

ABSTRACT
Most mycolic acid-containing actinobacteria and some proteobacteria use steroids as growth substrates, but the catabolism of the last two steroid rings has yet to be elucidated. In Mycobacterium tuberculosis, this pathway includes virulence determinants and has been proposed to be encoded by the KstR2-regulated genes, which include a predicted coenzyme A (CoA) transferase gene (ipdAB) and an acyl-CoA reductase gene (ipdC). In the presence of cholesterol, ΔipdC and ΔipdAB mutants of either M. tuberculosis or Rhodococcus jostii strain RHA1 accumulated previously undescribed metabolites: 3aα-H-4α(carboxyl-CoA)-5-hydroxy-7aβ-methylhexahydro-1-indanone (5-OH HIC-CoA) and (R)-2-(2-carboxyethyl)-3-methyl-6-oxocyclohex-1-ene-1-carboxyl-CoA (COCHEA-CoA), respectively. A ΔfadE32 mutant of Mycobacterium smegmatis accumulated 4-methyl-5-oxo-octanedioic acid (MOODA). Incubation of synthetic 5-OH HIC-CoA with purified IpdF, IpdC, and enoyl-CoA hydratase 20 (EchA20), a crotonase superfamily member, yielded COCHEA-CoA and, upon further incubation with IpdAB and a CoA thiolase, yielded MOODA-CoA. Based on these studies, we propose a pathway for the final steps of steroid catabolism in which the 5-member ring is hydrolyzed by EchA20, followed by hydrolysis of the 6-member ring by IpdAB. Metabolites accumulated by ΔipdF and ΔechA20 mutants support the model. The conservation of these genes in known steroid-degrading bacteria suggests that the pathway is shared. This pathway further predicts that cholesterol catabolism yields four propionyl-CoAs, four acetyl-CoAs, one pyruvate, and one succinyl-CoA. Finally, a ΔipdAB M. tuberculosis mutant did not survive in macrophages and displayed severely depleted CoASH levels that correlated with a cholesterol-dependent toxicity. Our results together with the developed tools provide a basis for further elucidating bacterial steroid catabolism and virulence determinants in M. tuberculosis.
KEYWORDS: CoA thioester, Mycobacterium tuberculosis, catabolism, cholesterol, ring opening
IMPORTANCE
Bacteria are the only known steroid degraders, but the pathway responsible for degrading the last two steroid rings has yet to be elucidated. In Mycobacterium tuberculosis, this pathway includes virulence determinants. Using a series of mutants in M. tuberculosis and related bacteria, we identified a number of novel CoA thioesters as pathway intermediates. Analysis of the metabolites combined with enzymological studies establishes how the last two steroid rings are hydrolytically opened by enzymes encoded by the KstR2 regulon. Our results provide experimental evidence for novel ring-degrading enzymes, significantly advance our understanding of bacterial steroid catabolism, and identify a previously uncharacterized cholesterol-dependent toxicity that may facilitate the development of novel tuberculosis therapeutics.
INTRODUCTION
Steroids have a variety of important physiological roles across all domains of life. In eukaryotes, they serve as critical components of cell membranes, as signaling molecules, and in mammals, they absorb dietary fat. However, the only known organisms that can catabolize steroids and utilize them as growth substrates are bacteria (1). Bacterial steroid catabolism can be either aerobic, as occurs in most mycolic acid-containing actinobacteria and some proteobacteria (1–3), or anaerobic, as in some proteobacteria (4). Aerobic catabolism has been studied for many decades, due in part to its potential to transform low-value steroids into high-value ones (5). Most recently, the cholesterol catabolic pathway of Mycobacterium tuberculosis has been studied due to its role in virulence (2, 6, 7): this catabolism is required for the survival of M. tuberculosis in macrophages and is a potential target for novel therapeutics that are urgently needed to treat tuberculosis (8). Despite the intensified research, many aspects of steroid catabolism remain unclear.
Based on studies in Rhodococcus jostii RHA1, M. tuberculosis, and Comamonas testosteroni TA441, the aerobic catabolism of steroids largely follows the structural elements of the steroid molecule: the alkyl side chain when present, rings A and B, and rings C and D, respectively (3, 9, 10). Side-chain degradation resembles the β-oxidation of fatty acids (11) proceeding via coenzyme A (CoA) thioester intermediates to generate propionyl- and acetyl-CoA. Ring A/B degradation includes oxygenases that catalyze the 9,10 cleavage of the steroid nucleus and the 4,5-extradiol cleavage of ring A, respectively (12–14). In M. tuberculosis and other actinobacteria, genes encoding cholesterol uptake and side-chain and ring A/B degradation are transcriptionally regulated by KstR, a TetR family repressor (15), and side-chain and ring A/B degradation occur concurrently to at least some extent (16). In all aerobic steroid-degrading bacteria characterized to date, catabolism yields 3aα-H-4α(3′-propanoate)-7aβ-methylhexahydro-1,5-indanedione (HIP) (17, 18), a 13-carbon catabolite containing intact rings C and D or a derivative thereof (Fig. 1A).
FIG 1 .

HIP catabolic genes in representative actinobacteria and proteobacteria. (A) The aerobic catabolism and anaerobic catabolism of cholesterol and other steroids appear to converge at HIP-CoA. (B) The HIP catabolic gene clusters of M. tuberculosis (Mtb) H37Rv and S. denitrificans DSMZ18526. Homologous genes are colored the same. For gene annotation, see Table 1.
In mycobacteria and rhodococci, HIP catabolism is specified by a series of enzymes encoded by ~15 genes that are transcriptionally regulated by a second TetR family repressor, KstR2 (10, 15) (Fig. 1B). However, in contrast to the catabolism of the steroid side chain and rings A and B, the catabolism of HIP remains largely uncharacterized. Fifty years ago, Lee and Sih proposed that HIP catabolism proceeds via the cleavage of ring C, involving either a β-oxidative reaction or a Baeyer-Villiger type mono-oxygenation (18). Subsequently, Hashimoto and Hayakawa (1977) reported the accumulation of 4-methyl-5-oxooctanedioic acid (MOODA) during the growth of a streptomycete on HIP (19). Homologs of the enzymes encoded by the KstR2 regulon have been found in all steroid-degrading bacteria characterized to date (Table 1). Actinobacteria that contain multiple steroid catabolic pathways appear to complete this catabolism using a single HIP catabolic pathway (9). Interestingly, recent bioinformatic and transcriptomic studies indicate that the aerobic and anaerobic steroid degradation pathways of Steroidobacter denitrificans also converge at HIP (4) (Fig. 1B). These studies in diverse bacteria suggest that a single HIP catabolic pathway is used in the bacterial catabolism of steroids. Nevertheless, the only characterized HIP catabolic enzyme to date is FadD3, which initiates catabolism by catalyzing the thioesterification of HIP in M. tuberculosis (17) and Pseudomonas putida DOC21 (20). In Actinobacteria, the reaction product HIP-CoA is the effector of KstR2 (10). Bioinformatic analyses suggest the involvement of at least two rounds of β-oxidation in the subsequent catabolism of HIP (17).
TABLE 1 .
Annotation of the KstR2 regulon
| Genea | Identification no. forb: |
Annotation of gene product | Best hitc | % amino acid identityd | ||||
|---|---|---|---|---|---|---|---|---|
| H37Rv | RHA1 | M. smegmatis | CNB-2 | S. denitrificans | ||||
| Rv3548c | RS22710 | 5999 | 1286 | 00355 | Short-chain-type dehydrogenase/reductase | P22414 | 42 | |
| Rv3549c | RS22705 | 6000 | 1330 | 12450e | Short-chain-type dehydrogenase/reductase | A6CQL2 | 34 | |
| echA20 | Rv3550 | RS27700 | 6001 | 1280 | 00335 | HIEC-CoA hydrolase | P76082 (1,4-dihydroxy-2-naphthoyl-CoA synthase [MenB]) | 28 |
| ipdA | Rv3551 | RS22695 | 6002 | 1276 | 00310 | COCHEA-CoA hydrolase, α subunit | Q59111 (glutaconate CoA-transferase, α subunit) | 26 |
| ipdB | Rv3552 | RS22690 | 6003 | 1277 | 00315 | COCHEA-CoA hydrolase, β subunit | Q59111 (glutaconate CoA-transferase, β subunit) | 25 |
| ipdC | Rv3553 | RS22685 | 6004 | 1279 | 00330 | 5-OH HIC-CoA reductase | Q9FBC5 [enoyl-(ACP) reductase II (FabK)] | 30 |
| fadA6 | Rv3556c | RS22430 | 6008 | 1283 | 00350 | β-Keto CoA thiolase | I6XHI4 (3-oxo-acyl CoA thiolase) | 38 |
| kstR2 | Rv3557c | RS22425 | 6009 | HIP-CoA repressorf | ||||
| ipdF | Rv3559c | RS22420 | 6011 | 1289 | 00370 | 5-Oxo HIC-CoA oxidase | Q9LBG2 (levodione reductase) | 39 |
| fadE30 | Rv3560c | RS22415 | 6012 | 1288 | 00365 | Acyl-CoA dehydrogenase | I6YCA3 | 31 |
| fadD3 | Rv3561 | RS22410 | 6013 | 1360 | 09100e | HIP-CoA synthetasef | ||
| fadE31 | Rv3562 | RS22400 | 6014 | 1281 | 00340 | Acyl-CoA dehydrogenase | I6YCA3 | 29 |
| fadE32 | Rv3563 | RS22395 | 6015 | 1282 | 00345 | MOODA-CoA dehydrogenase | I6YCA3 | 23 |
| fadE33 | Rv3564 | RS22390 | 6016 | 1287 | 00360 | Acyl-CoA dehydrogenase | I6YCA3 | 23 |
Name assigned based on the present study.
Identification numbers for the corresponding genes in M. tuberculosis H37Rv, R. jostii RHA1, M. smegmatis, C. testosteroni CNB-2, and Steroidobacter denitrificans DSM 18526. For simplicity, “Msmeg_” was omitted from identification numbers for M. smegmatis, “CtCNB2_” was omitted from those for C. testosteroni CNB-2, and “ACG33_” was omitted from those for S. denitrificans DSM 18526.
Accession numbers of functionally characterized best hits in the NCBI database are shown, with alternate protein names included for some entries.
Amino acid sequence identity of the M. tuberculosis enzyme and its experimentally characterized best hit based on full sequence alignment.
Not clustered with other ring C and D catabolic genes.
M. tuberculosis enzyme characterized.
Transposon mutagenesis studies of the cholesterol catabolic genes in M. tuberculosis indicate that some of the intermediates of HIP catabolism are toxic to the bacterial cell (21). In these studies, fadD3 insertion mutants were recovered at a reasonably high frequency in cholesterol- versus glycerol-containing media, consistent with the ability of a fadD3 deletion mutant to grow on the first half of the cholesterol molecule (17). In contrast, mutants of each of eight other KstR2 regulon genes, predicted to encode enzymes that act downstream of FadD3, including ipdA (Rv3551) and ipdC (Rv3553), were recovered at much lower frequencies. This result is consistent with the lack of the encoded enzymes inducing some form of toxicity in the presence of cholesterol. Genes of the KstR2 regulon predicted to be essential for virulence in transposon mutagenesis studies in macrophages and mice include ipdA, ipdB, fadA6, fadE30, and fadE32 (22, 23). The ipdA gene is most consistently implicated in these studies, and ipdAB mutants have been patented as a live vaccine based on studies in Rhodococcus equi (24). R. equi appears to be fairly unique in having a second copy of some KstR2 regulon genes, including ipdAB and fadA6 (24).
Herein, we elucidate key steps of HIP catabolism, including the order of opening of rings C and D. We generated a series of mutants in the KstR2-regulated genes of three mycolic acid-containing actinobacteria: M. tuberculosis, Mycobacterium smegmatis, and R. jostii RHA1. We used liquid chromatography-coupled mass spectrometry (LC-MS) to interrogate these mutants for the accumulation of intracellular CoA metabolites under different growth conditions. This led to the resolution of previously undescribed CoA thioesters that represent various HIP metabolites, including ring C/D-opened intermediates. The identity of key compounds was determined using 1H- and 13C-nuclear magnetic resonance (NMR) and chemical synthesis. We used purified enzymes from actinobacteria and a proteobacterium to generate CoA thioesters that were observed in the mutants. Finally, we investigated the phenotype of a ΔipdAB mutant of M. tuberculosis in macrophages to understand its importance in intracellular survival. The results enable us to propose a bacterial HIP catabolism pathway.
RESULTS
The ipdABC genes are required for growth on cholesterol and HIP.
The KstR2 regulon has been strongly implicated in the catabolism of HIP in mycolic acid-containing actinobacteria (10). Because FadD3, encoded by the KstR2 regulon, initiates HIP catabolism, we hypothesized that the other regulon-encoded enzymes act downstream of FadD3. To further elucidate the steps of the HIP catabolic pathway, we initially focused on ipdAB and ipdC in M. tuberculosis (Rv3551 to Rv3553) and R. jostii RHA1 (RHA1_RS22695-22685).
An ΔipdAB mutant constructed in M. tuberculosis Erdman did not grow on cholesterol (Fig. 2A), but grew as the wild type (WT) on glycerol (Fig. 2B). The growth defect on cholesterol was restored through complementation. Because M. tuberculosis mutants can exhibit unexplained strain differences (25), we verified that an equivalent ΔipdAB mutant constructed in M. tuberculosis CDC1551 had the same phenotype as the ΔipdAB M. tuberculosis Erdman strain (data not shown). Finally, a ΔipdAB mutant of R. jostii RHA1 exhibited a similar phenotype (see Fig. S1 in the supplemental material): it did not grow on either HIP or cholesterol and grew normally on pyruvate (Fig. S1). The growth defects on cholesterol and HIP were restored through complementation with ipdAB of M. tuberculosis.
FIG 2 .

Growth of ΔipdAB M. tuberculosis. WT M. tuberculosis Erdman (black), ΔipdAB M. tuberculosis (red), or ΔipdAB::ipdAB M. tuberculosis (blue) cells were grown on (A) 0.5 mM cholesterol, (B) on 0.2% glycerol, or (C) in phorbol myristate acetate (PMA)-differentiated THP-1 cells (MΦ). Data represent the mean from biological triplicates.
Growth and CoA metabolites of R. jostii RHA1 strains. (A to D) Growth of WT::pTip-Qc2 (blue), ΔipdAB::pTip-Qc2 (red), ΔipdAB::pTipCoL51 (red, dashed), ΔipdC::pTipQc2 (green), and ΔipdC::pTipRv3553 (green, dashed) on (A) 10 mM pyruvate, (B) 1 mM cholesterol, (C) 1.5 mM HIP, and (D) 1 mM HIP plus 10 mM pyruvate. (E) Depletion of HIP by RHA1 strains, color coded as in growth curves as measured by GC-MS and reported as percentage of initial levels. Data are the mean from triplicates. Error bars show standard deviations. (F) LC-MS chromatograms of CoA metabolites extracted from WT (blue) and ΔipdAB (red) RHA1 incubated with cholesterol. Numbers correspond to CoASH (1), acetyl-CoA (2), propionyl-CoA (3), and COCHEA-CoA (4). IS, internal standard. Data for panel D were acquired using a BioScreen C (Growth Curves USA). Download FIG S1, TIF file, 0.3 MB (336.5KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
An ΔipdC mutant of R. jostii RHA1 also did not grow on either cholesterol or HIP but grew normally on pyruvate (Fig. 3; Fig. S1). The growth defect on cholesterol and HIP was restored through complementation with M. tuberculosis ipdC (ipdCMtb). Gas chromatography-coupled mass spectrometry (GC-MS) established that while cholesterol and HIP were depleted in the wild-type and complemented strains, they were not detectably depleted by the ipdC mutant (Fig. S1E). Similar results were obtained in M. tuberculosis Erdman: the ΔipdC mutant did not grow on cholesterol but grew normally on glycerol (see Fig. S2 in the supplemental material). Moreover, this phenotype was restored through complementation with an integrative plasmid harboring ipdC.
FIG 3 .
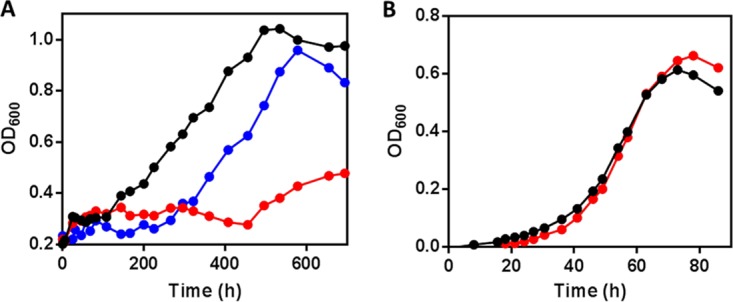
Growth of ΔipdC R. jostii RHA1. WT RHA1::pTipQC2 (black), ΔipdC RHA1::pTipQC2 (red), or ΔipdC RHA1::pTipRv3553 (blue) cells were grown on (A) 1 mM cholesterol or (B) 10 mM sodium pyruvate. OD600, optical density at 600 nm.
Growth and CoA metabolites of ΔipdC M. tuberculosis. WT (black), ΔipdC (red), and ΔipdC::ipdC (blue) M. tuberculosis Erdman were grown on (A) 1 mM cholesterol, (B) 0.2% glycerol, or (C) 0.5 mM cholesterol and 0.2% glycerol. (D) CoA metabolome of ΔipdC (red) and WT M. tuberculosis (blue) incubated with 0.5 mM cholesterol. Arrows indicate the peaks corresponding to the 5-OH HIC-CoA in the ΔipdC R. jostii RHA1 CoA metabolome. Download FIG S2, TIF file, 0.3 MB (354.4KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Growth in macrophages.
Transposon mutagenesis studies suggest that ipdA is essential for M. tuberculosis survival in macrophages (23). Moreover, the gene is essential for survival of R. equi in foals (24). We therefore tested growth of ΔipdAB M. tuberculosis in phorbol myristate acetate (PMA)-differentiated THP-1 cells (Fig. 2C). WT M. tuberculosis increased >350-fold over 7 days, corresponding to a doubling time of 19.6 h. The mutant increased ~10-fold over this time, corresponding to a doubling time of 46.5 h, while complementation restored intracellular replication to 131-fold. These results are consistent with M. tuberculosis catabolizing cholesterol during intracellular growth (8).
Accumulation of cholesterol catabolites in the ipd mutants.
In an attempt to identify the respective substrates of IpdAB and IpdC, we investigated the occurrence of metabolites in the RHA1 mutants. GC-MS analyses revealed that, when incubated with cholesterol, ΔipdC R. jostii RHA1 accumulated small amounts of a metabolite with an m/z of 356 (Fig. 4A). No metabolites were detected in the culture supernatant when cells of ΔipdAB R. jostii RHA1 were incubated with cholesterol (Fig. 4A).
FIG 4 .

Accumulation of cholesterol-derived metabolites from ΔipdAB and ΔipdC strains. (A) GC-MS traces of culture supernatants of ΔipdC, ΔipdAB, ΔfadD3 ipdAB, and WT R. jostii RHA1 incubated with cholesterol. Peaks 1 and 2 correspond to TMS-5α-OH HIC and TMS-HIP, respectively. (B and C) CoA metabolome of cholesterol-incubated cells of (B) WT (blue) and ΔipdC (red) RHA1 or (C) WT (blue) and ΔipdAB (red) M. tuberculosis. The major unique peaks in the ΔipdC and ΔipdAB metabolomes correspond to 5αOH-HIC-CoA and COCHEA-CoA, respectively (inset). Lighter-shaded curves in panels B and C are based on the 768→261 transition observed in free CoASH as well as CoA thioesters subjected to in-source fragmentation.
We hypothesized that the failure to detect significant amounts of extracellular metabolites in the supernatants of cholesterol-incubated ΔipdC and ΔipdAB mutants was due to the accumulation of intracellular CoA-thioesterified metabolites that are not readily excreted. To test this hypothesis, we extracted the CoA thioesters from cells and analyzed them using liquid chromatography (LC)-MS. LC was performed using a pentafluorophenyl (PFP) resin to maximize resolution of the CoA thioesters. M. tuberculosis, R. jostii RHA1, and M. smegmatis cells incubated with various substrates contained CoASH, acetyl-CoA, propionyl-CoA, and/or succinyl-CoA irrespective of the growth substrate (see Table S2 in the supplemental material). The identity of these metabolites was based on their m/z values and their retention time (Rt) on the PFP column with respect to synthetic standards. Their concentrations were quantified relative to p-coumaroyl-CoA, the internal standard. Some strains also contained small amounts of dephospho-CoASH, depending on the substrate, as has been reported in other cells (26).
Strains, plasmids, and oligonucleotides used in this study. Download TABLE S1, DOCX file, 0.1 MB (60.6KB, docx) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
List and characterization of CoA thioesters in M. tuberculosis, R. jostii RHA1, M. smegmatis, and ΔipdAB mutants. Download TABLE S2, DOCX file, 0.1 MB (21KB, docx) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
When incubated in the presence of cholesterol, the ipd mutants accumulated CoA thioesters that were not detected in either the wild-type strains or the ipd mutants incubated with glycerol or pyruvate (see Fig. S3 in the supplemental material). More specifically, cholesterol-incubated cells of ΔipdC M. tuberculosis and R. jostii RHA1 contained significant amounts of two CoA thioesters with m/z values of 962, one of which was more abundant than the other (Fig. 4B; Fig. S2D). The main CoA thioester that accumulated in cholesterol-incubated cells of ΔipdAB M. tuberculosis and R. jostii RHA1 eluted with a Rt of 22.7 min and had an m/z value of 976 (Fig. 4C; Fig. S1F).
CoA thioesters and metabolites produced by M. smegmatis strains. (A) WT and (B) ΔipdAB cells were incubated with cholesterol (blue) and glycerol (gray). Numbers represent CoASH (1), acetyl-CoA (2), succinyl-CoA (3), propionyl-CoA (4), unidentified CoA thioester of 838 m/z (5), unidentified CoA thioester of 852 m/z (6), and COCHEA-CoA (7). (C) GC-MS of culture supernatants of cholesterol-incubated M. smegmatis strains. Cells of the indicated strains were incubated with 0.5 mM cholesterol. Insets show structures of TMS-derivatized 5α-OH HIC and 2-(2-carboxyethyl)-3-methyl-6-oxocyclohex-1-ene-1. *, unidentified compounds present in all M. smegmatis extracts that are unrelated to cholesterol catabolism. Download FIG S3, TIF file, 0.4 MB (384.6KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Identification of CoA metabolites in the ipd mutants.
We predicted that the metabolite with an m/z value of 962 that accumulated in ΔipdC R. jostii RHA1 and M. tuberculosis could be produced by the β-oxidative cleavage of acetyl-CoA from HIP-CoA and reduction of the 5-oxo group to yield 3aα-H-4α(3′-carboxyl-CoA)-5-hydroxy-7aβ-methylhexahydro-1-indanone (5-OH HIC-CoA) (Fig. 4B). To identify the metabolites that accumulated in the ΔipdC mutants, the 5α and 5β isomers of 5-OH HIC were synthesized and confirmed by NMR (see the supplemental material). Each isomer was thioesterified to yield 5α- and 5β-OH HIC-CoA, respectively, which were then purified by high-performance liquid chromatography (HPLC). The high-resolution [M+H]+ m/z value and MS3 fragmentation pattern (of the [M+H]+ −507 fragment), of synthetic 5-OH HIC-CoA corresponded to those of the most abundant CoA thioesters in cholesterol-incubated ΔipdC mutants. The presence of both α and β diastereomers was confirmed by the Rt values of their corresponding standards. Interestingly, the m/z value of the metabolite that accumulated in the supernatant of cholesterol-incubated ΔipdC R. jostii RHA1 corresponds to that of 5-OH HIC (Fig. 4A).
The most abundant CoA thioester in cholesterol-incubated ΔipdAB mutants accumulated in sufficient quantities to allow its isolation from the R. jostii RHA1 mutant for further characterization. The metabolite was identified as 2-(2-carboxyethyl)-3-methyl-6-oxocyclohex-1-ene-1-carboxyl-CoA (COCHEA-CoA) based on high-resolution mass spectrometry (976.1960 m/z), 1H-NMR, correlation spectroscopy (COSY)-NMR, total COSY (TOCSY)-NMR, heteronuclear multiple bond correlation (HMBC)-NMR, and heteronuclear single quantum coherence (HSQC)-NMR (see the supplemental material). Most diagnostically, the C-8 methyl protons appear as a doublet, establishing that C-6 bears a hydrogen and that ring D is open. C-1″ is thioesterified based on C-3′ having the same 13C-NMR chemical shift as in 2-(2-carboxyethyl)-3-methyl-6-oxocyclohex-1-ene-1 (27).
Enzymatic transformation of 5-OH HIC-CoA.
To further elucidate the catabolism of HIP, we purified various KstR2 regulon-encoded enzymes, including IpdAB, IpdC, IpdF, EchA20, and FadA6. We used these preparations to evaluate their abilities to transform 5-OH HIC-CoA in vitro. We initially sought to work exclusively with the M. tuberculosis homologs. However, we were unable to obtain all of the M. tuberculosis homologs in stable, soluble forms despite testing various host strains and expression conditions. In some cases, the RHA1 homolog was more stable. For IpdC, we obtained the best preparations using the homolog from P. putida Doc21, a bile acid-degrading bacterium. The gene encoding P. putida IpdC (IpdCDoc21), DC0014-19, is the reciprocal best hit of ipdCMtb and occurs in a predicted operon similar in structure to that of S. denitrificans, which contains ipdAB (Fig. 1B). SDS-PAGE analyses of the various protein preparations are provided in Fig. S5 in the supplemental material. The subscripts “Mtb,” “RHA1,” and “Doc21” identify the parent species or strain of each enzyme. Transformation experiments were performed by incubating synthetic 5-OH HIC-CoA with various enzymes and characterizing the reaction products using LC-MS.
Incubation of 5α-OH HIC-CoA with IpdFMtb and IpdCDoc21 yielded a compound with an m/z value of 958 (Fig. 5A and B; red trace), consistent with the oxidation of the 5-OH group of HIC and the introduction of a double bond. Interestingly, the two enzymes did not detectably transform 5β-OH HIC-CoA (data not shown). Based on its mass and the structure of the downstream metabolite, COCHEA-CoA, we provisionally identified the IpdF/IpdC transformation product as (7aS)-7a-methyl-1,5-dioxo-2,3,5,6,7,7a-hexahydro-1H-indene-4-carboxyl-CoA (HIEC-CoA). This assignment is consistent with the function of the next step of the pathway, as discussed below. However, NMR data are required for a definitive identification. Neither IpdFMtb nor IpdCDoc21 transformed 5-OH HIC-CoA in the absence of the other enzyme. Moreover, as described below, ΔipdF M. smegmatis accumulated the same major metabolite as the ΔipdC mutants, 5α-OH HIC-CoA. Therefore, we were unable to determine the order of reaction of IpdF and IpdC. However, incubation of 5-OH HIP-CoA with IpdFMtb yielded HIP-CoA, demonstrating that this enzyme catalyzes oxidation of the 5-OH.
FIG 5 .

LC-MS analyses of the transformation of 5-OH HIC-CoA by purified enzymes. The left panels show HPLC traces of reaction mixtures containing 100 μM 5-OH HIC-CoA, 125 μM NAD+, 50 μM CoASH, 5 μM flavin mononucleotide (FMN) (10 mM phosphate [pH 7.5]) and (A) no enzyme (control), (B) IpdFMtb and IpdCDoc21, (C) IpdFMtb, IpdCDoc21, and EchA20RHA1, or (D) IpdFMtb, IpdCDoc21, EchA20RHA1, IpdABRHA1, and FadA6Mtb. LC-MS analyses of the reaction products identified the major HPLC peaks. The major peaks are color coded with fragmentation patterns in the right-hand panels and correspond to (peak 1) 5α-OH HIC-CoA (962 m/z), (peak 2) HIEC-CoA (958 m/z), (peak 3) COCHEA-CoA (976 m/z), and (peak 4) MOODA-CoA (952 m/z). Other LC peaks correspond to acetyl-CoA (810 m/z) and FMN (labeled “F”).
Incubation of 5-OH HIC-CoA with IpdFMtb, IpdCDoc21, and EchA20RHA1 yielded a compound whose Rt and m/z values were identical to those of COCHEA-CoA (Fig. 5C; green trace), the major metabolite that accumulated in the ΔipdAB mutants.
Finally, incubation of 5-OH HIC-CoA with IpdFMtb, IpdCDoc21, EchA20RHA1, IpdABRHA1, and FadA6Mtb yielded a compound with an m/z value of 952 (Fig. 5D; blue trace). Hydrolysis of the CoA thioester yielded a compound that GC-MS revealed to be 4-methyl-5-oxo-octanedioc acid (MOODA), which accumulated in a ΔfadE32 mutant of M. smegmatis when incubated with cholesterol (Fig. 6). MOODA-CoA has a predicted m/z value of 952. Consistent with such a role, the enzymatic transformation of COCHEA-CoA to MOODA-CoA required CoASH and yielded stoichiometric amounts of acetyl-CoA (Fig. 5D).
FIG 6 .

Cholesterol-derived metabolite of ΔfadE32 M. smegmatis. (A) GC-MS traces of culture supernatants of cholesterol-grown ΔfadE32 (red), ΔfadE32::msmeg_6015 (blue), and WT (black) M. smegmatis. The major metabolite observed in the mutant was MOODA (inset). (B) GC-MS trace of the product following hydrolysis of a metabolite of 952 m/z in 1 M NaOH. (C) MOODA purified from ΔfadE32 M. smegmatis incubated with cholesterol.
Bioinformatic analysis of HIP catabolic enzymes.
To better understand the activities of IpdC, IpdF, EchA20, IpdAB, and FadA6, we performed bioinformatics analyses (Table 1). Among characterized homologs, IpdF shares 39% amino acid sequence identity with levodione reductase from Corynebacterium aquaticum M-13 (28), which catalyzes the NADH-dependent reduction of a ring ketone. For its part, IpdC shares 30% amino acid sequence identity with FabK from Streptococcus pneumoniae, an enoyl-acyl ACP reductase that catalyzes double-bond reduction (29), the reverse of the predicted IpdC reaction. Like FabK, purified IpdCDoc21 contained a flavin (data not shown). Overall, these analyses are consistent with the ability of IpdF and IpdC to catalyze the transformation of 5α-OH HIC-CoA to HIEC-CoA.
EchA20 is one of 21 EchAs in M. tuberculosis. EchAs are members of the crotonase superfamily that are predicted to catalyze the hydration of enoyl-CoAs (30), of which HIEC-CoA, the substrate of EchA20, is an example. A phylogenetic analysis revealed that among M. tuberculosis EchAs, only EchA20 clustered with MenB (see Fig. S4 in the supplemental material), although it shares ~28% amino acid sequence identity with each of the proteins EchA8, EchA18, and MenB. EchA8 and EchA18 are uncharacterized. However, MenB is a 1,4-dihydroxy-2-naphthoyl-CoA synthase that catalyzes an intramolecular Claisen condensation (Dieckmann cyclization) in menaquinone biosynthesis (31). This reaction is essentially the reverse of the reaction catalyzed by EchA20. EchA20 also shares 24% amino acid sequence identity with BadI of Rhodopseudomonas palustris (Table 1). BadI, a β-ketocyclohexanecarboxyl-CoA hydrolase involved in the anaerobic catabolism of benzoate (32), catalyzes a hydrolytic ring-opening reaction similar to that of EchA20. Overall, the bioinformatic analyses indicate that EchA20 catalyzes the hydrolytic ring opening of HIEC-CoA to COCHEA-CoA via a reverse Dieckmann cyclization. Nevertheless, it is unclear whether other M. tuberculosis EchAs catalyze similar reactions.
Phylogenetic tree of enoyl-CoA hydratases (EchAs) and MenB in M. tuberculosis. A cluster containing EchA20 and MenBMtb is highlighted with a red oval. Download FIG S4, TIF file, 0.1 MB (86.6KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Electrophoretic analyses of gene deletion mutants and purified proteins. (A) PCR confirmation of ΔipdAB in R. jostii RHA1 and M. tuberculosis, ΔipdC in RHA1 and M. tuberculosis, and ΔipdF, ΔechA20, and ΔfadE32 in M. smegmatis using the listed primer sets (Table S1). (B) SDS-PAGE loaded, from left to right, with 0.5 μg each of MBP-IpdCDOC21, IpdFMtb, EchA20RHA1, IpdABRHA1, and FadA6Mtb. Purified proteins are flanked by molecular weight standards. Download FIG S5, TIF file, 0.4 MB (418.6KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
IpdAB shares 24% amino acid sequence identity with glutaconate CoA transferase (GCT) of Acidaminococcus fermentans (33), a type I CoA transferase. Most type I CoA transferases characterized to date catalyze the transfer of CoA between short acyl chains (33–36). To test whether IpdABRHA1 had such activity, we incubated the enzyme with acetyl-CoA, propionyl-CoA, or succinyl-CoA in the presence of acetate, propionate, and succinate. In none of these instances was any CoA transferase activity detected. Finally, FadA6 shares 38% amino acid sequence identity with FadA5, a β-ketoacyl-CoA thiolase involved in cholesterol side-chain degradation (37) (Table 1), consistent with it catalyzing the thiolysis of a COCHEA-CoA ring-opened product containing a β-keto thioester moiety, to MOODA-CoA and acetyl-CoA.
Validation of HIP catabolism using additional mutants.
To obtain further evidence for the HIP catabolic pathway suggested by analyses of the ipd mutants and the enzymatic transformations of 5-OH HIC-CoA, we deleted the following KstR2-regulated genes in M. smegmatis MC2155 and analyzed the ΔipdF, ΔipdAB, ΔechA20, and ΔfadE32 mutants (msmeg_6011, msmeg_6002-6003, msmeg_6001, and msmeg_6015, respectively). All four mutants were defective for growth on HIP: the ΔfadE32 strain grew more slowly on HIP, while the other three did not grow at all (Fig. 7A). The growth defect of each mutant on HIP was complemented by the M. tuberculosis or M. smegmatis gene supplied in trans. GC-MS analysis of the culture supernatants revealed that the ΔipdF and ΔechA20 mutants accumulated small amounts of 5-OH HIC (Fig. S3C). Moreover, the ΔfadE32 mutant accumulated a metabolite whose trimethylsilane (TMS) derivative had an m/z value of 346 (Fig. 6). The metabolite that accumulated in the supernatant of the cholesterol-grown ΔfadE32 mutant was purified and was identified as MOODA based on 1H-, COSY-, HMBC-, and HSQC-NMR analysis (SI).
FIG 7 .

Characterization of KstR2 regulon mutants of M. smegmatis. Growth of ΔechA20, ΔfadE32, ΔipdF, and ΔipdAB M. smegmatis mutants on (A) 1.5 mM HIP or (C) 1 mM HIP plus 0.2% glycerol. Curves show WT (black), KstR2 regulon mutants (red), and corresponding complements (blue) and are the means from three biological replicates. (B) CoA metabolomes of mutants. The numbers correspond to CoASH (1), acetyl-CoA (2), HIEC-CoA (3), 5β-OH HIC-CoA (4), 5α-OH HIC-CoA (5), COCHEA-CoA (6), and unknown CoA thioester of 992 m/z (7). IS, p-coumaroyl-CoA internal standard. Lighter-shaded curves indicate the 768→261 transition (Fig. 4).
The ΔipdAB mutant of M. smegmatis differed from those of R. jostii RHA1 and M. tuberculosis in that it accumulated two major metabolites in the supernatant when incubated with cholesterol. These had m/z values of 254 and 326 when derivatized with TMS reagent (Fig. S3C). The former was purified, and based on 1H-NMR, was identified as an analog of COCHEA lacking the C1 carboxyl (SI). A similar result was reported in an ipdAB deletion mutant of Comamonas testosteroni (27).
Cholesterol-incubated cells of the ΔipdF, ΔechA20, and ΔfadE32 mutants were analyzed for CoA thioesters. The profile of the ΔipdF mutant was similar to that of the ΔipdC mutant, containing a significant amount of 5α-OH HIC-CoA and a lesser amount of 5β-OH HIC-CoA (Fig. 7B). This is consistent with the enzymatic studies inasmuch as neither IpdC nor IpdF alone significantly transformed 5-OH HIC-CoA. The CoA metabolome of ΔechA20 M. smegmatis also contained significant quantities of the 5-OH HIC-CoA (Fig. 7B). However, it also contained a small amount of a metabolite whose retention time and m/z value (958) corresponded to that of the transformation product of 5-OH HIC-CoA by IpdC and IpdF (Fig. 7B). The CoA metabolome of ΔipdAB M. smegmatis was very similar to those of the corresponding R. jostii RHA1 and M. tuberculosis mutants (Fig. 7B). Finally, the CoA metabolome of the ΔfadE32 mutant was indistinguishable from that of WT M. smegmatis.
HIP-dependent toxicity.
The failure of the ΔipdAB and ΔipdC mutants to grow on cholesterol (Fig. 2A, 3A, and 7A) is in marked contrast to the phenotype of ΔfadD3 RHA1, which grows on cholesterol to ~50% the yield of the wild type (17). More specifically, the failure of the ipd mutants to grow on cholesterol despite the fact that the encoded enzymes act downstream of FadD3 suggests that the ipd deletions induce some form of toxicity. To explore this further, KstR2 regulon mutants were grown on a second carbon source in the presence of HIP. Interestingly, the ΔfadE32 and ΔipdF mutants grew on other carbon sources in the presence of HIP (Fig. 7C), while the ΔipdAB, ΔipdC, and ΔechA20 mutants did not (Fig. 7C and 8A; Fig. S1D and S2C). The inability to catabolize a secondary carbon source in the presence of HIP indicates that there is a HIP-dependent toxicity in some of the mutants, similar to the cholesterol-dependent toxicity observed for the ipdAB and ipdC mutants described above. One possible form of cholesterol (or HIP)-dependent toxicity is the accumulation of propionyl-CoA, which can be relieved by supplementation with vitamin B12 (38). However, supplementation of the ΔipdAB, ΔipdC, and ΔechA20 mutants with vitamin B12 did not relieve cholesterol-dependent toxicity, indicating that the basis of toxicity is independent of propionyl-CoA in these mutants. However, in analyzing the CoA metabolites of these mutants, we noted that the ΔipdAB, ΔipdC, and ΔechA20 mutants contained significantly lower levels (<20%) of CoASH compared to the WT when cells were incubated with cholesterol (Fig. 8B). In contrast, the ΔfadE32 and ΔipdF mutants contained statistically similar CoASH levels to the WT under these conditions. Indeed, the respective levels of CoASH and cholesterol-derived CoA thioesters appeared to be inversely related. For example, when incubated with cholesterol, 5-OH HIC-CoA and COCHEA-CoA accounted for 84% ± 2% and 94% ± 1% of the total CoA detected in cells of ΔipdC and ΔipdAB RHA1, respectively, indicating that sequestration of CoASH by cholesterol-derived CoA-thioesters may be the basis of toxicity.
FIG 8 .

Cholesterol-dependent toxicity. (A) Growth of WT (black), ΔipdAB (red), and ΔipdAB::ipdAB (blue) M. tuberculosis grown on 7H9 medium containing 0.5 mM cholesterol and 0.2% glycerol. The data represent the average from biological triplicates. (B) The relative abundance of CoASH (768→261) was normalized to the internal standard (p-coumaroyl-CoA [914→407]) in KstR2 regulon mutants. *, P < 0.05 compared to the WT strain. Error bars represent standard deviations. The numbers of replicates were as follows: 5, 5, 5, and 3 for WT, ΔipdAB, ΔipdC, and ΔfadD3 ΔipdAB R. jostii RHA1, respectively; 2, 1, and 1 for WT, ΔipdAB, and ΔipdC M. tuberculosis (Mtb), respectively; and 4, 5, 1, 4, and 1 for WT, ΔipdAB, ΔechA20, ΔipdF, and ΔfadE32 M. smegmatis, respectively.
DISCUSSION
The mutant data, enzymological transformations, and bioinformatic analyses presented herein support a model for HIP degradation in which cleavage of ring D precedes that of ring C (Fig. 9). More specifically, we propose a pathway for HIP catabolism in which the propionyl side chain is first degraded via β-oxidation to yield 5-OH HIC-CoA. This is then transformed to HIEC-CoA by IpdF and IpdC, before undergoing two successive ring cleavage reactions: EchA20-catalyzed hydrolysis of ring D followed by IpdAB-catalyzed hydrolysis of ring C. Thiolysis of the ring C-opened product, potentially by FadA6 or another thiolase, yields MOODA-CoA which is then oxidized to 2Δ-MOODA-CoA by an acyl-CoA dehydrogenase (ACAD) comprised in whole or in part by FadE32. Although the fate of 2Δ-MOODA-CoA is unclear, we propose that it undergoes a final round of β-oxidation to yield 2-methyl-β-ketoadipyl-CoA (MβKA-CoA). This could then be cleaved to propionyl-CoA and succinyl-CoA in a manner analogous to the cleavage of β-ketoadipyl-CoA to succinyl-CoA and acetyl-CoA in the final step of the β-ketoadipate pathway used in the bacterial catabolism of aromatic compounds (39). While several aspects of the HIP pathway have yet to be elucidated, three key metabolites have been definitively characterized: 5-OH HIC-CoA, COCHEA-CoA, and MOODA. Moreover, the data support the proposed physiological roles of four enzymes: IpdF, IpdC, EchA20, and IpdAB. The identity of the thiolase is less clear because FadA5 could be substituted for FadA6 (results not shown).
FIG 9 .

Proposed HIP catabolic pathway. NMR-confirmed metabolites are in blue. Metabolites for which MS data were obtained are in black. Other metabolites are in gray. *, the current study established that IpdF has this activity, but its physiological relevance is unclear. **, the role of FadE30 assigned previously (24).
Among the enzymes whose functions were assigned, only that of IpdAB was unexpected with respect to the bioinformatic analyses (Table 1). More specifically, no type I CoA transferase has been reported to catalyze a retro-aldol hydrolysis. Nevertheless, two lines of evidence indicate that IpdAB is not a CoA transferase. First, IpdABRHA1 did not catalyze the transfer of CoA between short-chain acyl substrates, in contrast to other type I CoA transferases characterized to date (33–36). Second, sequence alignments indicate that the catalytically essential glutamate in the β subunit of type I CoA transferases is not conserved in IpdAB, corresponding to Gly57β in IpdABRHA1. This glutamate, which is Glu54β in GCT, forms an anhydride with CoA in the transferase reaction (33). Finally, although inclusion of IpdABRHA1 in the reaction mixture containing IpdFMtb, IpdCDoc21, and EchA20RHA1 did not detectably alter the reaction product, COCHEA-CoA, in the absence of IpdABRHA1, the reaction only proceeded to ~10% completion. This suggests that interactions between the KstR2-encoded enzymes may accelerate the reactions.
The proposed catabolic pathway provides an important framework for further characterizing various aspects of steroid metabolism, including the identity of specific metabolites, such as HIEC-CoA and MβKA-CoA, as well as enzymatic steps, such as those catalyzed by Rv3548c and Rv3549c, encoded by the KstR2 regulon. The model further suggests the identities of the fadE-encoded ACADs that act on HIP-CoA and MOODA-CoA, respectively. Another unknown aspect of the pathway is the significance of the IpdF-catalyzed reaction: it is unclear why the 5-oxo group would be reduced and then reoxidized. Finally, this pathway also predicts that cholesterol feeds into central metabolism via four propionyl-CoAs, four acetyl-CoAs, one pyruvate, and one succinyl-CoA. Notably, propionyl-CoA, a potentially toxic metabolite (38), is derived from all three parts of cholesterol: the side chain, rings A and B, and rings C and D.
The HIP catabolic genes are conserved in steroid-degrading bacteria for which genome sequence data are available, suggesting that the pathway is employed not only in the degradation of steroids other than cholesterol (2, 3, 9), but also in the anaerobic degradation of steroids (4). Indeed, differences in the HIP catabolic gene cluster in diverse bacteria appear to reflect the different steroid-catabolizing capabilities of the strains. For example, in bacteria that catabolize cholate or other bile acids, the HIP catabolic gene cluster contains echA13 (RHA1_RS22405 in R. jostii RHA1) (1, 9, 10). A homolog of EchA20, EchA13 is proposed to remove the hydroxyl of 7β-OH HIP, generated from cholate degradation (10, 20), and is not present in M. tuberculosis, which does not degrade cholate. Similarly, the HIP catabolic gene cluster of S. denitrificans DSM 18526, which is upregulated during the aerobic and anaerobic catabolism of testosterone (4), lacks a homolog of fadD3/stdA3 (Fig. 1). Instead, this strain contains a homolog elsewhere in the genome: ACG33_09100 shares 53% amino acid sequence identity with StdA3 of P. putida DOC21 (20). The genomic context of fadD3 in S. denitrificans DSM 18526 may reflect the possibility that the β-oxidation of steroid rings A and B yields HIP-CoA directly, obviating the need for FadD3 in anaerobic steroid catabolism.
Recent interest in bacterial steroid degradation has been fueled in large part by its role in the pathogenesis of M. tuberculosis (8, 40). Disruption of cholesterol catabolic genes generates both attenuated and avirulent strains of M. tuberculosis due to the predicted accumulation of toxic cholesterol-derived metabolites (7, 41, 42). Our findings corroborate this hypothesis. Deletion of ipdAB and ipdC in M. tuberculosis yielded strains that failed to grow on glycerol in the presence of cholesterol and, in the case of ipdAB, significantly slowed the growth in THP-1-derived macrophages. These strains displayed distinct differences in the concentration and identity of CoA thioesters and CoASH. This cholesterol-dependent toxicity of the mutants may be due to the sequestration of CoASH, making it unavailable for other cellular processes. Interestingly, disruption of the ratio between acetyl-CoA and propionyl-CoA in M. tuberculosis during growth on cholesterol has been reported to result in a toxic phenotype (43). Intriguingly, reduction in CoASH in strains displaying cholesterol-dependent toxicity typically coincided with reduced acetyl-CoA levels in our CoA metabolic data (data not shown). Consistent with the sequestration hypothesis, strains that grow in the presence of cholesterol (ΔfadD3 R. jostii RHA1, ΔfadE32 M. smegmatis, and ΔipdF M. smegmatis) failed to accumulate significant levels of unique, cholesterol-derived CoA thioesters (17). The lack of toxicity in the ΔipdF and ΔfadE32 mutants could reflect the ease of hydrolysis of the corresponding CoA thioesters (e.g., MOODA-CoA to MOODA) and/or the presence of compensatory enzymatic activities (e.g., other cellular dehydrogenases could catalyze oxidation of the 5-OH HIC-CoA hydroxyl). Although additional data are required to test these hypotheses, the observed toxicity rationalizes the effectiveness of the ipdAB mutant as a vaccine in R. equi and indicates which HIP degradation enzymes would be good targets.
MATERIALS AND METHODS
Additional materials and methods are provided in Text S1 in the supplemental material.
Supplemental materials and methods. Download TEXT S1, DOCX file, 0.2 MB (167.9KB, docx) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Preparation of CoA metabolites.
Cells were grown in 900 ml pyruvate or glycerol minimal medium as described above. Cells were harvested at mid-log phase, washed with fresh medium lacking growth substrate, and then suspended in 100 ml growth medium supplemented with either 0.5 mM cholesterol, 20 mM pyruvate (R. jostii RHA1), or 0.2% glycerol (M. smegmatis and M. tuberculosis). Biotransformants were incubated for 48 h at 30°C at 200 rpm for RHA1 and 37°C at 200 rpm for M. smegmatis and 37°C in roller bottles for M. tuberculosis. Cells were cooled on ice, harvested by centrifugation, washed once with ice-cold minimal medium, and then stored at −80°C until use.
CoA thioesters were extracted from cell pellets using a modified protocol described for eukaryotic cells (44). Preparations were kept on ice or 4°C unless otherwise noted. Frozen cell pellets were suspended in 4 ml of acetonitrile-isopropanol (3:1 vol/vol) containing 15 to 50 nmol p-coumaroyl-CoA. Cells were disrupted using a FastPrep-24 bead beater (6 × 40 s at 6.5 m/s, with 5-min pauses on ice between rounds). After the first three rounds, 0.1 M KH2PO4 (pH 6.7) was added to a final concentration of ~25 mM KH2PO4. The supernatant was recovered by centrifugation (15,000 × g for 30 min), filtered through a 0.2-µm-pore regenerated cellulose membrane (Phenomenex), and acidified with 0.25 ml glacial acetic acid per ml of extract. The acidified extract was applied to a 100 mg 2-(2-pyridyl)ethyl-functionalized silica column (Supelco, 54127-U), equilibrated using 1 ml “equilibration” solution (acetonitrile-isopropanol-water-acetic acid [9:3:4:4 vol/vol:vol/vol]) at −20°C. The resin was washed with 2 ml equilibration solution at −20°C before CoA metabolites were eluted with 2 ml methanol–0.25 M ammonium acetate (pH 7) (4:1 vol/vol) at −20°C. Methanol was evaporated under N2, and then the sample was flash frozen in liquid N2 and lyophilized overnight. The lyophilized sample was suspended in 0.6 ml methanol, deposited on a Phree column (Phenomenex) to remove phospholipids, and recovered by centrifugation (500 × g for 10 min). This sample eluate was dried under N2, suspended in 0.2 ml methanol, and stored at −80°C. Immediately prior to LC-MS analysis (described below), the cellular extracts was diluted 10-fold in 0.1 M ammonium acetate (pH 4.5) and filtered in a 0.2-µm-pore polytetrafluoroethylene (PTFE) filter.
CoA metabolite profiling using MRM.
CoA thioesters were detected in cellular extracts using an Agilent 6460 triple quadrupole (QQQ) mass spectrometer operated in positive-ion mode and connected to an 80- by 0.25-mm Luna 3-μm PFP(2) (Phenomenex) analytical column through a 15- by 0.25-mm PFP(2) trapping column. CoA thioesters were separated using a gradient of 100 mM ammonium acetate (pH 4.5) into 20 mM ammonium acetate (pH 4.5) in 98% methanol over 30 min, operated at 3 μl min−1. A mixture of CoA thioester standards was run prior to each CoA metabolome to verify column performance and multiple reaction monitoring (MRM) sensitivity (see Fig. S6A in the supplemental material). Collision energy dissociation (CID) and fragmentor voltages were selected based on signal optimization using CoA thioester standards (Fig. S6). The MRM transitions recorded for each CoA metabolome are described below.
LC-MS of CoA thioester standards. (A) Peaks correspond to 25 pmol CoASH (light blue), acetyl-CoA (purple), propionyl-CoA (dark blue), 5α-OH HIC-CoA (yellow), HIP-CoA (green), and p-coumaroyl-CoA (red). (B) Representative standard curves for authentic CoA thioesters. Data points correspond to [M+H]+ → [M+H −507]+ (blue) and [M+H]+ → 428 (red) transitions. Lines represent best fit linear regression as follows: CoASH (768→261, y = 667.34x − 1,699.3, R2 = 0.9633; 768→428, y = 350.09x + 157.39, R2 = 0.9892), acetyl-CoA (810→303, y = 1,167.7x − 2,786.8, R2 = 0.9803; 810→428, y = 254.43x − 365.49, R2 = 0.9742), HIP-CoA (988→481, y = 447.29x − 1,099, R2 = 0.9734; 988→428, y = 97.542x − 192.62, R2 = 0.9735), p-coumaroyl-CoA (914→407, y = 428.8x + 19.13, R2 = 0.9786). (C) Collision energy dissociation (CID) optimization. Peak intensities of 50 pmol authentic CoA thioester standards for [M+H]+ → [M+H −507]+ (blue) and [M+H]+ → 428 (red) transitions over different CID voltages. Download FIG S6, TIF file, 0.4 MB (424.1KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
MS/MS-based untargeted analysis.
To ensure that no CoA thioesters were missed using our targeted analysis method, representative CoA metabolomes were analyzed using LC-tandem MS (MS/MS) as previously described (26). Briefly, cellular extracts were diluted 1:49 with acetonitrile-water (3:97 vol/vol) supplemented with 0.1% formic acid and then injected onto a Zorbax SB300-C18 150- by 0.075-mm column (Agilent Technologies) operated at 0.3 μl min−1 and eluted using a 10-min gradient from 3 to 97% acetonitrile. Mass spectra were recorded in positive-ion mode on an Agilent 6550 time of flight (ToF) mass spectrophotometer using a scanned mass range of 50 to 1,100 Da. Species were determined to be CoA thioesters based on the characteristic [M+H]+ −507 and 428 m/z fragments.
High-resolution MS.
High-resolution MS, MS2, and MS3 analyses of CoA thioesters were performed in positive-ion mode on a Bruker Impact-II Q-ToF mass spectrometer equipped with a 150- by 0.25-mm Luna 3-μm PFP(2) (Phenomenex) column. CoA thioesters were eluted using a gradient of 100 mM ammonium acetate in 2% methanol and 20 mM ammonium acetate in 98% methanol. The mass spectrometer was calibrated daily.
Analysis of CoA metabolomic data.
Peak integration, retention time, and the signal-to-noise (S/N) ratio were calculated using MassHunter Qualitative Analysis B.06.00 (Agilent Technologies). Peaks were defined as having an S/N ratio of >3. Analysis of CoA metabolomic data was completed using the [M+H]+ → [M+H −507]+ m/z transitions due to the higher signal intensity compared to the [M+H]+ → 428 m/z transitions, although the latter transition was confirmed for each CoA thioester characterized. CoA thioester levels were normalized to the internal standard (p-coumaroyl-CoA) prior to calculating their relative concentrations and proportion of the total cellular CoA pool. A complete summary of [M+H]+ → [M+H −507]+ transitions for each mutant is provided in Table S3 in the supplemental material.
List of targeted MRMs followed for each CoA metabolome analyzed by LC-MS. Download TABLE S3, DOCX file, 0.1 MB (20.6KB, docx) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ACKNOWLEDGMENTS
This research was supported by an Operating Grant from the Canadian Institutes for Health Research to L.D.E. and a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada to V.S. I.C. received a postdoctoral fellowship from the Fonds de Recherche en Santé du Québec and the Michael Smith Foundation for Health Research. A.M.C. was supported by a CIHR doctoral fellowship. L.E. is the recipient of a Canada Research Chair.
Footnotes
Citation Crowe AM, Casabon I, Brown KL, Liu J, Lian J, Rogalski JC, Hurst TE, Snieckus V, Foster LJ, Eltis LD. 2017. Catabolism of the last two steroid rings in Mycobacterium tuberculosis and other bacteria. mBio 8:e00321-17. https://doi.org/10.1128/mBio.00321-17.
REFERENCES
- 1.Bergstrand LH, Cardenas E, Holert J, Van Hamme JD, Mohn WW. 2016. Delineation of steroid-degrading microorganisms through comparative genomic analysis. mBio 7:e00166-16. doi: 10.1128/mBio.00166-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, Eltis LD. 2007. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A 104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horinouchi M, Hayashi T, Kudo T. 2012. Steroid degradation in Comamonas testosteroni. J Steroid Biochem Mol Biol 129:4–14. doi: 10.1016/j.jsbmb.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 4.Yang FC, Chen YL, Tang SL, Yu CP, Wang PH, Ismail W, Wang CH, Ding JY, Yang CY, Yang CY, Chiang YR. 2016. Integrated multi-omics analyses reveal the biochemical mechanisms and phylogenetic relevance of anaerobic androgen biodegradation in the environment. ISME J 10:1967–1983. doi: 10.1038/ismej.2015.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donova MV, Egorova OV. 2012. Microbial steroid transformations: current state and prospects. Appl Microbiol Biotechnol 94:1423–1447. doi: 10.1007/s00253-012-4078-0. [DOI] [PubMed] [Google Scholar]
- 6.Pandey AK, Sassetti CM. 2008. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A 105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yam KC, D’Angelo I, Kalscheuer R, Zhu H, Wang JX, Snieckus V, Ly LH, Converse PJ, Jacobs WR Jr, Strynadka N, Eltis LD. 2009. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog 5:e1000344. doi: 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB, Memmott C, Crowe AM, Eltis LD, Perola E, Deininger DD, Wang T, Locher CP, Russell DG. 2015. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog 11:e1004679. doi: 10.1371/journal.ppat.1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohn WW, Wilbrink MH, Casabon I, Stewart GR, Liu J, van der Geize R, Eltis LD. 2012. Gene cluster encoding cholate catabolism in Rhodococcus spp. J Bacteriol 194:6712–6719. doi: 10.1128/JB.01169-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casabon I, Zhu SH, Otani H, Liu J, Mohn WW, Eltis LD. 2013. Regulation of the KstR2 regulon of Mycobacterium tuberculosis by a cholesterol catabolite. Mol Microbiol 89:1201–1212. doi: 10.1111/mmi.12340. [DOI] [PubMed] [Google Scholar]
- 11.Martin CK. 1977. Microbial cleavage of sterol side chains. Adv Appl Microbiol 22:29–58. doi: 10.1016/S0065-2164(08)70159-X. [DOI] [PubMed] [Google Scholar]
- 12.Gibson DT, Wang KC, Sih CJ, Whitlock H Jr.. 1966. Mechanisms of steroid oxidation by microorganisms. IX. On the mechanism of ring A cleavage in the degradation of 9,10-seco steroids by microorganisms. J Biol Chem 241:551–559. [PubMed] [Google Scholar]
- 13.Horinouchi M, Yamamoto T, Taguchi K, Arai H, Kudo T. 2001. Meta-cleavage enzyme gene tesB is necessary for testosterone degradation in Comamonas testosteroni TA441. Microbiology 147:3367–3375. doi: 10.1099/00221287-147-12-3367. [DOI] [PubMed] [Google Scholar]
- 14.van der Geize R, Hessels GI, van Gerwen R, van der Meijden P, Dijkhuizen L. 2002. Molecular and functional characterization of kshA and kshB, encoding two components of 3-ketosteroid 9alpha-hydroxylase, a class IA monooxygenase, in Rhodococcus erythropolis strain SQ1. Mol Microbiol 45:1007–1018. doi: 10.1046/j.1365-2958.2002.03069.x. [DOI] [PubMed] [Google Scholar]
- 15.Kendall SL, Burgess P, Balhana R, Withers M, ten Bokum A, Lott JS, Gao C, Uhia-Castro I, Stoker NG. 2010. Cholesterol utilization in mycobacteria is controlled by two TetR-type transcriptional regulators: kstR and kstR2. Microbiology 156:1362–1371. doi: 10.1099/mic.0.034538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capyk JK, Casabon I, Gruninger R, Strynadka NC, Eltis LD. 2011. Activity of 3-ketosteroid 9α-hydroxylase (KshAB) indicates cholesterol side chain and ring degradation occur simultaneously in Mycobacterium tuberculosis. J Biol Chem 286:40717–40724. doi: 10.1074/jbc.M111.289975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casabon I, Crowe AM, Liu J, Eltis LD. 2013. FadD3 is an acyl-CoA synthetase that initiates catabolism of cholesterol rings C and D in actinobacteria. Mol Microbiol 87:269–283. doi: 10.1111/mmi.12095. [DOI] [PubMed] [Google Scholar]
- 18.Lee SS, Sih CJ. 1967. Mechanisms of steroid oxidation by microorganisms. XII. Metabolism of hexahydroindanpropionic acid derivatives. Biochemistry 6:1395–1403. doi: 10.1021/bi00857a023. [DOI] [PubMed] [Google Scholar]
- 19.Hashimoto S, Hayakawa S. 1977. Microbiological degradation of bile acids. Metabolites formed from 3-(3a alpha-hexahydro-7a beta-methyl-1,5-dioxoindan-4 alpha-yl) propionic acid by Streptomyces rubescens. Biochem J 164:715–726. doi: 10.1042/bj1640715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrientos A, Merino E, Casabon I, Rodriguez J, Crowe AM, Holert J, Philipp B, Eltis LD, Olivera ER, Luengo JM. 2015. Functional analyses of three acyl-CoA synthetases involved in bile acid degradation in Pseudomonas putida DOC21. Environ Microbiol 17:47–63. [DOI] [PubMed] [Google Scholar]
- 21.Griffin JE, Gawronski JD, DeJesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A 100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rengarajan J, Bloom BR, Rubin EJ. 2005. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A 102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Geize R, Grommen AW, Hessels GI, Jacobs AA, Dijkhuizen L. 2011. The steroid catabolic pathway of the intracellular pathogen Rhodococcus equi is important for pathogenesis and a target for vaccine development. PLoS Pathog 7:e1002181. doi: 10.1371/journal.ppat.1002181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maksymiuk C, Ioerger T, Balakrishnan A, Bryk R, Rhee K, Sacchettini J, Nathan C. 2015. Comparison of transposon and deletion mutants in Mycobacterium tuberculosis: the case of rv1248c, encoding 2-hydroxy-3-oxoadipate synthase. Tuberculosis 95:689–694. doi: 10.1016/j.tube.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Q, Zhang S, Berthiaume JM, Simons B, Zhang GF. 2014. Novel approach in LC-MS/MS using MRM to generate a full profile of acyl-CoAs: discovery of acyl-dephospho-CoAs. J Lipid Res 55:592–602. doi: 10.1194/jlr.D045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horinouchi M, Hayashi T, Kudo T, Koshino H. 2006. Manufacture of 3-(2-carboxyethyl)-4-methyl-2-cyclohexenone and derivatives with Comamonas mutant. Jpn Kokai Tokkyo Koho Japanese patent JP 2006271380 A 20061012.
- 28.Sogabe S, Yoshizumi A, Fukami TA, Shiratori Y, Shimizu S, Takagi H, Nakamori S, Wada M. 2003. The crystal structure and stereospecificity of levodione reductase from Corynebacterium aquaticum M-13. J Biol Chem 278:19387–19395. doi: 10.1074/jbc.M208146200. [DOI] [PubMed] [Google Scholar]
- 29.Saito J, Yamada M, Watanabe T, Iida M, Kitagawa H, Takahata S, Ozawa T, Takeuchi Y, Ohsawa F. 2008. Crystal structure of enoyl-acyl carrier protein reductase (FabK) from Streptococcus pneumoniae reveals the binding mode of an inhibitor. Protein Sci 17:691–699. doi: 10.1110/ps.073288808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamed RB, Batchelar ET, Clifton IJ, Schofield CJ. 2008. Mechanisms and structures of crotonase superfamily enzymes—how nature controls enolate and oxyanion reactivity. Cell Mol Life Sci 65:2507–2527. doi: 10.1007/s00018-008-8082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li HJ, Li X, Liu N, Zhang H, Truglio JJ, Mishra S, Kisker C, Garcia-Diaz M, Tonge PJ. 2011. Mechanism of the intramolecular Claisen condensation reaction catalyzed by MenB, a crotonase superfamily member. Biochemistry 50:9532–9544. doi: 10.1021/bi200877x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pelletier DA, Harwood CS. 1998. 2-Ketocyclohexanecarboxyl coenzyme A hydrolase, the ring cleavage enzyme required for anaerobic benzoate degradation by Rhodopseudomonas palustris. J Bacteriol 180:2330–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacob U, Mack M, Clausen T, Huber R, Buckel W, Messerschmidt A. 1997. Glutaconate CoA-transferase from Acidaminococcus fermentans: the crystal structure reveals homology with other CoA-transferases. Structure 5:415–426. doi: 10.1016/S0969-2126(97)00198-6. [DOI] [PubMed] [Google Scholar]
- 34.Corthésy-Theulaz IE, Bergonzelli GE, Henry H, Bachmann D, Schorderet DF, Blum AL, Ornston LN. 1997. Cloning and characterization of Helicobacter pylori succinyl CoA:acetoacetate CoA-transferase, a novel prokaryotic member of the CoA-transferase family. J Biol Chem 272:25659–25667. doi: 10.1074/jbc.272.41.25659. [DOI] [PubMed] [Google Scholar]
- 35.Sramek SJ, Frerman FE. 1975. Purification and properties of Escherichia coli coenzyme A-transferase. Arch Biochem Biophys 171:14–26. doi: 10.1016/0003-9861(75)90002-8. [DOI] [PubMed] [Google Scholar]
- 36.Heider J. 2001. A new family of CoA-transferases. FEBS Lett 509:345–349. doi: 10.1016/S0014-5793(01)03178-7. [DOI] [PubMed] [Google Scholar]
- 37.Schaefer CM, Lu R, Nesbitt NM, Schiebel J, Sampson NS, Kisker C. 2015. FadA5 a thiolase from Mycobacterium tuberculosis: a steroid-binding pocket reveals the potential for drug development against tuberculosis. Structure 23:21–33. doi: 10.1016/j.str.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griffin JE, Pandey AK, Gilmore SA, Mizrahi V, McKinney JD, Bertozzi CR, Sassetti CM. 2012. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem Biol 19:218–227. doi: 10.1016/j.chembiol.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harwood CS, Parales RE. 1996. The beta-ketoadipate pathway and the biology of self-identity. Annu Rev Microbiol 50:553–590. doi: 10.1146/annurev.micro.50.1.553. [DOI] [PubMed] [Google Scholar]
- 40.Ouellet H, Johnston JB, de Montellano PR. 2011. Cholesterol catabolism as a therapeutic target in Mycobacterium tuberculosis. Trends Microbiol 19:530–539. doi: 10.1016/j.tim.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nesbitt NM, Yang X, Fontán P, Kolesnikova I, Smith I, Sampson NS, Dubnau E. 2010. A thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infect Immun 78:275–282. doi: 10.1128/IAI.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu Y, van der Geize R, Besra GS, Gurcha SS, Liu A, Rohde M, Singh M, Coates A. 2010. 3-ketosteroid 9alpha-hydroxylase is an essential factor in the pathogenesis of Mycobacterium tuberculosis. Mol Microbiol 75:107–121. doi: 10.1111/j.1365-2958.2009.06957.x. [DOI] [PubMed] [Google Scholar]
- 43.Lee W, VanderVen BC, Fahey RJ, Russell DG. 2013. Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J Biol Chem 288:6788–6800. doi: 10.1074/jbc.M112.445056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Minkler PE, Kerner J, Ingalls ST, Hoppel CL. 2008. Novel isolation procedure for short-, medium-, and long-chain acyl-coenzyme A esters from tissue. Anal Biochem 376:275–276. doi: 10.1016/j.ab.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Growth and CoA metabolites of R. jostii RHA1 strains. (A to D) Growth of WT::pTip-Qc2 (blue), ΔipdAB::pTip-Qc2 (red), ΔipdAB::pTipCoL51 (red, dashed), ΔipdC::pTipQc2 (green), and ΔipdC::pTipRv3553 (green, dashed) on (A) 10 mM pyruvate, (B) 1 mM cholesterol, (C) 1.5 mM HIP, and (D) 1 mM HIP plus 10 mM pyruvate. (E) Depletion of HIP by RHA1 strains, color coded as in growth curves as measured by GC-MS and reported as percentage of initial levels. Data are the mean from triplicates. Error bars show standard deviations. (F) LC-MS chromatograms of CoA metabolites extracted from WT (blue) and ΔipdAB (red) RHA1 incubated with cholesterol. Numbers correspond to CoASH (1), acetyl-CoA (2), propionyl-CoA (3), and COCHEA-CoA (4). IS, internal standard. Data for panel D were acquired using a BioScreen C (Growth Curves USA). Download FIG S1, TIF file, 0.3 MB (336.5KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Growth and CoA metabolites of ΔipdC M. tuberculosis. WT (black), ΔipdC (red), and ΔipdC::ipdC (blue) M. tuberculosis Erdman were grown on (A) 1 mM cholesterol, (B) 0.2% glycerol, or (C) 0.5 mM cholesterol and 0.2% glycerol. (D) CoA metabolome of ΔipdC (red) and WT M. tuberculosis (blue) incubated with 0.5 mM cholesterol. Arrows indicate the peaks corresponding to the 5-OH HIC-CoA in the ΔipdC R. jostii RHA1 CoA metabolome. Download FIG S2, TIF file, 0.3 MB (354.4KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains, plasmids, and oligonucleotides used in this study. Download TABLE S1, DOCX file, 0.1 MB (60.6KB, docx) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
List and characterization of CoA thioesters in M. tuberculosis, R. jostii RHA1, M. smegmatis, and ΔipdAB mutants. Download TABLE S2, DOCX file, 0.1 MB (21KB, docx) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
CoA thioesters and metabolites produced by M. smegmatis strains. (A) WT and (B) ΔipdAB cells were incubated with cholesterol (blue) and glycerol (gray). Numbers represent CoASH (1), acetyl-CoA (2), succinyl-CoA (3), propionyl-CoA (4), unidentified CoA thioester of 838 m/z (5), unidentified CoA thioester of 852 m/z (6), and COCHEA-CoA (7). (C) GC-MS of culture supernatants of cholesterol-incubated M. smegmatis strains. Cells of the indicated strains were incubated with 0.5 mM cholesterol. Insets show structures of TMS-derivatized 5α-OH HIC and 2-(2-carboxyethyl)-3-methyl-6-oxocyclohex-1-ene-1. *, unidentified compounds present in all M. smegmatis extracts that are unrelated to cholesterol catabolism. Download FIG S3, TIF file, 0.4 MB (384.6KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic tree of enoyl-CoA hydratases (EchAs) and MenB in M. tuberculosis. A cluster containing EchA20 and MenBMtb is highlighted with a red oval. Download FIG S4, TIF file, 0.1 MB (86.6KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Electrophoretic analyses of gene deletion mutants and purified proteins. (A) PCR confirmation of ΔipdAB in R. jostii RHA1 and M. tuberculosis, ΔipdC in RHA1 and M. tuberculosis, and ΔipdF, ΔechA20, and ΔfadE32 in M. smegmatis using the listed primer sets (Table S1). (B) SDS-PAGE loaded, from left to right, with 0.5 μg each of MBP-IpdCDOC21, IpdFMtb, EchA20RHA1, IpdABRHA1, and FadA6Mtb. Purified proteins are flanked by molecular weight standards. Download FIG S5, TIF file, 0.4 MB (418.6KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental materials and methods. Download TEXT S1, DOCX file, 0.2 MB (167.9KB, docx) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
LC-MS of CoA thioester standards. (A) Peaks correspond to 25 pmol CoASH (light blue), acetyl-CoA (purple), propionyl-CoA (dark blue), 5α-OH HIC-CoA (yellow), HIP-CoA (green), and p-coumaroyl-CoA (red). (B) Representative standard curves for authentic CoA thioesters. Data points correspond to [M+H]+ → [M+H −507]+ (blue) and [M+H]+ → 428 (red) transitions. Lines represent best fit linear regression as follows: CoASH (768→261, y = 667.34x − 1,699.3, R2 = 0.9633; 768→428, y = 350.09x + 157.39, R2 = 0.9892), acetyl-CoA (810→303, y = 1,167.7x − 2,786.8, R2 = 0.9803; 810→428, y = 254.43x − 365.49, R2 = 0.9742), HIP-CoA (988→481, y = 447.29x − 1,099, R2 = 0.9734; 988→428, y = 97.542x − 192.62, R2 = 0.9735), p-coumaroyl-CoA (914→407, y = 428.8x + 19.13, R2 = 0.9786). (C) Collision energy dissociation (CID) optimization. Peak intensities of 50 pmol authentic CoA thioester standards for [M+H]+ → [M+H −507]+ (blue) and [M+H]+ → 428 (red) transitions over different CID voltages. Download FIG S6, TIF file, 0.4 MB (424.1KB, tif) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
List of targeted MRMs followed for each CoA metabolome analyzed by LC-MS. Download TABLE S3, DOCX file, 0.1 MB (20.6KB, docx) .
Copyright © 2017 Crowe et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.