

Abstract
We describe the rational design of a linked, bis-thiourea catalyst with enhanced activity relative to monomeric analogs in a representative enantioselective anion-abstraction reaction. Mechanistic insights guide development of this linking strategy to favor substrate activation though the intramolecular cooperation of two thiourea subunits while avoiding nonproductive aggregation. The resulting catalyst platform overcomes many of the practical limitations that have plagued hydrogen-bond donor catalysis and enables use of catalyst loadings as low as 0.05 mol %. Computational analyses of possible anion-binding modes provide detailed insight into the precise mechanism of anion-abstraction catalysis with this pseudo-dimeric thiourea.
Graphical abstract
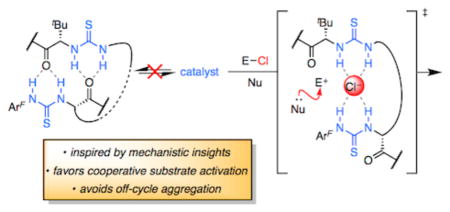
Chiral dual hydrogen-bond (H-bond) donors such as urea, thiourea, and squaramide derivatives comprise an important class of enantioselective catalysts for reactions proceeding through ion-pair intermediates.1,2 While these air- and moisture-stable organocatalysts have enabled the development of numerous, highly enantioselective transformations, these methods typically suffer from several practical constraints that impede widespread application. These include requirements for high catalyst loadings (5–20 mol %), long reaction times (≥ 24 h), and dilute reaction conditions (≤0.1 M in substrate) to achieve maximal levels of enantioinduction.3
In an effort to understand the basis for these limitations and thereby develop strategies to overcome them, we recently undertook a detailed mechanistic study of a representative transformation involving anion-abstraction catalysis by a chiral amido-thiourea (1, Figure 1A).2c,4,5 This analysis revealed that thiourea catalyst 1 forms nonproductive dimeric aggregates in the resting state; these aggregates must dissociate before two molecules of catalyst recombine with substrates to promote rate- and enantioselectivity-determining C–C bond formation (Figure 1B).4a We hypothesized that appropriate linkage of the monomers should result in stabilization of the dominant transition-state complex and, therefore, enhanced catalytic activity. The linked units would also need to adopt a relative orientation closely resembling that accessed by the untethered monomers in order to promote reactions with similar levels of enantioselectivity. Analogous linking strategies have afforded reactivity enhancements in other systems displaying a bimolecular dependence on chiral catalyst.6 However, this case poses an additional challenge, because the tether must also disfavor unproductive ground-state aggregation. Herein, we describe the mechanism-guided development of a highly active bis-thiourea catalyst for anion-abstraction catalysis. In addition to providing a platform for future catalyst design, the linking strategy validates computational analyses shedding light on the cooperative mechanism of anion abstraction.
Figure 1.

(A–C.) Anion-abstraction catalysis involving the cooperative action of two amido-thiourea catalysts. Ar = 4-fluorophenyl, ArF = 3,5-bis(trifluoromethyl)phenyl.
While the cooperative action of thioureas such as 1 in anion-abstraction catalysis has been well established, uncertainty remains regarding the precise mode of electrophile activation by two catalyst units. Two different cooperative arrangements, which we term “2H-” and “4H-” anion abstraction (Figure 1C), are consistent with experimental data and kinetically indistinguishable.7 Furthermore, preliminary computational analyses of [1]2•Cl− complexes using density functional theory (DFT) did not reveal a clear energetic preference for one over the other.4 We postulated that dimeric catalysts designed to enable Cl− abstraction through only one of these two arrangements could help reveal which of the two activation modes is operative in the enantioselective alkylation of α-chloroisochroman and in related anion-binding pathways promoted by this class of chiral H-bond donor. Given that these catalysts have been shown to form [cat]2•NMe4Cl complexes with a 4H-Cl−-binding geometry in the solid state (Figure 2A),4,8 we chose to target linkage strategies that would favor the 4H-activation mode. Introduction of a methyl substituent in the arylpyrrolidine component of 1 has been demonstrated to confer improved reactivity and enantioselectivity by minimizing competing reaction pathways,8 and accordingly, we used this modified structure (5a) as the starting point for further catalyst design.
Figure 2.

(A–C.) Identification of a linking strategy for bis-thiourea anion-abstraction catalysts. Carbon atoms represented in silver (for 5), slate blue (for 6), or light pink (for 7). Linkage sites highlighted in cyan. Carbon-bound hydrogen atoms omitted for clarity. N = blue, O = red, F = lime, Si = gold, S = yellow, Cl = magenta. Computations performed with B3LYP-D3(BJ)/6-31G(d,p)/PCM(toluene)//B3LYP/6-31G(d). Corrected free energies reported relative to the lowest energy transition state identified with each catalyst. See Supporting Information for additional discussion.
In order to link two units of 5a in a manner that might stabilize the reactive conformation of the resulting dimer selectively, we sought to identify linkage points that would allow access to the 4H-Cl−-bound complex, but disfavor formation of the inactive, self-aggregated complex.9 The structures of both the nonproductive aggregate (5a•5a) and a 4H-Cl−-bound complex ([5a]2•NMe4Cl) have been characterized by X-ray diffraction.4,8,10 We noted that, in the solid state, the distance between the backbone tert-butyl group of one molecule and the aryl thiourea of the other is considerably greater in the nonproductive aggregate than in the Cl−-bound complex (Figure 2A). This observation suggested that linkage through these sites might fulfill the intended purpose. We deemed that an appropriate linker between these two sites would need to meet the following stringent criteria: 1) it should cause minimal structural and electronic perturbation to the key catalyst features identified in the original optimization efforts;2c 2) it must be long enough to allow the same 4H-binding arrangement that is accessible to the unlinked monomers; but, 3) it must be short enough to prevent intramolecular formation of the nonproductive aggregate.
The relative disposition of the 5b units in the lowest energy rate- and enantiodetermining 4H-alkylation transition structure predicted using DFT was found to be very similar to that in the Cl−-bound complex characterized in the solid state.8,11–13 We used the computed transition structure to evaluate a variety of hypothetical linked structures according to the criteria outlined above. Through this analysis, pseudo-dimeric thiourea catalyst 6b, which bears a 5-atom spacer between the amino acid side chain of one thiourea unit and the aryl group of another, was modeled and found to access 4H-transition structures that overlay almost perfectly with the transition structure calculated with 5b (Figure 2C, left). In contrast, 7b, which possesses a linker just one methylene unit shorter, was predicted to be incapable of achieving similar conformations (Figure 2C, right). Both catalysts were selected for experimental assessment.
Bis-thioureas 6a and 7a, along with their monomeric analogs 5c and 5d, were synthesized and compared with 1 and 5a as catalysts for the alkylation of α-chloroisochroman with silyl ketene acetal 3b. While modification of the tert-leucine side chain (as in 5c) has a negligible effect on enantioselectivity, replacement of the trifluoromethyl group with an ester (as in 5d) does have a slightly detrimental impact (Figure 3A). Nonetheless, catalyst 6a displays markedly enhanced reactivity, affording high conversion with a low catalyst loading and short reaction time, while preserving the enantioselectivity observed with 5a. In contrast, catalyst 7a exhibits very similar reactivity to the monomeric analogs, consistent with the prediction that the 4-atom linker is too short to enable intramolecular formation of the optimal, cooperative transition structure.
Figure 3.

A. Alkylation of α-chloroisochroman with 3b catalyzed by mono- (1 and 5a,c,d at 0.5 mol %) and bis-thioureas (6a and 7a at 0.25 mol %) at low loading. Relative rates at 15% conversion. B. Alkylation exhibits a first-order dependence on catalyst 6a with dramatic rate acceleration over catalyst 1. See ref. 4a. Rates at 30% conversion. C. The enantiomeric excess of product 4a shows a linear dependence on the ee of bis-thiourea 6a (0.25 mol %); this stands in contrast to the nonlinear dependence on the ee of catalyst 1 (0.50 mol %).
The improved efficiency of catalyst 6a allows us to address several of the practical limitations presented by the conditions optimized for catalyst 1. With just 0.1 mol % 6a, the alkylation of α-chloroisochroman with silyl ketene acetal 3a can be run on preparatory scale under relatively concentrated conditions (0.5 M initial substrate concentration) to afford 4a with excellent yield and enantioselectivity after just 3 hours (Scheme 1). This stands in stark contrast to the original conditions, in which a 24-hour reaction time with 10 mol % 1 at 0.1 M is required to achieve good yield with the same level of enantioinduction.2c
Scheme 1.

Improved Volumetric Throughput on Preparatory Scale
The improved efficiency observed with pseudo-dimeric catalyst 6a provides circumstantial evidence for the validity of the cooperative mechanism for anion-abstraction catalysis outlined in Figure 1B. In order to establish rigorously whether the two linked thiourea subunits act cooperatively within the same molecule, the kinetic dependence of alkylation rate on catalyst concentration was determined by reaction progress kinetic analysis using in situ infrared spectroscopy (Figure 3B).4a,14 The reaction exhibits a strict first-order dependence on catalyst at low [6a], under conditions where a second-order dependence on [1] is observed.4a The increased activity of 6a relative to monomeric catalyst 1 is therefore magnified at very low catalyst loadings, and as little as 0.05 mol % 6a can be employed while maintaining useful reaction rates. Finally, an absence of a nonlinear effect is observed with catalyst 6a at low catalyst concentrations (Figure 3C).15,16 Taken together, these results provide strong evidence that the thiourea subunits in catalyst 6a cooperate in an intramolecular fashion to promote oxocarbenium alkylation.
Furthermore, the successful development of a highly active pseudo-dimeric catalyst designed around the 4H-activation mode suggests that the 4H-mechanism is most likely operative under the conditions described above. To evaluate whether the 2H-activation mode is also accessible to the dimeric catalysts, the intermediate cat•oxocarbenium•Cl− complexes and the transition structures for oxocarbenium alkylation promoted by the 2H- and 4H-arrangements of catalyst 6b were examined computationally. Across a variety of functionals, both the intermediate and transition structures in which the bis-thiourea was forced to access a 2H-geometry were predicted to be significantly higher in energy than the alternative 4H-structures. (See Supporting Information for additional discussion.)
In conclusion, the design of highly active and efficient anion abstraction catalyst 6a represents the culmination of a series of mechanistic insights. The mechanism-driven design strategy has afforded a linked, bis-thiourea catalyst system that overcomes many of the practical limitations traditionally associated with H-bond-donor-mediated anion-abstraction catalysis. Key insights enabled formation of a pseudo-dimeric complex for cooperative activation of a chloroether electrophile by a 4H-mechanism while disfavoring nonproductive ground-state aggregation. The high efficiencies enabled by this catalyst design hold promise for application of dual hydrogen-bond-donor-mediated anion-abstraction catalysis to historically challenging transformations. In this vein, the development of highly stereoselective reactions with tertiary alkyl chloride and glycosyl chloride electrophiles is the focus of our ongoing attention.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM-43214) and through fellowships to C.R.K. (NSF, DGE1144152), D.L. (NSERC PDF), N.S.R. (Boehringer Ingelheim), and D.D.F. (Eli Lilly). The authors thank Dr. Shao-Liang Zheng (Harvard X-Ray Laboratory) for collection and refinement of X-ray crystallographic data, Dr. Robert Knowles (Prince-ton) for early crystallographic analyses, Dr. Alison E. Wendlandt and Andrew J. Bendelsmith (Harvard) for synthetic assistance, and Daniel A. Strassfeld (Harvard) for helpful discussion.
Footnotes
Funding Sources
No competing financial interest has been declared.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website. Experimental procedures; spectroscopic data; kinetic data (tabulated and graphical); discussion, geometries, and energies of calculated stationary points; summary of alternative linking strategies (PDF). X-ray crystallographic structures for synthetic intermediates and select urea and thiourea catalysts (CCDC 1478174, 1482963–1482976, 1501460) (CIF)
References
- 1.For reviews, see: Zhang Z, Schreiner PR. Chem Soc Rev. 2009;38:1187–1198. doi: 10.1039/b801793j.Brak K, Jacobsen EN. Angew Chem Int Ed. 2012;52:534–561. doi: 10.1002/anie.201205449.Phipps RJ, Hamilton GL, Toste FD. Nature Chem. 2012;4:603–614. doi: 10.1038/nchem.1405.Beckendorf S, Asmus S, Mancheño OG. Chem Cat Chem. 2012;4:926–936.Mahlau M, List B. Angew Chem Int Ed. 2013;52:518–533. doi: 10.1002/anie.201205343.Seidel D. Synlett. 2014;25:783–794.
- 2.For select examples of enantioselective anion-binding catalysis with dual H-bond donors, see: Taylor MS, Tokunaga N, Jacobsen EN. Angew Chem Int Ed. 2005;44:6700–6704. doi: 10.1002/anie.200502277.Raheem IT, Thiara PV, Peterson EA, Jacobsen EN. J Am Chem Soc. 2007;129:13404–13405. doi: 10.1021/ja076179w.Reisman SE, Doyle AG, Jacobsen EN. J Am Chem Soc. 2008;130:7198–7199. doi: 10.1021/ja801514m.Klausen RS, Jacobsen EN. Org Lett. 2009;11:887–890. doi: 10.1021/ol802887h.De CK, Klauber EG, Seidel D. J Am Chem Soc. 2009;131:17060–17061. doi: 10.1021/ja9079435.Xu H, Zuend SJ, Woll MG, Tao Y, Jacobsen EN. Science. 2010;327:986–990. doi: 10.1126/science.1182826.Knowles RR, Lin S, Jacobsen EN. J Am Chem Soc. 2010;132:5030–5032. doi: 10.1021/ja101256v.Brown AR, Kuo WH, Jacobsen EN. J Am Chem Soc. 2010;132:9286–9288. doi: 10.1021/ja103618r.Burns NZ, Witten MG, Jacobsen EN. J Am Chem Soc. 2011;133:14578–14581. doi: 10.1021/ja206997e.Lin S, Jacobsen EN. Nature Chem. 2012;4:817–824. doi: 10.1038/nchem.1450.Schafer AG, Wieting JM, Fisher TJ, Mattson AE. Angew Chem Int Ed. 2013;52:11321–11324. doi: 10.1002/anie.201305496.Metz AE, Ramalingam K, Kozlowski MC. Tetrahedron Lett. 2015:5180–5184. doi: 10.1016/j.tetlet.2015.07.058.Mittal N, Lippert KM, De CK, Klauber EG, Emge TJ, Schreiner PR, Seidel D. J Am Chem Soc. 2015;137:5748–5758. doi: 10.1021/jacs.5b00190.Hardman–Baldwin AM, Visco MD, Wieting JM, Stern C, Kondo S, Mattson AE. Org Lett. 2016;18:3766–3769. doi: 10.1021/acs.orglett.6b01783.
- 3.A few noteworthy exceptions have been reported. For select examples of highly efficient urea or thiourea-catalyzed transformations, see: Kotke M, Schreiner PR. Tetrahedron. 2006;62:434–439.Kotke M, Schreiner PR. Synthesis. 2007:779–790.Zuend SJ, Coughlin MP, Lalonde MP, Jacobsen EN. Nature. 2009;461:968–970. doi: 10.1038/nature08484.Birrell JE, Desrosiers JN, Jacobsen EN. J Am Chem Soc. 2011;133:13872–13875. doi: 10.1021/ja205602j.
- 4.(a) Ford DD, Lehnherr D, Kennedy CR, Jacobsen EN. J Am Chem Soc. 2016;138:7860–7863. doi: 10.1021/jacs.6b04686. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ford DD, Lehnherr D, Kennedy CR, Jacobsen EN. ACS Catal. 2016;6:4616–4620. doi: 10.1021/acscatal.6b01384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For representative reports in which novel anion-binding catalysts were validated using the alkylation of α-chloroisochroman with silyl ketene acetal nucleophiles as a benchmark reaction, see: Kniep F, Jungbauer SH, Zhang Q, Walter SM, Schindler S, Schnapperelle I, Herdtweck E, Huber SM. Angew Chem Int Ed. 2013;52:7028–7032. doi: 10.1002/anie.201301351.Jungbauer SH, Huber SM. J Am Chem Soc. 2015;137:12110–12120. doi: 10.1021/jacs.5b07863.
- 6.(a) Konsler RG, Karl J, Jacobsen EN. J Am Chem Soc. 1998;120:10780–10781. [Google Scholar]; (b) Ready JM, Jacobsen EN. J Am Chem Soc. 2001;123:2687–2688. doi: 10.1021/ja005867b. [DOI] [PubMed] [Google Scholar]; (c) Ready JM, Jacobsen EN. Angew Chem Int Ed. 2002;41:1374–1377. doi: 10.1002/1521-3773(20020415)41:8<1374::aid-anie1374>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; (d) Denmark SE, Fu J. J Am Chem Soc. 2003;125:2208–2216. doi: 10.1021/ja021280g. [DOI] [PubMed] [Google Scholar]; (e) Nakano K, Hashimoto S, Nozaki K. Chem Sci. 2010;1:369–373. [Google Scholar]; (f) Kalow J, Doyle AG. J Am Chem Soc. 2011;133:16001–16012. doi: 10.1021/ja207256s. [DOI] [PubMed] [Google Scholar]; (g) Klimczyk S, Misale A, Huang X, Maulide N. Angew Chem Int Ed. 2015;54:10365–10369. doi: 10.1002/anie.201503851. [DOI] [PubMed] [Google Scholar]
- 7.For examples in which intramolecular H-bonding has been proposed to increase the “2H” anion- or neutral Lewis base-binding ability of a urea or thiourea catalyst, see: Jones CR, Pantoş GD, Morrison AJ, Smith MD. Angew Chem Int Ed. 2009;48:7391–7394. doi: 10.1002/anie.200903063.Probst N, Madarász Á, Valkonen A, Pápai I, Rissanen K, Neuvonen A, Pihko PM. Angew Chem Int Ed. 2012;51:8495–8499. doi: 10.1002/anie.201203852.(c) ref 2m.
- 8.While 1 exists as a slowly interconverting mixture of amide rotamers, 5a exists exclusively in the more active and enantioselective (Z)-rotameric form. This constraint has been demonstrated to lead to improved activity and enantioselectivity in the alkylation of α-chloroisochroman. See: Lehnherr D, Ford DD, Bendelsmith AJ, Kennedy CR, Jacobsen EN. Org Lett. 2016;18:3241–3217. doi: 10.1021/acs.orglett.6b01435.
- 9.The extent of this challenge was further illustrated by the poor efficiencies of symmetrically linked catalysts. See Supporting Information for details.
- 10.The solution-state structures of the nonproductive aggregates 1•1 and 5a•5a have also been determined via DFT-aided 2D NMR analysis; see refs. 4a, 8. These structures are similar to the solid-state structures and position the backbone tert-butyl group of one molecule distal from the aryl thiourea of the other.
- 11.Catalyst 5b provides a significantly reduced computational cost relative to catalyst 5a. For experimental validation, see ref. 8.
- 12.Frisch MJ, et al. Gaussian 09, Revision D.02. Gaussian Inc; Wallingford, CT: 2009. see Supporting Information for full citation.
- 13.(a) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (c) Miehlich B, Savin A, Stoll H, Preuss H. Chem Phys Lett. 1989;157:200–206. [Google Scholar]; (d) Frisch MJ, Pople JA, Binkley JS. J Chem Phys. 1984;80:3265–3269. [Google Scholar]; (e) Grimme S, Antony J, Ehrlich S, Krieg H. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 14.(a) Blackmond DG. Angew Chem Int Ed. 2005;44:4302–4320. doi: 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]; (b) Blackmond DG. J Am Chem Soc. 2015;137:10852–10866. doi: 10.1021/jacs.5b05841. [DOI] [PubMed] [Google Scholar]
- 15.(a) Puchot C, Samuel O, Duñach E, Zhao S, Agami C, Kagan HB. J Am Chem Soc. 1986;108:2353–2357. doi: 10.1021/ja00269a036. [DOI] [PubMed] [Google Scholar]; (b) Guillaneux D, Zhao S, Samuel O, Rainford D, Kagan HB. J Am Chem Soc. 1994;116:9430–9439. [Google Scholar]; (c) Satyanarayana T, Abraham S, Kagan HB. Angew Chem Int Ed. 2009;48:456–494. doi: 10.1002/anie.200705241. [DOI] [PubMed] [Google Scholar]
- 16.A nonlinear effect would be expected if 6a were undergoing self-aggregation to a measurable extent under the catalytic conditions, or if 6a achieves cooperative anion abstraction through an intermolecular pathway. In the latter scenario, the magnitude of the nonlinear effect would depend on the degree of stereochemical communication between the thiourea subunits in the ee-determining transition structure.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.