,16,17]. The major limitation of genetic models is the concern that such mouse models represent a form of NAFLD that may be distinct from that associated with metabolic syndrome in humans. Lastly, a number of hepatotoxins (e.g. carbon tetrachloride (CCl4) and LPS in the context of high fat diet) have also been used to model hepatic stellate cell activation and the development of fibrosis [15]. The development and use of animal models that can reliably reflect human disease would facilitate our understanding of the pathophysiology of NAFLD and the development of therapeutic agents.
Currently, there are no FDA approved drugs for the treatment of NAFLD. Although lifestyle modifications with weight loss are an effective form of treatment, for many individuals this is not attainable and is subject to high rates of recidivism [18]. With our current understanding of the underlying pathophysiology of NAFLD, agents that correct metabolic abnormalities or have insulin sensitizing properties have been investigated. One of the most commonly targeted class of compounds act as ligands of the nuclear receptor transcription factor family known as the peroxisome proliferator-activated receptors (PPARs). There are three PPAR isoforms: alpha (α), beta/delta (β/δ), and gamma (γ), which are differentially expressed in various tissues [19,20]. PPARα is expressed ubiquitously, but is largely present in the liver. PPARβ/δ is expressed mainly in skeletal muscle and to a lesser degree in adipose tissue and skin. PPARγ is highly expressed in adipose tissue [19,20,21,22]. (Table 1).
Table 1.
PPAR expression and respective ligands.
| Primary tissue | Ligands | |
|---|---|---|
| PPARα | Liver | Wy-14643 Fenofibrate Clofibrate Gemfibrozil |
| PPARδ | Skeletal muscle | GW0742 GW501516 MBX-8025 |
| PPARγ | Adipose | TZDs (rosiglitazone, pioglitazone) INT131 |
|
Dual agonists PPARα/δ PPARα/γ |
GFT505 Bezafibrate Glitazars (saroglitazar) |
Several endogenous PPAR ligands have been suggested including free fatty acids, eicosanoids, and various complex lipids [23,24]. Exogenous ligands include environmental and pharmaceutical molecules that can activate one or all of the PPAR family receptors to varying degrees [25,26,27,28,29]. Once ligand-bound, PPARs form a heterodimer with retinoid X receptor (RXR) to bind response elements that regulate the expression of genes encoding enzymes or proteins involved in beta oxidation, fatty acid uptake, adipogenesis, and adipocyte differentiation [19,30,31,32]. Although PPAR was originally named for the ability of ligands to induce hepatic peroxisome proliferation and resulting hepatocellular carcinoma [30], this phenomenon is specific to rodents [33,34]. Given the PPARs’ critical role as a master regulator of lipid and glucose metabolism in multiple cell types, it is not surprising that this family of nuclear receptors has been the target of drug development for the treatment of metabolic diseases including NAFLD.
Below, we discuss existing evidence for the efficacy of PPAR ligands as potential therapeutics for this emerging public health problem. Due to the focus of this review on NAFLD and NASH, we have emphasized papers that have specifically looked at this aspect of hepatic pathophysiology. We have also limited our scope to studies using direct ligands and excluded papers with use of natural products or endogenous lipids that may act as PPAR ligands. Lastly, we apologize for any papers omitted due to oversight or space limitations.
3. PPARα
PPARα is expressed ubiquitously, but is most highly expressed in the liver. It plays a critical role in the regulation of fatty acid uptake, beta oxidation, ketogenesis, bile acid synthesis, and triglyceride turnover [19,32,35,36]. In addition to its role in the regulation of metabolism, PPARα is also thought to have anti-inflammatory effects through complex regulation of NF-κB [37]. As discussed below, several papers have examined the effects of PPARα in regulating hepatic metabolism and insulin sensitivity in liver using both knockout mice and a variety of synthetic PPARα ligands.
The administration of a high fat diet is often associated with increased hepatic expression of PPARα and PPARα target genes involved in fatty acid oxidation in wild-type mice and it has been suggested that this is an adaptive or protective response by PPARα [38,39,40,41]. In humans with NAFLD, hepatic expression of PPARα is decreased, but was found to increase in parallel with NAFLD histological improvement secondary to lifestyle intervention or bariatric surgery [42]. Mice with genetic deletion of PPARα are viable and in the context of a high fat diet, PPARα−/− mice accumulated more hepatic triglycerides with a significantly higher NAFLD activity score (NAS) compared to WT controls [38,43,44]. PPARα knockout mice fed a high fat diet have increased markers of oxidative stress, inflammation, and cell death [44,45].
When Ip and colleagues fed wild type and PPARα−/− mice an MCD diet for five weeks it was found that PPARα−/− mice developed more severe steatohepatitis compared to wild type mice. Furthermore, one of the most potent PPARα agonists, Wy-14643 (Table 2), prevented MCD diet-induced intrahepatic triglyceride accumulation and liver injury in wild type mice, but had no effect on PPARα−/− mice [46]. PPARα activation is thought to prevent triglyceride accumulation by increasing fatty acid turnover and catabolism as Wy-14643 administration resulted in increased gene expression of acyl-CoA oxidase, liver fatty acid binding protein, L-bifunctional enzyme, and peroxisomal ketothiolase [46]. A subsequent study by Ip and colleagues also showed that administration of Wy-14,643 was able to reverse established steatohepatitis induced by an MCD diet [47]. Together, this suggests that Wy-14,643 can prevent and reverse MCD diet-induced NASH in a PPARα-dependent manner, likely via increased fatty acid oxidation [46,47]. In conjunction with improved histological findings, Wy-14643 treatment was also associated with decreased number of activated hepatic stellate cells. However, its effect on the expression of genes implicated in hepatic fibrosis is less clear since there was no change in TGF-β1, TIMP-1, MMP-2, or connective tissue growth factor, but there was a significant decrease TIMP-2 and MMP-13 [47]. Thus, although histological evaluation following treatment with Wy-14643 was consistent with substantially decreased fibrosis in mice, the mechanism by which this occurs remains unclear.
Table 2.
Clinical studies evaluating the efficacy of PPAR agonists in the treatment of NAFLD.
| Histological features | |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Ligand | Treatment duration | Placebo-controlled | Serum ALT or GGT | Steatosis | Inflammation | Fibrosis | |
| PPARα | Clofibrate [51] | 12months | No | No change | No change | No change | No change |
| Fenofibrate [52] | 48weeks | No | Decreased | No change | No change | No change | |
| Gemfibrozil [53] | 4weeks | No# | Decreased | NA | NA | NA | |
|
| |||||||
| PPARδ | GW501516 [63] | 2weeks | Yes | Decreased | NA | NA | NA |
| MBX-8025 [67] | 8weeks | Yes | Decreased | NA | NA | NA | |
|
| |||||||
| PPARγ | Rosiglitazone [88] | 48weeks | No | Decreased | Improved | Improved | No changeˆ |
| Rosiglitazone [91] | 48weeks | No | Decreased | Improved | Improved | Improved | |
| Rosiglitazone [89,90] | 1,2,3 years | Yes | Decreased | Improved | No change | No change | |
| Pioglitazone [101] | 6months | Yes | Decreased | Improved | Improved | No change | |
| Pioglitazone [102] | 12months | Yes | Decreased | No change | No change* | No change** | |
| Pioglitazone [103] | 96weeks | Yes | Decreased | Improved | Improved | No change | |
| Pioglitazone [104] | 18months | Yes | Decreased | Improved | Improved | No change*** | |
|
| |||||||
| Dual agonist | GFT505 [118] | 8weeks | Yes | Decreased | NA | NA | NA |
Abbreviations: NA, not available; ALT, alanine aminotransferase; GGT, gamma-glutamyl transferase.
When a placebo is present, comparison of histological features and serum markers of liver injury is between the treatment group and the placebo group. When a placebo group is not present, comparison refers to before and after treatment within the same group.
comparison group received no treatment but not placebo
improvement noted in perisinusoidal fibrosis, but not overall fibrosis score
No improvement in inflammation, but there was decreased hepatocyte ballooning and Mallory bodies
p=0.05
statistically significant decrease in fibrosis score but the number of patients who demonstrated improvement in fibrosis was not statistically significant
The fibrates are a class of less potent, compared to Wy-14,643, but clinically relevant PPARα agonists that have also been evaluated in experimental models and in studies in humans. In humanized APOE2 knockin (APOE2KI) mice fed a western diet containing high levels of sucrose and cholesterol as a model of steatohepatitis, treatment with fenofibrate resulted in decreased hepatic steatosis, hepatic macrophage accumulation, inflammatory gene expression, and upregulation of genes involved in beta oxidation [48]. Fenofibrate also reduced hepatic steatosis and inflammation in rats fed a high fat and fructose diet [49]. Similarly, in rats given fructose to induce hepatic steatosis, fenofibrate reduced liver inflammation and lipid accumulation [50]. While the use of fibrates has been shown to prevent and reverse NASH findings in rodents, conclusions about fibrate effectiveness in clinical studies are less clear. In a small study involving 16 patients with biopsy-confirmed NASH, twelve months of clofibrate treatment resulted in no change in baseline ALT or histological findings [51]. Similarly, a small pilot study involving 16 NAFLD patients treated with fenofibrate for 48 weeks showed lower plasma ALT concentration, but there was no significant improvement in histological findings compared to baseline liver biopsies [52]. Basarangoglu and colleagues conducted a larger study involving 46 patients with NASH. They found that four weeks of gemfibrozil treatment resulted in an improvement in serum ALT concentration compared to the non-placebo control group. However, histological endpoints were not assessed [53]. Thus, given the available clinical data, PPARα agonists may lower plasma ALT concentrations, but there is no evidence that they produce histological improvement of NASH in humans.
Differences in the efficacy of PPARα agonists between rodent and human studies may be explained by dissimilarities in PPARα tissue expression patterns [54,55,56]. Compared to humans, rodents have a ten-fold higher expression of PPARα in the liver [54,55]. There also may be species-specific differences in PPARα biology since use of PPARα ligands in rodents is associated with peroxisome proliferation and hepatocellular carcinoma, but this has not been recapitulated in humans [33,34,57]. Clinically approved agonists are also much less potent than many of the experimental compounds and there may also be differences in the relative doses given. Therefore, although disappointing, it is not entirely surprising that rodent and human NAFLD treatment studies using PPARα agonists have yielded different results.
3. PPARδ
The second isoform of the PPAR family is known as PPARβ/δ, but henceforth will be referred to as PPARδ, as this is most common in higher organisms. The PPARδ isoform is expressed in skeletal muscle, adipose tissue, and skin, but it is most highly expressed in muscle, where it is involved in regulating mitochondrial metabolism and fatty acid beta oxidation [58,59]. In the liver, PPARδ is well expressed in hepatocytes, but is also expressed in Kupffer cells and hepatic stellate cells, suggesting a potential role in inflammation and fibrosis [19,20,58]. There is very little work using PPARδ knockout mice to study NAFLD specifically. However, it has been shown that PPARδ null mice are more susceptible to CCl4-induced hepatic fibrosis [60,61].
Several rodent studies have evaluated the effects of treatment with the PPARδ agonist, GW501516. This compound has been shown to protect mice against developing diet-induced obesity on high fat diet. This finding may be due to higher rates of energy expenditure since oxygen consumption rates were increased, which is consistent with a role for PPARδ in skeletal muscle fatty acid oxidation. GW501516 treatment also improved insulin sensitivity and prevented the accumulation of hepatic lipids in mice fed a high fat diet [58]. However, the caveat to these observations is that mice treated with GW501516 were leaner compared to controls and thus, the improvements in hepatic steatosis and insulin sensitivity may all be secondary to the lean phenotype. The effects of GW501516 were also investigated by Nagasawa and colleagues using the MCD diet in mice. Treatment was shown to decrease hepatic triglyceride content but was not associated with decreased plasma ALT concentration and only modest histological improvement in steatosis and inflammation. Although GW501516 treatment was associated with increased hepatic expression of genes involved in fatty acid beta oxidation such as acyl-CoA oxidase, carnitine palmitoyltransferase-1, and liver fatty acid binding protein, its effect on the expression of inflammatory cytokines was variable and it was not associated with increased adiponectin levels [62]. A major limitation of this study was the absence of fibrosis in the control animals fed an MCD diet, which prohibits the ability to assess for treatment effects on a critical component of progressive NASH. Limited, early stage clinical trials assessing the efficacy of GW501516 for treating NAFLD have been conducted. A two week pilot trial of with six subjects randomized to each arm (PPARδ agonist or placebo) showed that, in addition to improved lipid profiles, treatment with PPARδ agonist reduced circulating liver enzyme concentrations and liver fat content as measured by MRI [63]. However, histological evaluation was not performed. This study was limited by the small number of patients, short duration, and extensive exclusion criteria, reducing generalizability of the results. Of note, in 2007, further clinical development of GW501516 for all uses was abandoned due to development of cancer in preclinical models [59,64,65].
The therapeutic potential of other PPARδ agonists have also been evaluated. In the OLETF (Otsuka Long Evans Tokushima Fatty) rat model, treatment with GW0742, an experimental PPARδ agonist, improved insulin signaling, reduced hepatic steatosis, and decreased expression of inflammatory genes. Though the final body weight of the diabetic rats that were treated with PPARδ agonist was not statistically different from the untreated diabetic rats, the treated rats gained significantly less weight than the untreated rats [66]. Hence, it is possible that the improvements may be secondary to decreased weight gain. Additionally, GW074 has been examined in mice treated with CCl4 to induce liver toxicity and fibrosis. While treatment with GW0742 resulted in improvement in fibrosis in wild type mice, this effect was not seen in PPARδ null mice demonstrating a PPARδ-dependent mechanism of action to alleviate fibrosis in response to this stimulus [60,61].
Lastly, a novel PPARδ agonist, MBX-8025, was evaluated in a small, randomized, double-blind, placebo-controlled study. The study included overweight subjects with dyslipidemia and found that treatment with MBX-8025 resulted in favorable lipid profiles and decreased GGT as a measure of liver injury. However, other indicators of NAFLD or liver injury were not measured thus limiting interpretation of its effectiveness in the treatment of NAFLD [67]. Overall, at this time, data regarding the effectiveness of any PPARδ agonist for the treatment of NAFLD remains too limited to formulate conclusions. With regards to safety concerns, the role of PPARδ in carcinogenesis remains controversial as there are conflicting studies in in vitro studies as well as preclinical and clinical studies [59,64].
4. PPARγ
PPARγ is most highly expressed in adipose tissue, where it serves an essential role in the regulation of adipocyte differentiation, adipogenesis, and lipid metabolism [68]. It should also be noted that hepatic PPARγ expression is robustly induced in NAFLD patients and experimental models [69,70,71,72]. In fact, PPARγ deletion in mouse hepatocytes has been shown to be protective against development of steatosis [73,74,75], but this also exacerbates insulin resistance [74]. It is likely that increased PPARγ activity in liver of mice leads to activation of an adipogenic gene expression program and storage of lipid in liver. As discussed below, it is likely that this does not translate to human physiology or occur with PPARγ ligand administration in humans.
Thiazolidinediones (TZDs) are the most widely investigated PPARγ agonists. TZDs represent a class of clinically-used insulin-sensitizing drugs, which currently includes rosiglitazone and pioglitazone. PPARγ activation by TZDs results in increased production of various adipokines, including adiponectin, which enhances hepatic fatty acid oxidation [1,76,77]. PPARγ activation also promotes fat storage in adipocytes and decreases adipose tissue lipolysis thereby decreasing the concentration of fatty acids presented to the liver. In addition to its metabolic effects, TZDs are also thought to decrease inflammation and cytokine production in patients with metabolic syndrome [78,79]. Various studies conducted in rodents have clearly demonstrated improvement in metabolic profiles with improved insulin sensitivity after treatment with TZDs. When histological data is available, TZDs appear to also improve some features of NASH such as steatosis and inflammation, but this is not universal, especially in the histologic evaluation of fibrosis [80,81,82]. Below, we have detailed experimental evidence for both the clinically-used TZDs and other PPARγ agonists.
Of the TZDs, rosiglitazone is the most potent PPARγ ligand [83]. In many rodent models, use of rosiglitazone has little effect on, or actually exacerbates, hepatic steatosis [74,84]. It is possible that the strong activation of PPARγ by rosiglitazone in liver of steatotic mice drives the expression of genes promoting fat storage. In other models, however, rosiglitazone administration reduced hepatic steatosis [85]. Even in studies where rosiglitazone exacerbated steatosis, use of this ligand almost universally resulted in reduced inflammation and prevention or reversal of fibrosis and hepatic stellate cell activation. These include studies conducted in mice with steatohepatitis secondary to an MCD diet [82,86], treated with CCl4 [87] or in LDLR−/− mice on a high fat diet [85]. Furthermore, as discussed below, clinical studies utilizing PPARγ agonists have not detected worsening of steatosis. These discrepancies once again highlight the difference between rodent and human PPARs and the limitations of applying preclinical data to humans.
Rosiglitazone has been evaluated in humans for treatment of NAFLD and NASH. One trial, conducted without a placebo control group, involved patients with biopsy-proven NASH to investigate the effects of 48 weeks of rosiglitazone. Approximately half of the patients had impaired glucose tolerance or diabetes at the start of the trial. After treatment, ALT levels decreased significantly and both HOMA-IR and QUICKI statistically improved. Histological evaluation revealed decreased steatosis, hepatocyte ballooning, and notably, 45% of patients no longer met criteria for NASH diagnosis. Although there was an improvement in zone 3 perisinusoidal fibrosis, the global fibrosis score showed no statistically significant change. Unfortunately, despite improved histological findings and improved insulin sensitivity, rosiglitazone was associated with significant weight gain in the majority of patients. Also disappointing, within six months of discontinuation of rosiglitazone, ALT levels returned to pretreatment values [88].
The FLIRT trial was a randomized, placebo-controlled trial evaluating the efficacy of rosiglitazone. Compared to placebo, patients treated with rosiglitazone were more likely to demonstrate improvement in hepatic steatosis and normalization of serum ALT. However, there was no significant improvement in other histological features such as fibrosis, hepatocyte ballooning and inflammation [89]. In its extension trial, FLIRT2, 44 patients (22 of whom were originally on placebo and 18 of whom were originally on rosiglitazone) received rosiglitazone for an additional two years [90]. For patients who were assigned to the treatment group in the FLIRT trial, two additional years of rosiglitazone treatment produced no additional benefit in measured steatosis and again there was no significant improvement in NAS, inflammation, fibrosis, or hepatocyte ballooning [90]. Thus, it is unlikely that inadequate duration of treatment is responsible for its lack of efficacy as there was no effect on inflammation or fibrosis even in the group of patients who received three years of treatment. Interestingly, as with improvement in steatosis, maximum improvement in insulin sensitivity appears to occur after one year of treatment with rosiglitazone [90]. Torres and colleagues examined the effects of rosiglitazone in a randomized, open label trial. This trial sought to compare the effects of rosiglitazone alone compared to rosiglitazone plus metformin and rosiglitazone plus losartan. All three groups demonstrated improvement in ALT and histological features from baseline. Unfortunately, none of the groups were significantly different from each other, suggesting that the addition of supplementary agents (at least in the form of metformin and losartan) is ineffective [91]. Although Torres and colleagues demonstrated histological improvement in steatosis, inflammation and fibrosis in all three groups, these effects were not compared to a placebo group.
Pioglitazone is a less potent PPARγ agonist, but clinically this agent is very effective and associated with fewer side effects [83,92,93,94,95,96]. The role of pioglitazone in NAFLD has been evaluated in rodent studies using a variety of high fat diets and has been shown to be very effective [80,97,98,99]. Pioglitazone is also efficacious in preventing the development of steatohepatitis in the MCD diet model. Compared to mice fed an MCD diet without pioglitazone, there was a significant decrease in serum ALT and total hepatic lipid content and an increase in adiponectin expression with pioglitazone treatment [99]. In a choline-deficient L-amino acid defined (CDAA) diet-induced liver fibrosis model, treatment with pioglitazone decreased hepatic triglyceride content and prevented [80] and reversed [97] hepatic fibrosis. Concomitant in vitro studies illustrated decreased activation of hepatic stellate cells treated with pioglitazone [80]. Using diet-induced and CCl4-induced hepatic fibrosis, Leclercq and colleagues showed that pioglitazone, when administered early in the course of injury, reduced fibrosis. However, later administration was not associated with improvement. Furthermore, pioglitazone may not have an effect on fibrosis secondary to bile duct ligation [100]. This suggests that pioglitazone does not have all-encompassing effects on suppressing hepatic stellate cell activation and fibrosis and that fibrosis formation and resolution is regulated by a complex and dynamic extracellular matrix milieu. The effectiveness of this compound may depend not only the cause of injury but also duration and severity of injury at the time of treatment initiation.
There have been numerous clinical trials with pioglitazone over the years. Belfort and colleagues conducted a placebo-controlled trial of pioglitazone in 55 NASH patients with impaired glucose tolerance or type 2 diabetes. Compared to placebo, patients receiving pioglitazone had improved glycemic control and glucose tolerance, lower ALT concentrations, higher adiponectin concentrations, and reduced hepatic fat content. In histological outcomes, both groups demonstrated improvement in inflammation score following treatment, but only the pioglitazone group demonstrated improvement in ballooning necrosis, steatosis and fibrosis following treatment. Of note, the fibrosis score between the placebo group and the pioglitazone group was not statistically different after treatment. The authors speculated that the duration of treatment, as well as limitations of biopsy specimens, due to the patchy nature of NAFLD, were potentially limiting factors [101].
Subsequently, a larger randomized, placebo-controlled trial treated NASH patients for 12 months with pioglitazone. Of note, diabetic patients were excluded from this study. Compared to placebo, there was statistically significant improvement in findings of hepatocellular injury (i.e., hepatocyte ballooning, apoptosis, necrosis) and Mallory bodies, but there was no significant improvement in steatosis or inflammation. Although there was a trend towards improved fibrosis, this did not reach statistical significance [102]. The PIVENS trial was a randomized, placebo-controlled, double-blind clinical trial investigating the role of pioglitazone or vitamin E for nondiabetic adults with biopsy proven NASH. Patients (247 adult men and women) were recruited and subjects received treatment for 96 weeks. Although pioglitazone significantly improved some histological features such as steatosis and inflammation, there was no benefit over placebo in the fibrosis score or the rate of improvement in NASH [103]. As a significant percentage of patients with NASH also have diabetes and diabetes represents a risk factor for rapid progression of fibrosis, the findings could be explained by the inclusion of only nondiabetic subjects in the studies.
A large randomized, controlled trial investigating the effects of pioglitazone in prediabetic and type 2 diabetic patients with NASH was recently published. After 18 months of treatment, patients assigned to the pioglitazone arm were statistically more likely to achieve resolution of NASH [104], although the effect size was modest. While there was a modest decrease in fibrosis score, pioglitazone treatment patients were not more likely show improvement in fibrosis. Additionally, consistent with previous trials involving TZDs, patients in the pioglitazone arm gained significantly more weight than those in the placebo arm [104]. Only approximately 50% of the patients enrolled in this study had diabetes and did not include a subgroup analysis, thus, the applicability of these findings remains limited.
To summarize the findings with TZDs, a recent meta-analysis sought to systematically evaluate randomized placebo-controlled trials utilizing TZDs in the treatment of patients with NASH. Four high quality randomized, placebo-controlled trials were identified and the authors concluded that compared to placebo, treatment with TZDs resulted in significant decrease in serum ALT and improvement in steatosis, inflammation, and hepatocyte ballooning; the change in fibrosis was not statistically significant [105]. When only the three studies evaluating the effectiveness of pioglitazone were included, the improvement in fibrosis became statistically significant [105] potentially suggesting that compared to rosiglitazone, pioglitazone has superior effects on reversing hepatic fibrosis. However, it should be noted that the effect was marginal. Another recent systematic review assessed three studies using pioglitazone and one study using rosiglitazone to evaluate the relationship between adiponectin levels and histological changes in TZD. The effects on other histological parameters were variable, but there was an inverse relationship between histological score and adiponectin levels, which were increased by TZDs in all three studies. This suggests that adiponectin may play a key role in histological improvement of NASH and the authors propose that development of selective PPARγ modulators (SPPARMs) that increase adiponectin levels can improve NASH without the side effects typically seen with the canonical TZDs [106]. Given these results in rodents, a clinical trial to evaluate the effects of the PPARγ-sparing TZD (MSDC-0602) on NASH has been initiated (https://clinicaltrials.gov/ct2/show/NCT02784444). For example, we have recently evaluated a TZD with very limited ability to activate PPARγ that has beneficial effects on insulin sensitivity and adiponectin concentrations in blood [107] and prevents and reverses fibrosis in a mouse model of NASH (McCommis et al., submitted). As discussed above, the less potent PPARγ agonist, pioglitazone may be superior to rosiglitazone for suppressing fibrosis. Though antithetical to the dogma that TZDs function as PPARγ agonists, this could suggest that there are also PPARγ-independent effects of TZDs on these parameters [108].
To summarize, clinical trials utilizing TZDs have indicated significant improvement in hepatic steatosis and inflammation. The ability to reverse fibrosis is less impressive, but may be significant in pioglitazone as opposed to rosiglitazone. However, concerns regarding weight gain and other dose-limiting side effects remain.
5. Dual agonists
Because the PPAR isoforms have large, relatively non-selective ligand binding pockets and significant sequence homology, PPAR agonists with binding affinity for multiple isoforms, termed dual agonists or pan agonists, have been identified and may represent interesting therapeutic targets.
Several PPARα/γ dual agonists, termed glitazars, have been developed and shown to improve insulin resistance, dyslipidemia [109,110], and fatty liver [111] in rodent studies. However, these drugs were abandoned before reaching markets due to cardiovascular [112,113] and renal [114] side effects [65]. It is possible that this is a class effect that will limit the utility of these drugs for treating fatty liver disease. Nevertheless, a novel PPARα/γ agonist (saroglitazar) has been approved for use in India and there is a registered prospective, randomized clinical trial comparing saroglitazar with pioglitazone in NAFLD patients (GLAZED) (https://clinicaltrials.gov/ct2/show/NCT02265276). The study began in October 2014 and was projected to end in September 2015. No published study results are currently available.
Bezafibrate is a structural member of the fibrate class of PPARα agonists that is believed to act as a pan-agonist with binding affinity for PPARα and PPARδ and to a lesser extent, PPARγ [115]. Bezafibrate has been evaluated in rodent models and demonstrated to prevent the development of steatohepatitis induced by an MCD deficient diet. Treatment led to decreased ALT, hepatic triglyceride and lipoperoxide content, and increased plasma adiponectin concentration. Bezafibrate also increased expression of genes involved in fatty acid metabolism and oxidation, such as acyl-CoA oxidase, carnitine palmitoyltransferase-1, and liver fatty acid binding protein. It has been suggested that its beneficial effects are mediated through increased insulin sensitivity, fatty acid oxidation, and decreased inflammation [62]. The same group published similar findings in MCD diet-fed KK-A(y) mice [116]. Bezafibrate reduced hepatic inflammation and fibrosis and suppressed expression of inflammatory cytokine and profibrogenic genes in vivo and in stimulated stellate cells in culture. To our knowledge, the effects of bezafibrate on NASH in humans have not been specifically studied.
There have also been a few studies evaluating a novel dual PPARα/δ agonist, GFT505, for efficacy in treating NASH. Using various rodent models of NASH, including western diet-fed ApoE2KI mice, carbon tetrachloride, and db/db mice fed an MCD diet, treatment with GFT505 demonstrated improvement in histologic measures of NASH, decreased hepatic triglyceride content, and diminished expression of inflammatory cytokine, and fibrosis markers [117]. Interestingly, the use of GFT505 also prevented steatohepatitis and decreased expression of inflammatory and fibrosis genes in western diet fed hApoE2 KI/PPARα KO mice, demonstrating that these effects can be mediated independently of PPARα. However, GFT505 treatment did not improve plasma triglyceride or free fatty acid levels in the knockouts, thus suggesting both PPARα-independent and -dependent effects [117]. The contribution of PPARδ to the PPARα-independent effects is not entirely clear. GFT505 has also been used in humans to evaluate potential metabolic benefits. In a double blind, placebo-controlled trial involving 47 patients with prediabetes, Cariou and colleagues administered GFT505 and found a significant reduction in GGT and ALT compared to placebo control. However, improvement in NAFLD was not a defined primary outcome and no liver biopsies were obtained. Thus, the clinical significance in the reduction of ALT and GGT remains unclear [118,119]. The investigation of PPAR dual or pan-agonists as a potential treatment option for NAFLD is still in its early stages, but preliminary data suggest possible beneficial effects.
6. Conclusion
NAFLD represents a significant unmet medical need that is emerging as a major public health problem. Left unchecked, it can progress to cirrhosis and hepatocellular carcinoma. As the public health crisis of obesity worsens, NAFLD is being increasingly recognized and diagnosed. In the absence of treatment, a substantial percentage of patients will develop significant liver disease, which is associated with increased morbidity and mortality. Studies conducted in rodents and early clinical trials in humans suggest that PPAR agonists are protective against NASH through various mechanisms including inducing expression of genes involved in beta oxidation, decreasing inflammation, decreasing oxidative stress, and increasing the secretion of beneficial adipokines like adiponectin. Although PPARα agonists seemed promising in animal studies, they were not effective in clinical trials. PPARδ agonists may also be promising but data from human trials is extremely limited. PPARγ agonists have been more successful in improving histological outcomes including fibrosis in clinical trials. However, the widespread use of thiazolidinediones is limited by side effects such as weight gain, edema, bone loss, and increased risk of bladder cancer and cardiovascular complications. Lastly, the use of agonists that activate more than one PPAR has also yielded promising results, but with the caveat that side effects with dual agonists have been noted. It remains unclear what is the model profile and ratio of PPAR agonism for each isoform as well. Thus, an ideal agent remains to be discovered and validated.
Highlights.
Nonalcoholic fatty liver disease is the most common cause of liver disease.
Without intervention NAFLD can progress to end-stage liver disease.
No therapeutic agents are available for the treatment of NASH.
Peroxisome proliferator-activated receptors regulate lipid and glucose metabolism.
We discuss studies evaluating the efficacy of PPAR agonists for treatment of NASH.
Acknowledgments
Dr. Finck is supported by NIH grants R01 DK078187 and R01 DK104735. Dr. Liss is supported by the NIH training grant T32 DK077653.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brunt EM, Wong VW, Nobili V, Day CP, Sookoian S, Maher JJ, Bugianesi E, Sirlin CB, Neuschwander-Tetri BA, Rinella ME. Nonalcoholic fatty liver disease. Nat Rev Dis Primers. 2015;1:15080. doi: 10.1038/nrdp.2015.80. [DOI] [PubMed] [Google Scholar]
- 2.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–2273. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- 3.Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118:1388–1393. doi: 10.1542/peds.2006-1212. [DOI] [PubMed] [Google Scholar]
- 4.Younossi ZM, Blissett D, Blissett R, Henry L, Stepanova M, Younossi Y, Racila A, Hunt S, Beckerman R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology. 2016 doi: 10.1002/hep.28785. [DOI] [PubMed] [Google Scholar]
- 5.Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 6.Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 7.Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology. 2011;141:1249–1253. doi: 10.1053/j.gastro.2011.06.061. [DOI] [PubMed] [Google Scholar]
- 8.Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005;42:132–138. doi: 10.1016/j.jhep.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 9.Patton HM, Yates K, Unalp-Arida A, Behling CA, Huang TT, Rosenthal P, Sanyal AJ, Schwimmer JB, Lavine JE. Association between metabolic syndrome and liver histology among children with nonalcoholic Fatty liver disease. Am J Gastroenterol. 2010;105:2093–2102. doi: 10.1038/ajg.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 12.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, Vogt TF, Hobbs HH, Cohen JC. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46:352–356. doi: 10.1038/ng.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chalasani N, Guo X, Loomba R, Goodarzi MO, Haritunians T, Kwon S, Cui J, Taylor KD, Wilson L, Cummings OW, Chen YD, Rotter JI, N. Nonalcoholic Steatohepatitis Clinical Research Genome-wide association study identifies variants associated with histologic features of nonalcoholic Fatty liver disease. Gastroenterology. 2010;139:1567–1576. 1576e1561–1566. doi: 10.1053/j.gastro.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.London RM, George J. Pathogenesis of NASH: animal models. Clin Liver Dis. 2007;11:55–74. viii. doi: 10.1016/j.cld.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 16.Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8:35–44. doi: 10.1038/nrgastro.2010.191. [DOI] [PubMed] [Google Scholar]
- 17.Maher JJ. Modeling fatty liver disease in animals: Is there an optimal approach, and is the effort worthwhile? Hepatology. 2016 doi: 10.1002/hep.28823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, Friedman SL, Diago M, Romero-Gomez M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology. 2015;149:367–378 e365. doi: 10.1053/j.gastro.2015.04.005. quiz e314–365. [DOI] [PubMed] [Google Scholar]
- 19.Cave MC, Clair HB, Hardesty JE, Falkner KC, Feng W, Clark BJ, Sidey J, Shi H, Aqel BA, McClain CJ, Prough RA. Nuclear receptors and nonalcoholic fatty liver disease. Biochim Biophys Acta. 2016;1859:1083–1099. doi: 10.1016/j.bbagrm.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta. 2012;1821:809–818. doi: 10.1016/j.bbalip.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 21.Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015;62:720–733. doi: 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- 22.Motojima K, Passilly P, Peters JM, Gonzalez FJ, Latruffe N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor alpha and gamma activators in a tissue- and inducer-specific manner. J Biol Chem. 1998;273:16710–16714. doi: 10.1074/jbc.273.27.16710. [DOI] [PubMed] [Google Scholar]
- 23.Yu K, Bayona W, Kallen CB, Harding HP, Ravera CP, McMahon G, Brown M, Lazar MA. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J Biol Chem. 1995;270:23975–23983. doi: 10.1074/jbc.270.41.23975. [DOI] [PubMed] [Google Scholar]
- 24.Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23:351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Takeuchi S, Matsuda T, Kobayashi S, Takahashi T, Kojima H. In vitro screening of 200 pesticides for agonistic activity via mouse peroxisome proliferator-activated receptor (PPAR)alpha and PPARgamma and quantitative analysis of in vivo induction pathway. Toxicol Appl Pharmacol. 2006;217:235–244. doi: 10.1016/j.taap.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 26.Corton JC. Evaluation of the role of peroxisome proliferator-activated receptor alpha (PPARalpha) in mouse liver tumor induction by trichloroethylene and metabolites. Crit Rev Toxicol. 2008;38:857–875. doi: 10.1080/10408440802209796. [DOI] [PubMed] [Google Scholar]
- 27.Laughter AR, Dunn CS, Swanson CL, Howroyd P, Cattley RC, Corton JC. Role of the peroxisome proliferator-activated receptor alpha (PPARalpha) in responses to trichloroethylene and metabolites, trichloroacetate and dichloroacetate in mouse liver. Toxicology. 2004;203:83–98. doi: 10.1016/j.tox.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 28.Fisher CD, Jackson JP, Lickteig AJ, Augustine LM, Cherrington NJ. Drug metabolizing enzyme induction pathways in experimental non-alcoholic steatohepatitis. Arch Toxicol. 2008;82:959–964. doi: 10.1007/s00204-008-0312-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sznaidman ML, Haffner CD, Maloney PR, Fivush A, Chao E, Goreham D, Sierra ML, LeGrumelec C, Xu HE, Montana VG, Lambert MH, Willson TM, Oliver WR, Jr, Sternbach DD. Novel selective small molecule agonists for peroxisome proliferator-activated receptor delta (PPARdelta)–synthesis and biological activity. Bioorg Med Chem Lett. 2003;13:1517–1521. doi: 10.1016/s0960-894x(03)00207-5. [DOI] [PubMed] [Google Scholar]
- 30.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 31.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 32.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 33.Holden PR, Tugwood JD. Peroxisome proliferator-activated receptor alpha: role in rodent liver cancer and species differences. J Mol Endocrinol. 1999;22:1–8. doi: 10.1677/jme.0.0220001. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez FJ, Shah YM. PPARalpha: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology. 2008;246:2–8. doi: 10.1016/j.tox.2007.09.030. [DOI] [PubMed] [Google Scholar]
- 35.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci. 2004;61:393–416. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanden Berghe W, Vermeulen L, Delerive P, De Bosscher K, Staels B, Haegeman G. A paradigm for gene regulation: inflammation, NF-kappaB and PPAR. Adv Exp Med Biol. 2003;544:181–196. doi: 10.1007/978-1-4419-9072-3_22. [DOI] [PubMed] [Google Scholar]
- 38.Patsouris D, Reddy JK, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology. 2006;147:1508–1516. doi: 10.1210/en.2005-1132. [DOI] [PubMed] [Google Scholar]
- 39.Kim S, Sohn I, Ahn JI, Lee KH, Lee YS, Lee YS. Hepatic gene expression profiles in a long-term high-fat diet-induced obesity mouse model. Gene. 2004;340:99–109. doi: 10.1016/j.gene.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 40.Gregoire FM, Zhang Q, Smith SJ, Tong C, Ross D, Lopez H, West DB. Diet-induced obesity and hepatic gene expression alterations in C57BL/6J and ICAM-1-deficient mice. Am J Physiol Endocrinol Metab. 2002;282:E703–713. doi: 10.1152/ajpendo.00072.2001. [DOI] [PubMed] [Google Scholar]
- 41.Redonnet A, Groubet R, Noel-Suberville C, Bonilla S, Martinez A, Higueret P. Exposure to an obesity-inducing diet early affects the pattern of expression of peroxisome proliferator, retinoic acid, and triiodothyronine nuclear receptors in the rat. Metabolism. 2001;50:1161–1167. doi: 10.1053/meta.2001.26759. [DOI] [PubMed] [Google Scholar]
- 42.Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, Lefebvre P, Taskinen MR, Van Hul W, Mertens I, Hubens G, Van Marck E, Michielsen P, Van Gaal L, Staels B. PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J Hepatol. 2015;63:164–173. doi: 10.1016/j.jhep.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 43.Stienstra R, Mandard S, Patsouris D, Maass C, Kersten S, Muller M. Peroxisome proliferator-activated receptor alpha protects against obesity-induced hepatic inflammation. Endocrinology. 2007;148:2753–2763. doi: 10.1210/en.2007-0014. [DOI] [PubMed] [Google Scholar]
- 44.Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J Nutr. 2011;141:603–610. doi: 10.3945/jn.110.135210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stienstra R, Duval C, Muller M, Kersten S. PPARs, Obesity, and Inflammation. PPAR Res. 2007;2007:95974. doi: 10.1155/2007/95974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38:123–132. doi: 10.1053/jhep.2003.50307. [DOI] [PubMed] [Google Scholar]
- 47.Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39:1286–1296. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 48.Shiri-Sverdlov R, Wouters K, van Gorp PJ, Gijbels MJ, Noel B, Buffat L, Staels B, Maeda N, van Bilsen M, Hofker MH. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J Hepatol. 2006;44:732–741. doi: 10.1016/j.jhep.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 49.Zhang N, Lu Y, Shen X, Bao Y, Cheng J, Chen L, Li B, Zhang Q. Fenofibrate treatment attenuated chronic endoplasmic reticulum stress in the liver of nonalcoholic fatty liver disease mice. Pharmacology. 2015;95:173–180. doi: 10.1159/000380952. [DOI] [PubMed] [Google Scholar]
- 50.Abd El-Haleim EA, Bahgat AK, Saleh S. Resveratrol and fenofibrate ameliorate fructose-induced nonalcoholic steatohepatitis by modulation of genes expression. World J Gastroenterol. 2016;22:2931–2948. doi: 10.3748/wjg.v22.i10.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laurin J, Lindor KD, Crippin JS, Gossard A, Gores GJ, Ludwig J, Rakela J, McGill DB. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology. 1996;23:1464–1467. doi: 10.1002/hep.510230624. [DOI] [PubMed] [Google Scholar]
- 52.Fernandez-Miranda C, Perez-Carreras M, Colina F, Lopez-Alonso G, Vargas C, Solis-Herruzo JA. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis. 2008;40:200–205. doi: 10.1016/j.dld.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 53.Basaranoglu M, Acbay O, Sonsuz A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J Hepatol. 1999;31:384. doi: 10.1016/s0168-8278(99)80243-8. [DOI] [PubMed] [Google Scholar]
- 54.Hertz R, Bar-Tana J. Peroxisome proliferator-activated receptor (PPAR) alpha activation and its consequences in humans. Toxicol Lett. 1998;102–103:85–90. doi: 10.1016/s0378-4274(98)00290-2. [DOI] [PubMed] [Google Scholar]
- 55.Palmer CN, Hsu MH, Griffin KJ, Raucy JL, Johnson EF. Peroxisome proliferator activated receptor-alpha expression in human liver. Mol Pharmacol. 1998;53:14–22. [PubMed] [Google Scholar]
- 56.Rakhshandehroo M, Hooiveld G, Muller M, Kersten S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS One. 2009;4:e6796. doi: 10.1371/journal.pone.0006796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat Rev Cancer. 2012;12:181–195. doi: 10.1038/nrc3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, Watanabe M, Magoori K, Ioka RX, Tachibana K, Watanabe Y, Uchiyama Y, Sumi K, Iguchi H, Ito S, Doi T, Hamakubo T, Naito M, Auwerx J, Yanagisawa M, Kodama T, Sakai J. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci U S A. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mackenzie LS, Lione L. Harnessing the benefits of PPARbeta/delta agonists. Life Sci. 2013;93:963–967. doi: 10.1016/j.lfs.2013.10.022. [DOI] [PubMed] [Google Scholar]
- 60.Shan W, Palkar PS, Murray IA, McDevitt EI, Kennett MJ, Kang BH, Isom HC, Perdew GH, Gonzalez FJ, Peters JM. Ligand activation of peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) attenuates carbon tetrachloride hepatotoxicity by downregulating proinflammatory gene expression. Toxicol Sci. 2008;105:418–428. doi: 10.1093/toxsci/kfn142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shan W, Nicol CJ, Ito S, Bility MT, Kennett MJ, Ward JM, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-beta/delta protects against chemically induced liver toxicity in mice. Hepatology. 2008;47:225–235. doi: 10.1002/hep.21925. [DOI] [PubMed] [Google Scholar]
- 62.Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, Yamazaki Y, Kuroda J, Shibata N. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 63.Riserus U, Sprecher D, Johnson T, Olson E, Hirschberg S, Liu A, Fang Z, Hegde P, Richards D, Sarov-Blat L, Strum JC, Basu S, Cheeseman J, Fielding BA, Humphreys SM, Danoff T, Moore NR, Murgatroyd P, O’Rahilly S, Sutton P, Willson T, Hassall D, Frayn KN, Karpe F. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes. 2008;57:332–339. doi: 10.2337/db07-1318. [DOI] [PubMed] [Google Scholar]
- 64.Peters JM, Gonzalez FJ, Muller R. Establishing the Role of PPARbeta/delta in Carcinogenesis. Trends Endocrinol Metab. 2015;26:595–607. doi: 10.1016/j.tem.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sahebkar A, Chew GT, Watts GF. New peroxisome proliferator-activated receptor agonists: potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin Pharmacother. 2014;15:493–503. doi: 10.1517/14656566.2014.876992. [DOI] [PubMed] [Google Scholar]
- 66.Lee MY, Choi R, Kim HM, Cho EJ, Kim BH, Choi YS, Naowaboot J, Lee EY, Yang YC, Shin JY, Shin YG, Chung CH. Peroxisome proliferator-activated receptor delta agonist attenuates hepatic steatosis by anti-inflammatory mechanism. Exp Mol Med. 2012;44:578–585. doi: 10.3858/emm.2012.44.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bays HE, Schwartz S, Littlejohn T, 3rd, Kerzner B, Krauss RM, Karpf DB, Choi YJ, Wang X, Naim S, Roberts BK. MBX-8025, a novel peroxisome proliferator receptor-delta agonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J Clin Endocrinol Metab. 2011;96:2889–2897. doi: 10.1210/jc.2011-1061. [DOI] [PubMed] [Google Scholar]
- 68.Zhu Y, Alvares K, Huang Q, Rao MS, Reddy JK. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J Biol Chem. 1993;268:26817–26820. [PubMed] [Google Scholar]
- 69.Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, Rao MS, Gonzalez FJ, Reddy JK. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J Biol Chem. 2003;278:498–505. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- 70.Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, Nicol CJ, Vinson C, Gonzalez FJ, Reitman ML. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278:34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- 71.Pettinelli P, Videla LA. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J Clin Endocrinol Metab. 2011;96:1424–1430. doi: 10.1210/jc.2010-2129. [DOI] [PubMed] [Google Scholar]
- 72.Nakamuta M, Kohjima M, Morizono S, Kotoh K, Yoshimoto T, Miyagi I, Enjoji M. Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med. 2005;16:631–635. [PubMed] [Google Scholar]
- 73.Moran-Salvador E, Lopez-Parra M, Garcia-Alonso V, Titos E, Martinez-Clemente M, Gonzalez-Periz A, Lopez-Vicario C, Barak Y, Arroyo V, Claria J. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011;25:2538–2550. doi: 10.1096/fj.10-173716. [DOI] [PubMed] [Google Scholar]
- 74.Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B, Jr, Reitman ML, Gonzalez FJ. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest. 2003;111:737–747. doi: 10.1172/JCI17223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hall AM, Soufi N, Chambers KT, Chen Z, Schweitzer GG, McCommis KS, Erion DM, Graham MJ, Su X, Finck BN. Abrogating monoacylglycerol acyltransferase activity in liver improves glucose tolerance and hepatic insulin signaling in obese mice. Diabetes. 2014;63:2284–2296. doi: 10.2337/db13-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, Nagaretani H, Matsuda M, Komuro R, Ouchi N, Kuriyama H, Hotta K, Nakamura T, Shimomura I, Matsuzawa Y. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50:2094–2099. doi: 10.2337/diabetes.50.9.2094. [DOI] [PubMed] [Google Scholar]
- 77.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 78.Esposito K, Ciotola M, Carleo D, Schisano B, Saccomanno F, Sasso FC, Cozzolino D, Assaloni R, Merante D, Ceriello A, Giugliano D. Effect of rosiglitazone on endothelial function and inflammatory markers in patients with the metabolic syndrome. Diabetes Care. 2006;29:1071–1076. doi: 10.2337/diacare.2951071. [DOI] [PubMed] [Google Scholar]
- 79.Esposito K, Ciotola M, Merante D, Giugliano D. Rosiglitazone cools down inflammation in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2006;26:1413–1414. doi: 10.1161/01.ATV.0000223874.94624.11. [DOI] [PubMed] [Google Scholar]
- 80.Kawaguchi K, Sakaida I, Tsuchiya M, Omori K, Takami T, Okita K. Pioglitazone prevents hepatic steatosis, fibrosis, and enzyme-altered lesions in rat liver cirrhosis induced by a choline-deficient L-amino acid-defined diet. Biochem Biophys Res Commun. 2004;315:187–195. doi: 10.1016/j.bbrc.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 81.Nan YM, Fu N, Wu WJ, Liang BL, Wang RQ, Zhao SX, Zhao JM, Yu J. Rosiglitazone prevents nutritional fibrosis and steatohepatitis in mice. Scand J Gastroenterol. 2009;44:358–365. doi: 10.1080/00365520802530861. [DOI] [PubMed] [Google Scholar]
- 82.Yu J, Zhang S, Chu ES, Go MY, Lau RH, Zhao J, Wu CW, Tong L, Zhao J, Poon TC, Sung JJ. Peroxisome proliferator-activated receptors gamma reverses hepatic nutritional fibrosis in mice and suppresses activation of hepatic stellate cells in vitro. Int J Biochem Cell Biol. 2010;42:948–957. doi: 10.1016/j.biocel.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 83.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 84.Garcia-Ruiz I, Rodriguez-Juan C, Diaz-Sanjuan T, Martinez MA, Munoz-Yague T, Solis-Herruzo JA. Effects of rosiglitazone on the liver histology and mitochondrial function in ob/ob mice. Hepatology. 2007;46:414–423. doi: 10.1002/hep.21687. [DOI] [PubMed] [Google Scholar]
- 85.Gupte AA, Liu JZ, Ren Y, Minze LJ, Wiles JR, Collins AR, Lyon CJ, Pratico D, Finegold MJ, Wong ST, Webb P, Baxter JD, Moore DD, Hsueh WA. Rosiglitazone attenuates age- and diet-associated nonalcoholic steatohepatitis in male low-density lipoprotein receptor knockout mice. Hepatology. 2010;52:2001–2011. doi: 10.1002/hep.23941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nan YM, Han F, Kong LB, Zhao SX, Wang RQ, Wu WJ, Yu J. Adenovirus-mediated peroxisome proliferator activated receptor gamma overexpression prevents nutritional fibrotic steatohepatitis in mice. Scand J Gastroenterol. 2011;46:358–369. doi: 10.3109/00365521.2010.525717. [DOI] [PubMed] [Google Scholar]
- 87.Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati-Baroni G, Ridolfi F, Trozzi L, Surrenti C, Casini A. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122:1924–1940. doi: 10.1053/gast.2002.33666. [DOI] [PubMed] [Google Scholar]
- 88.Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology. 2003;38:1008–1017. doi: 10.1053/jhep.2003.50420. [DOI] [PubMed] [Google Scholar]
- 89.Ratziu V, Giral P, Jacqueminet S, Charlotte F, Hartemann-Heurtier A, Serfaty L, Podevin P, Lacorte JM, Bernhardt C, Bruckert E, Grimaldi A, Poynard T, Group LS. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135:100–110. doi: 10.1053/j.gastro.2008.03.078. [DOI] [PubMed] [Google Scholar]
- 90.Ratziu V, Charlotte F, Bernhardt C, Giral P, Halbron M, Lenaour G, Hartmann-Heurtier A, Bruckert E, Poynard T, Group LS. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51:445–453. doi: 10.1002/hep.23270. [DOI] [PubMed] [Google Scholar]
- 91.Torres DM, Jones FJ, Shaw JC, Williams CD, Ward JA, Harrison SA. Rosiglitazone versus rosiglitazone and metformin versus rosiglitazone and losartan in the treatment of nonalcoholic steatohepatitis in humans: a 12-month randomized, prospective, open- label trial. Hepatology. 2011;54:1631–1639. doi: 10.1002/hep.24558. [DOI] [PubMed] [Google Scholar]
- 92.Juurlink DN, Gomes T, Lipscombe LL, Austin PC, Hux JE, Mamdani MM. Adverse cardiovascular events during treatment with pioglitazone and rosiglitazone: population based cohort study. BMJ. 2009;339:b2942. doi: 10.1136/bmj.b2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Starner CI, Schafer JA, Heaton AH, Gleason PP. Rosiglitazone and pioglitazone utilization from January 2007 through May 2008 associated with five risk-warning events. J Manag Care Pharm. 2008;14:523–531. doi: 10.18553/jmcp.2008.14.6.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Balkrishnan R, Arondekar BV, Camacho FT, Shenolikar RA, Horblyuk R, Anderson RT. Comparisons of rosiglitazone versus pioglitazone monotherapy introduction and associated health care utilization in medicaid-enrolled patients with type 2 diabetes mellitus. Clin Ther. 2007;29:1306–1315. Spec No. [PubMed] [Google Scholar]
- 95.Beysen C, Murphy EJ, Nagaraja H, Decaris M, Riiff T, Fong A, Hellerstein MK, Boyle PJ. A pilot study of the effects of pioglitazone and rosiglitazone on de novo lipogenesis in type 2 diabetes. J Lipid Res. 2008;49:2657–2663. doi: 10.1194/jlr.M800165-JLR200. [DOI] [PubMed] [Google Scholar]
- 96.de Vries CS, Russell-Jones DL. Rosiglitazone or pioglitazone in type 2 diabetes? BMJ. 2009;339:b3076. doi: 10.1136/bmj.b3076. [DOI] [PubMed] [Google Scholar]
- 97.Uto H, Nakanishi C, Ido A, Hasuike S, Kusumoto K, Abe H, Numata M, Nagata K, Hayashi K, Tsubouchi H. The peroxisome proliferator-activated receptor-gamma agonist, pioglitazone, inhibits fat accumulation and fibrosis in the livers of rats fed a choline-deficient, l-amino acid-defined diet. Hepatol Res. 2005;32:235–242. doi: 10.1016/j.hepres.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 98.Hsiao PJ, Hsieh TJ, Kuo KK, Hung WW, Tsai KB, Yang CH, Yu ML, Shin SJ. Pioglitazone retrieves hepatic antioxidant DNA repair in a mice model of high fat diet. BMC Mol Biol. 2008;9:82. doi: 10.1186/1471-2199-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Leclercq IA, Lebrun VA, Starkel P, Horsmans YJ. Intrahepatic insulin resistance in a murine model of steatohepatitis: effect of PPARgamma agonist pioglitazone. Lab Invest. 2007;87:56–65. doi: 10.1038/labinvest.3700489. [DOI] [PubMed] [Google Scholar]
- 100.Leclercq IA, Sempoux C, Starkel P, Horsmans Y. Limited therapeutic efficacy of pioglitazone on progression of hepatic fibrosis in rats. Gut. 2006;55:1020–1029. doi: 10.1136/gut.2005.079194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, Berria R, Ma JZ, Dwivedi S, Havranek R, Fincke C, DeFronzo R, Bannayan GA, Schenker S, Cusi K. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 102.Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, Austin AS, Freeman JG, Morgan L, Webber J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1176–1184. doi: 10.1053/j.gastro.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 103.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR, Nash CRN. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, Tio F, Hardies J, Darland C, Musi N, Webb A, Portillo-Sanchez P. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann Intern Med. 2016;165:305–315. doi: 10.7326/M15-1774. [DOI] [PubMed] [Google Scholar]
- 105.Boettcher E, Csako G, Pucino F, Wesley R, Loomba R. Meta-analysis: pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2012;35:66–75. doi: 10.1111/j.1365-2036.2011.04912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Polyzos SA, Mantzoros CS. Adiponectin as a target for the treatment of nonalcoholic steatohepatitis with thiazolidinediones: A systematic review. Metabolism. 2016;65:1297–1306. doi: 10.1016/j.metabol.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 107.Chen Z, Vigueira PA, Chambers KT, Hall AM, Mitra MS, Qi N, McDonald WG, Colca JR, Kletzien RF, Finck BN. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator-activated receptor gamma-sparing thiazolidinedione. J Biol Chem. 2012;287:23537–23548. doi: 10.1074/jbc.M112.363960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, Kletzien RF, Burgess SC, Finck BN. Loss of Mitochondrial Pyruvate Carrier 2 in the Liver Leads to Defects in Gluconeogenesis and Compensation via Pyruvate-Alanine Cycling. Cell Metab. 2015;22:682–694. doi: 10.1016/j.cmet.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brand CL, Sturis J, Gotfredsen CF, Fleckner J, Fledelius C, Hansen BF, Andersen B, Ye JM, Sauerberg P, Wassermann K. Dual PPARalpha/gamma activation provides enhanced improvement of insulin sensitivity and glycemic control in ZDF rats. Am J Physiol Endocrinol Metab. 2003;284:E841–854. doi: 10.1152/ajpendo.00348.2002. [DOI] [PubMed] [Google Scholar]
- 110.Harrity T, Farrelly D, Tieman A, Chu C, Kunselman L, Gu L, Ponticiello R, Cap M, Qu F, Shao C, Wang W, Zhang H, Fenderson W, Chen S, Devasthale P, Jeon Y, Seethala R, Yang WP, Ren J, Zhou M, Ryono D, Biller S, Mookhtiar KA, Wetterau J, Gregg R, Cheng PT, Hariharan N. Muraglitazar, a novel dual (alpha/gamma) peroxisome proliferator-activated receptor activator, improves diabetes and other metabolic abnormalities and preserves beta-cell function in db/db mice. Diabetes. 2006;55:240–248. [PubMed] [Google Scholar]
- 111.Ye JM, Iglesias MA, Watson DG, Ellis B, Wood L, Jensen PB, Sorensen RV, Larsen PJ, Cooney GJ, Wassermann K, Kraegen EW. PPARalpha/gamma ragaglitazar eliminates fatty liver and enhances insulin action in fat-fed rats in the absence of hepatomegaly. Am J Physiol Endocrinol Metab. 2003;284:E531–540. doi: 10.1152/ajpendo.00299.2002. [DOI] [PubMed] [Google Scholar]
- 112.Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA. 2005;294:2581–2586. doi: 10.1001/jama.294.20.joc50147. [DOI] [PubMed] [Google Scholar]
- 113.Henry RR, Lincoff AM, Mudaliar S, Rabbia M, Chognot C, Herz M. Effect of the dual peroxisome proliferator-activated receptor-alpha/gamma agonist aleglitazar on risk of cardiovascular disease in patients with type 2 diabetes (SYNCHRONY): a phase II, randomised, dose-ranging study. Lancet. 2009;374:126–135. doi: 10.1016/S0140-6736(09)60870-9. [DOI] [PubMed] [Google Scholar]
- 114.Hamren B, Ohman KP, Svensson MK, Karlsson MO. Pharmacokinetic-pharmacodynamic assessment of the interrelationships between tesaglitazar exposure and renal function in patients with type 2 diabetes mellitus. J Clin Pharmacol. 2012;52:1317–1327. doi: 10.1177/0091270011416937. [DOI] [PubMed] [Google Scholar]
- 115.Peters JM, Aoyama T, Burns AM, Gonzalez FJ. Bezafibrate is a dual ligand for PPARalpha and PPARbeta: studies using null mice. Biochim Biophys Acta. 2003;1632:80–89. doi: 10.1016/s1388-1981(03)00065-9. [DOI] [PubMed] [Google Scholar]
- 116.Nakano S, Nagasawa T, Ijiro T, Inada Y, Tamura T, Maruyama K, Kuroda J, Yamazaki Y, Kusama H, Shibata N. Bezafibrate prevents hepatic stellate cell activation and fibrogenesis in a murine steatohepatitis model, and suppresses fibrogenic response induced by transforming growth factor-beta1 in a cultured stellate cell line. Hepatol Res. 2008;38:1026–1039. doi: 10.1111/j.1872-034X.2008.00363.x. [DOI] [PubMed] [Google Scholar]
- 117.Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, Baron M, Lucas A, Tailleux A, Hum DW, Ratziu V, Cariou B, Hanf R. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2013;58:1941–1952. doi: 10.1002/hep.26461. [DOI] [PubMed] [Google Scholar]
- 118.Cariou B, Hanf R, Lambert-Porcheron S, Zair Y, Sauvinet V, Noel B, Flet L, Vidal H, Staels B, Laville M. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care. 2013;36:2923–2930. doi: 10.2337/dc12-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cariou B, Zair Y, Staels B, Bruckert E. Effects of the new dual PPAR alpha/delta agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care. 2011;34:2008–2014. doi: 10.2337/dc11-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]