

Abstract
The oral route is an attractive delivery route for the administration of DNA-based therapeutics, specifically for applications in gene therapy and DNA vaccination. However, oral DNA delivery is complicated by the harsh and variable conditions encountered throughout gastrointestinal (GI) transit, leading to degradation of the delivery vector and DNA cargo, and subsequent inefficient delivery to target cells. In this work, we demonstrate the development and optimization of a hybrid-dual particulate delivery system consisting of two natural biomaterials, zein (ZN) and chitosan (CS), to mediate oral DNA delivery. Chitosan-Zein Nano-in-Microparticles (CS-ZN-NIMs), consisting of core Chitosan/DNA nanoparticles (CS/DNA NPs) prepared by ionic gelation with sodium tripolyphosphate (TPP), further encapsulated in ZN microparticles, were formulated using a water-in-oil emulsion (W/O). The resulting particles exhibited high CS/DNA NP loading and encapsulation within ZN microparticles. DNA release profiles in simulated gastric fluid (SGF) were improved compared to un-encapsulated CS/DNA NPs. Further, site-specific degradation of the outer ZN matrix and release of transfection competent CS/DNA NPs occurred in simulated intestinal conditions with CS/DNA NP cores successfully mediating transfection in vitro. Finally, CS-ZN-NIMs encoding GFP delivered by oral gavage in vivo induced the production of anti-GFP IgA antibodies, demonstrating in vivo transfection and expression. Together, these results demonstrate the successful formulation of CS-ZN-NIMs and their potential to improve oral gene delivery through improved protection and controlled release of DNA cargo.
Keywords: Oral delivery, DNA delivery, Zein, Chitosan, Gene Therapy, DNA vaccination
Graphical Abstract
1. Introduction
Gene delivery involves the introduction of exogenous genetic material to somatic cells of patients with the subsequent expression of a therapeutic molecule. Because all basic physiological processes are regulated by gene expression, gene delivery strategies represent an enormous potential for therapeutic treatment of numerous diseases as well as an alternative method for vaccination [1]. Currently, gene delivery is accomplished by means of two main delivery systems: viral and nonviral systems. While viral delivery vectors are extremely efficient in delivering DNA in vitro and in vivo, there remains concerns with their toxicity, the potential of generating unwanted immune responses, and their limited DNA cargo capacity. Nonviral delivery systems, which typically make use of cationic polymers or lipids to electrostatically combine with, or physically encapsulate DNA, can overcome many of the toxicity and immune concerns of viral vectors. However, they suffer from low delivery efficiency, highlighting the need for nonviral vectors that improve transgene delivery while remaining non-toxic.
Of the many routes of administration for nonviral gene delivery systems, the oral route is preferable due to high patient compliance and the ease of administration and dosing [2, 3]. The oral route is of particular interest for the applications of gene therapy and DNA vaccination due to continuously cycling cells of the intestinal epithelium, which present a large cellular surface area for transfection with a therapeutic gene [4]. Additionally, the highly vascularized nature of the intestinal epithelium [5] facilitates both local (e.g. Crohn’s disease [6] and ulcerative colitis [7]) and systemic therapies (e.g. diabetes [8], hemophilia A [9, 10] and B [11], and hypoparathyroidism [12]). Because of the potential for treating both local and systemic diseases, gene therapy via oral delivery has been an area of intense focus in recent years [6, 11, 13, 14].
In addition to gene therapy, the oral route of delivery is also considered to be highly promising as an administration route for DNA vaccines. DNA vaccination involves the delivery of plasmid DNA containing a transgene encoding a disease-specific target antigen driven by a eukaryotic promoter, resulting in the intracellular production and subsequent sampling of the target antigen by professional antigen presenting cells (APCs) [15, 16]. DNA vaccines are an attractive alternative to traditional vaccine strategies, most of which are largely protein-based and often fail to generate complete cell and antibody-mediated immune responses [17]. Orally delivered DNA vaccines have the additional advantage of being able to target mucosal membranes (i.e. intestinal mucosa) to generate mucosal immunity [18] while also meeting the requirements for effective global mobilization (i.e. no dependence on “cold chain” storage and transportation).
Despite the enormous potential of oral gene therapy and DNA vaccination strategies, the efficient delivery of DNA-based therapeutics via the oral route is complicated by the harsh environment of the gastrointestinal (GI) tract (i.e., low pH, gastric enzymes and endogenous nucleases) and the low permeability of the intestinal epithelium. Numerous efforts at achieving oral delivery have been made employing several synthetic [12, 19, 20] and natural polymers [11, 21] as well as lipid-based formulations [22]. However, those systems often yield only modest transgene expression due to the instability of the delivery systems. In order to overcome stability issues, recent attempts at oral DNA delivery have focused on the use of dual material systems, taking advantage of differing material properties to protect the encapsulated payload through gastric transit and allow for efficient delivery at the site of interest [1, 6, 23, 24]. However, these systems make use of synthetic materials with very slow degradation rates that limit the release and subsequent delivery of the DNA cargo and require harsh solvents and fabrication techniques that may compromise DNA integrity.
Zein (ZN), the major prolamine from corn, is a natural biomaterial that has received recent attention for gene delivery and tissue engineering applications due to its inherent biocompatibility and degradability [25, 26]. Being amphiphilic in nature, ZN has the ability to self-assemble into a variety of micro and nanostructures including nanoparticles [27, 28], microparticles [29, 30], fibers [31, 32], and films [33, 34]. Moreover, ZN can be processed using simple techniques including coacervation [35], phase separation [36], emulsion with solvent evaporation [37], anti-solvent precipitation [38], and liquid-liquid dispersion [39], and can successfully encapsulate a variety of hydrophobic and hydrophilic compounds, including vitamins [40], antibiotics [41], essential oils [42], and, in our previous study, DNA [26].
ZN’s insolubility in aqueous conditions, resistance to gastric enzymes and acidic pH, and rapid degradation in the presence of intestinal enzymes, make it an ideal candidate for an oral delivery vehicle. To date, ZN has been investigated as an oral drug delivery system for reactive oxygen species scavengers [43], insulin [44], protein antigens [35], steroid drugs for inflammatory bowel disease [45], and antioxidants [46]. However, ZN is not suitable for gene delivery applications by itself due to its high stability in physiological conditions and subsequent slow release of encapsulated DNA leading to inadequate transfection levels [26]. The inability of ZN to mediate efficient transfection highlights the need for a secondary material to fully realize the potential of ZN as an oral gene delivery vehicle. Chitosan (CS), another natural polymer, has been widely used in oral nonviral gene delivery applications including gene therapy and DNA vaccination, and is uniquely suited for intestinal delivery due to its mucoadhesive nature and stability in intestinal conditions. However, CS is unstable in gastric conditions due to its solubility in acidic aqueous conditions [47], highlighting the need for a second, gastric-protective outer material. In this study, we report the development of a CS/DNA-ZN nano-in-microparticle (CS-ZN-NIM) dual-particulate oral DNA delivery system, consisting of inner CS/DNA NP cores further encapsulated in ZN microparticles, using a single water-in-oil (W/O) emulsion. We hypothesize that the outer ZN microparticle protects the DNA cargo though gastric transit, and upon reaching the small intestine, rapidly and completely degrades via enzymatic action to release chitosan/DNA nanoparticles (CS/DNA NPs) capable of being internalized by and transfecting cellular targets in the small intestine. We provide evidence of ZN-mediated protection of CS/DNA NPs from simulated gastric fluid (SGF), their release from ZN microparticles following simulated in vitro GI transit, and transgene expression mediated from CS-ZN-NIMs subjected to simulated GI fluid treatment. Furthermore, we provide evidence of in vivo transgene expression following oral delivery of CS-ZN-NIMs to mice, and subsequent activation of a mucosal immune response, highlighting the potential of this oral delivery system for use in DNA vaccination and gene therapy applications.
2. Materials and Methods
2.1 Materials, Cell lines, and Cell Culture
Chitosan oligosaccharide lactate (Avg MW 5000), pepsin from porcine gastric mucosa, chitosanase from Streptomyces sp., sodium tripolyphosphate (TPP), petroleum ether, lysozyme from chicken egg white and Whatman No. 5 qualitative filter papers were purchased from Sigma-Aldrich (St. Louis, MO). Pancreatin, USP grade from porcine pancreas was purchased from MP Biomedicals (Santa Ana, CA). F4400 ZN was purchased from Freeman Industries LLC (Tuckahoe, NY). Hoechst 33258 nuclei stain was purchased from Life Technologies (Eugene, OR). 32P radiolabeled deoxyadenosine triphosphate and Ultima Gold liquid scintillation cocktail was purchased from Perkin Elmer (California, USA). The DNA Nick Translation System for radiolabeling plasmid DNA was purchased from Invitrogen Life Technologies (Carlsbad, CA). Water-soluble tetrazolium salt (WST-1) cell proliferation reagent was purchased from Roche Life Sciences (Indianapolis, IN). Human embryonic epithelial kidney cells, HEK 293T (ATCC, Manassas, VA), were cultured in T-75 flasks in Dulbecco’s modified Eagle’s medium (DMEM, Gibco/Invitrogen, Carlsbad, CA) containing 4.5 g/L glucose and 2 mM L-glutamine (Gibco), and supplemented with 10% fetal bovine serum (FBS, Gibco), and 1% penicillin/streptomycin (Gibco) and 100 mM sodium pyruvate at a final concentration of 1%. Human colon carcinoma cells, Caco-2 (ATCC) were cultured in T-75 flasks in Eagle’s minimum essential medium (EMEM, ATCC) supplemented with 20% FBS (Gibco) and 1% penicillin/streptomycin (Gibco). Mouse macrophages, RAW 264.7, (ATCC) were cultured in T-25 flasks in DMEM (Gibco) supplemented with 10% FBS and 1% penicillin/streptomycin. ELISA flat-bottom immuno MaxiSorp 96 well plates were purchased from Fisher Scientific (Pittsburgh, PA). Recombinant green fluorescent protein (GFP) from A. Victoria was purchased from Abcam (Cambridge, MA). Alkaline phosphatase conjugated goat anti-mouse IgA antibody and p-nitrophenol phosphate one component microwell substrate were purchased from SouthernBiotech (Birmingham, AL).
2.2 Mice
Male BALB/cByJ mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice 6–8 weeks old were used in all experiments. Experimental animal procedures using mice were approved by and conducted in accordance with the Institutional Animal Care and Use Committee (IACUC) at the University of Nebraska-Lincoln.
2.3 Plasmid Preparation and 32P radiolabeling of plasmid
All transfection experiments were performed using the pEGFP-LUC (Clontech, Mountain View, CA) plasmid encoding for both firefly luciferase protein and green fluorescent protein under the direction of the CMV promoter. The plasmid was purified from E. coli using a Qiagen Giga kit (Valencia, CA) and stored in Tris-EDTA (TE) buffer solution (10 mM Tris, 1mM EDTA, pH 7.4) at −20° until use. Plasmid DNA (pEGFP-LUC) was radiolabeled with 32P α-dATP (3000 Ci/mmol, 250 μCi) using the Invitrogen Nick Translation System according to the manufacturer’s protocol with minor modifications. Briefly, a total of 6 μg of pEGFP-LUC plasmid (1 μg/μL) was diluted to 100 ng/μL in TE buffer, to which 6 nmol of dNTP mix minus dATP, 250 μCi of 32P α-dATP, ultrapure H2O, and DNA Polymerase I/DNase I mix were added. The reaction was allowed to run for 60 minutes at 15°C, at which point 10 μL stop buffer was added. The resulting labeled DNA was purified using a Qiagen MiniPrep kit and diluted with unlabeled pEGFP-LUC plasmid to a final concentration of 0.801 μg/μL
2.4 Formation of CS/DNA NPs
CS/DNA NPs encapsulating pEGFP-LUC were prepared using a modified version of the methods described by Calvo et al [48] based on the ionic gelation of CS with TPP. A 5 mg/mL solution of CS oligosaccharide lactate was made by dissolving the CS in ultrapure water. The resulting solution was filtered with a 0.22 μm syringe filter (EMD Millipore, Billerica, MA) to remove any impurities. Similarly, a 0.5 mg/mL TPP solution was prepared in ultrapure water. To prepare the nanoparticles, the stock 5.0 mg/mL CS solution was diluted in ultrapure water to achieve various chitosan concentrations (0.5 – 2.0 mg/mL). Plasmid DNA in TE buffer (10 mM Tris, 1mM EDTA, pH 7.4) was added to the TPP solutions, mixed thoroughly and then TPP/DNA solution was added dropwise to the CS solution. The resulting NP suspension was immediately vortexed for 15 seconds and then allowed to incubate at room temperature for 20 minutes. The amount of TPP solution used in the particle formulation was varied to achieve various CS:TPP ratios (w/w). For all characterization and transfection studies, the amount of DNA used in particle formation was held constant at 10% (w/w) of the amount of CS used in the formulation.
2.5 Characterization of CS/DNA NPs and determination of DNA encapsulation efficiency
The size and zeta potential of the CS/DNA NPs was determined by dynamic light scattering and Laser Doppler micro-electrophoresis, respectively, using a Zetasizer Nano ZS90 (Malvern Instruments Ltd, UK). Size measurements were taken at 25°C at a scattering angle of 90° and size reported as the Z-average diameter. Zeta potential measurements were also taken at 25°C using folded capillary cells with the measurement mode set to automatic. DNA encapsulation efficiency of the CS NPs was determined by measuring the amount of free DNA in the aqueous particle suspension after particle formation. Freshly prepared NPs were centrifuged at 10,000 x g for 20 minutes and the amount of DNA in the supernatant was measured using the Hoechst assay. Briefly, supernatant samples were diluted in 1X TNE buffer (10 mM Tris; 0.2 M NaCl; 1 mM EDTA; pH 7.4) to a final volume of 1 mL and mixed with 1 volume of a 200 ng/mL solution of Hoechst 33258 dye. After a 5-minute incubation at room temperature, fluorescence was measured using a modulus luminometer/fluorometer (Turner Biosystems, Sunnyvale, CA). The encapsulation efficiency of the CS NPs was determined by calculating the difference between the amount of measured free DNA and the initial amount of DNA used in the particle preparation according to equation 1:
| (1) |
2.6 Transfection efficiency of CS/DNA NPs
Transfection studies with CS/DNA NPs encapsulating pEGFP-LUC were performed using the human embryonic kidney cell line (HEK293T). Cells were seeded in 48 well plates at a seeding density of 33,000 cells/well in a total of volume of 300 μL of complete Dulbecco’s Modified Eagle’s Medium (DMEM) 18 hours prior to delivery of the CS/DNA NPs. The day of transfection, CS/DNA NPs were prepared as described above. CS/DNA NPs encapsulating 1 μg of DNA were diluted in an appropriate volume of serum free Opti-MEM to deliver a total volume of 75 μL/well and then added to each well. The NPs were allowed to incubate with the cells for 24 hours at which point transfection was assessed qualitatively using fluorescent microscopy. Transfection levels were quantified using the luciferase assay system (Promega, Madison, WI) to measure luciferase activity. Briefly, cells were lysed by adding 200 μL of 1X reporter lysis buffer (Promega) and the cell lysates were measured for luciferase activity using a luminometer with an integration time of 10 seconds. Measured relative light units (RLU) were normalized to total protein content measured by the BCA protein assay (Pierce).
2.7 Formation of CS-ZN NIMs
CS/DNA NPs were encapsulated in ZN microparticles using a W/O emulsion with solvent evaporation technique as described in [37] with some modifications. Briefly, CS/DNA NPs were formed as described above and collected by centrifugation at 10,000x g for 20 min to remove the particles from any unbound DNA and any free CS. The collected NPs were then completely re-suspended in 50 μL of ultrapure water and subsequently added to 1.5 mL of ZN solution (5–25% w/v) previously dissolved in 90% (v/v) ethanol and pH adjusted to 8–9. The CS/DNA NP-ZN suspension was immediately added to 150 mL of cold corn oil containing 0.75% span 80 (v/v) stirred at a constant rate of 800 rpm on a magnetic stirrer. After the microparticles had completely hardened, the particles were collected by vacuum filtration, washed repeatedly with petroleum ether to remove excess oil, and oven dried at 50°C overnight. Particles were stored at 4° until use.
2.8 Scanning electron microscopy of CS-ZN NIMs and particle size determination
CS-ZN NIMs formed as described above were oven dried for 24 hours and then stored under vacuum prior to scanning electron microscopy (SEM) imaging. The microparticles were mounted to double-sided carbon tape on brass stubs, and sputter coated with gold under argon atmosphere before SEM imaging (Nova NanoSEM 450) Micrographs were taken at a beam voltage of 5 kV and spot size of 3. Due to both the large size of the CS-ZN-NIMs and the interference with DLS measurements from ZN autofluorescence, the number-average diameter of the CS-ZN NIMs was determined from SEM image analysis. Briefly, particle formulations were prepared in triplicate and representative images of particles from each sample were analyzed using the ImageJ particle analysis tool. A minimum of 150 particles was analyzed for sizing purposes.
2.9 Determination of DNA loading and encapsulation in CS-ZN NIMs
Loading and encapsulation of CS/DNA NPs into ZN microparticles was determined using radiolabeled DNA due to the autofluorescence of ZN [26], which makes the use of DNA binding dyes (e.g. Hoechst, PicoGreen) for measuring DNA inaccurate. Briefly, CS/DNA NPs were formed as previously described at varying CS:DNA ratios (5, 10, and 25) with 32P α-dATP labeled pEGFP-LUC. The resulting CS/DNA NPs were encapsulated in ZN microparticles formed from varying ZN solutions (12, 15, and 25% w/v) as described above. The particles were filtered from the oil and added to 15 mL of Ultima Gold liquid scintillation cocktail and counts per minute were read using a Packard 1900 TR Liquid Scintillation Counter. The amount of DNA in the particles was determined using a standard curve with known amounts of DNA. Loading was calculated as μg of DNA/g of ZN particles. DNA encapsulation was calculated as the amount of DNA measured in the ZN particles divided by the initial amount of DNA used in particle preparation (equation 2):
| (2) |
2.10 Cell viability assay and CS-ZN-NIMs biocompatibility
The biocompatibility of the CS-ZN-NIMs was assessed using the WST-1 cell proliferation assay. Briefly, HEK293T, Caco-2, or RAW 264.7 cells were seeded at a density of 36,000, 25,000, and 280,000 cells per well, respectively, in 48 well plates. Eighteen hours after plating, 0.15 mg of CS-ZN-NIMs (formed using 12, 15, or 25% ZN) in serum-free Optimem were added to each well. Cells and particles were cocultured for 24 hours. Media and particles were then discarded and cells were washed with 200 μL of PBS. Each well received 336 μL of WST-1 reagent diluted at a 1:10 ratio in serum free DMEM (Gibco); cells and reagent were incubated for three hours at 37°C. After incubation, 150 μL of the WST-1 reagent from each well was transferred to a 96-well plate and absorbance at 430 and 690 nm was read using an Epoch Microplate spectrophotometer (BioTek, Winooski, VT). Empty wells containing only WST-1 reagent were used as a blank and absorbance values were normalized to cells that received no particles.
2.11 Gastric release of DNA from CS-ZN-NIMs and Uncoated CS/DNA NPs
The release profile of DNA from CS-ZN-NIMs was determined using 32P α-dATP labeled pEGFP-LUC due to the autofluorescence of ZN [26]. Briefly, CS-ZN-NIMs, prepared as described above, were incubated in 4 mL of SGF (0.034 M NaCl, 0.085 M HCl, 3.2 g/L pepsin, pH ~1.2) (Gastric Fluid, USP) for varying times (0–240 min) at 37°C with agitation. At various time points, particles were centrifuged at 500 g for 5 minutes; 2 mL of the supernatant were removed and collected into 20 mL scintillation vials containing 10 mL of Ultima Gold scintillation cocktail. The particles were then resuspended in 2 mL of fresh release media and the incubation continued. Upon completion of the release study, the particles were collected, and the DNA released at each time point, as well as the DNA remaining in the particles, were measured with a Packard 1900 TR Liquid Scintillation Counter. The amount of DNA in the samples was determined using a standard curve with known amounts of DNA and release was calculated using equation (3):
| (3) |
Due to issues with resuspension of CS/DNA NPs following SGF incubation, measuring release from the same sample over multiple time points was technically challenging. Therefore, release from CS/DNA NPs (without ZN) was determined by centrifuging and collecting entire samples at various time points. Particles were centrifuged at 10,000 x g for 20 min at 4°C and the supernatant discarded. The remaining CS/DNA NPs were subjected to a chitosanase/lysozyme digestion to release encapsulated DNA. 0.5 μL of chitosanase and 0.5 μL lysozyme (equivalent to 0.06 and 214 Units, respectively) in 400 μL of a 50 mM sodium acetate-acetic acid buffer at pH 5.5 were added to CS/DNA NPs and incubated at 37° C for 4 hours. Following the chitosanase/lysozyme digestion, the DNA in both the digested sample and supernatant was measured using the Hoechst assay as described above. Release from CS/DNA NPs was calculated using equation (4):
| (4) |
2.12 Intestinal enzyme mediated release of CS/DNA NPs from CS-ZN NIM and transfection profiles
To determine the mechanism of ZN degradation and release of CS/DNA NPs from CS-ZN-NIMs, a series of simulated intestinal fluid (SIF) incubations were performed with the particles prior to delivery to cells. Briefly, HEK293T cells were seeded at a density of 36,000 cells per well in a 48 well plate 18 hours prior to the addition of treated particles. The day of transfection, CS-ZN-NIMs encapsulating pEGFP-LUC, and formed using various ZN solutions (5, 7, 10, 12, 15 and 25% w/v), were incubated in either 10 mL of SIF pH 6.8 containing pancreatin (Intestinal Fluid, USP) at 37° or 10 mL of PBS at 37°C for 60 minutes with continuous agitation. After 60 minutes, or varying times (0, 15, 30, 60 minutes) in SIF or PBS, the CS-ZN-NIMs were filtered through a 2.5 μm cellulose filter to remove residual ZN and residual pancreatin. The filtrate was collected and centrifuged at 5200 rpm for 20 minutes to collect CS/DNA NP cores. These NPs were resuspended in Opti-MEM and 75 μL of NP suspension was added to each well of cells. Twenty-four hours after delivery of these “conditioned” NPs to cells, luciferase expression was quantified as described above.
2.13 In vitro simulation of oral delivery of CS-ZN-NIMs particles and transfection studies
To determine the ability of the ZN-CS-NIMs to protect the CS/DNA NPs through the gastric environment and subsequently release intact particles for delivery in the intestinal compartment, a series of in vitro simulations of oral delivery were performed in simulated gastric and intestinal fluids. For transfection studies, HEK293T cells were seeded in 48 well plates at a density of 36,000 cells/well 18 hours prior to oral delivery simulation. The day of transfection, dried CS-ZN-NIMs encapsulating pEGFP-LUC were first incubated in 10 mL of SGF at pH ~1.2 containing pepsin (Gastric Fluid, USP) for 15 min at 37°C. Particles were then collected by centrifugation at 5200 rpm for 10 min, the gastric fluid completely removed and the particles resuspended in 10 mL of SIF at pH 6.8 containing pancreatin (Intestinal Fluid, USP) and incubated for an additional 60 min at 37°C. SIF containing the CS-ZN-NIMs was then filtered as described above. The filtrate was collected and centrifuged at 5200 rpm for 20 min to collect the CS/DNA NPs. After centrifugation, particles were washed twice with PBS and then resuspended in 240 μL of serum-free Opti-MEM media. For delivery to cells, 75 μL of the CS/DNA NPs suspended in Opti-MEM were added directly to each well. Transfection was qualitatively assed 24 hours after transfection with fluorescent microscopy. Transfection levels were quantified using the luciferase assay system as described above.
2.14 Oral Delivery of CS-ZN-NIMs
The ability of the CS-ZN-NIMs to mediate transgene expression following oral delivery was evaluated in mice. For oral delivery, CS-ZN-NIMs (prepared using 12% ZN solution, CS:DNA ratio = 10, CS:TPP ratio = 8) encapsulating the pEGFP-LUC plasmid were formed as described above. Prior to CS-ZN-NIM oral delivery, 6–8 week old male BALB/c mice (Jackson Laboratories) were fasted overnight. The following day, CS-ZN-NIMs encapsulating either 35 or 100 μg of pEGFP-LUC plasmid DNA were suspended in saline and administered to mice (4 mice/treatment group) via oral gavage. Two mice received 20 μg of recombinant GFP adjuvanted with Addavax (Invivogen, San Diego, CA) via intraperitoneal injection (GFP:Addavax 1:1 (v/v) in a total volume of 500 μL) as a positive control for generating anti-GFP antibodies. A separate cohort of mice (3 mice/group) received either 35 or 100 μg of pEGFP-LUC plasmid DNA encapsulated in uncoated CS/DNA NPs formulated using the same conditions as those loaded into CS-ZN-NIMs (CS:DNA ratio = 10, CS:TPP ratio = 8). For this, CS/DNA NPs were formed as described above, centrifuged to remove any un-encapsulated DNA, resuspended in 200 μL saline, and delivered to mice via oral gavage.
2.15 Evaluation of antibody response to orally administered CS-ZN-NIMs: ELISA assay
To analyze antibody production in mice treated with either CS-ZN-NIMs, CS/DNA NPs, or recombinant GFP protein and adjuvant, fecal samples were collected three weeks after the initial oral administration of CS-ZN-NIMs. Fecal samples were weighed and diluted at 1:20 (w/v) in PBS. An ELISA assay was performed to quantify anti-GFP IgA titers. For the ELISA, 96 well plates (MaxiSorp, Nunc USA) were coated with 50 μL of recombinant GFP protein (10 μg/mL in 1X PBS) and allowed to adsorb overnight before the assay was performed. The following day, plates were blocked for one hour with blocking buffer (1X PBS, 2% FBS, 10 mM HEPES, pH 7.4). Fecal slurries were added at a starting dilution of 1:20 and subsequently diluted 1:2 to obtain a dilution curve. Samples were incubated for 2.5 hours, extensively washed, and incubated with alkaline phosphatase (AP)-conjugated goat anti-mouse IgA antibody (1:1000 dilution in blocking buffer) for an additional hour. After one hour, 50 μL of pNPP AP substrate reagent was added to each well and the reaction was allowed to develop for 30 minutes, at which point optical densities were measured at 405 nm using an Epoch Microplate spectrophotometer. Endpoint titers were obtained by calculating the reciprocal of the fecal sample dilution at the background optical density set using fecal slurries from a mouse immunized with irrelevant antigen.
2.16 Statistical Analyses
All experiments were performed between three and six times (noted in figure legends). Comparative analyses were completed using either a student’s t-test or one-way ANOVA followed by Tukey’s post-test for comparing multiple treatment conditions, both at a 95% confidence level using Prism software (GraphPad Prism 5, LaJolla, CA). All values are reported as mean ± standard error of the mean.
3. Results
3.1 Characterization of CS oligosaccharide/DNA NPs: size, charge, DNA encapsulation and transfection efficiency
The objective of this work was to develop CS-ZN-NIMs, which consist of inner CS/DNA NP cores designed for optimal transfection that are further encapsulated in a ZN microparticle to improve particle stability and DNA protection through the GI tract for oral gene delivery applications. Because the characteristics of the CS/DNA NP inner cores (e.g. size, charge, DNA encapsulation) will ultimately determine the transfection efficiency of the system, their complete characterization was essential. CS/DNA NPs formed with varying weight ratios of CS to TPP (from 4 to 14), and at initial CS solution concentrations of 0.5 mg/mL, 1.0 mg/mL, and 2.0 mg/mL were characterized for size and zeta potential (Figure 1A–C) and DNA encapsulation (Figure 1D–F). As the CS:TPP weight ratio increased, the overall diameter of the CS/DNA NPs increased from 197.56 to 307.47 nm for 0.5 mg/mL CS concentrations, from 233.6 to 378.6 nm for 1.0 mg/mL CS concentrations, and from 299 to 421 nm for 2.0 mg/mL CS concentrations (Figure 1A–C, respectively). Similar trends were observed for the zeta potential measurements, with values increasing from 14.5 to 48.1 mV, 28.9 to 44.7 mV, and 16.3 to 40.2 mV for 0.5 mg/mL CS, 1.0 mg/mL CS and 2.0 mg/mL CS, respectively (Figure 1A–C). The DNA encapsulation efficiency of CS/DNA NPs formed with varying CS:TPP weight ratios and CS concentrations of 0.5, 1.0, and 2.0 mg/mL was also investigated (Figure 1D–F). As CS:TPP ratio increased encapsulation efficiencies decreased from 91.34 to 61.74% for 0.5 mg/mL CS, 90.75 to 62.26% for 1.0 mg/mL CS, and 99% to 45.4% for 2.0 mg/mL CS as CS:TPP ratios increased (Figure 1D–F). CS/DNA NPs were also evaluated for their ability to mediate transgene expression in vitro. Transgene expression levels 24 hours after transfection of HEK293T cells with varying CS/DNA NP formulations showed similar trends for particles formed with 0.5, 1.0 and 2.0 mg/mL initial CS concentrations (Figure 1G–I). Overall, a trend of decreasing transgene expression was observed as the CS:TPP weight ratios increased from 4 to 14, with particles formed from 2.0 mg/mL CS concentration inducing a significantly higher transgene expression than particles formed from 0.5 and 1.0 mg/mL CS concentrations.
Figure 1.

Characterization of CS/DNA NPs formed via ionic gelation. CS/DNA NPs formed at varying CS:TPP weight ratios were characterized for size (circles) and charge (triangles) (A–C), DNA encapsulation efficiency (D–F), and transfection (G–I). The concentration of the CS solution used in particle formation was held constant at 0.5 mg/mL (A,D and G), 1.0 mg/mL (B, E and H), and 2.0 mg/mL (C, F, and I) for all CS:TPP ratios. All data represented as mean ± SEM (n = 6). Asterisks (*) denote significance compared to 0.5 mg/mL conditions (* p ≤ 0.05, ** p ≤ 0.01, and *** p ≤ 0.001). Plus signs (+) denote significance compared to 1.0 mg/mL conditions (++ p ≤ 0.01 and +++ p ≤ 0.001).
3.3 Formation and Size Characterization of CS-ZN-NIMs
CS/DNA NPs were encapsulated in ZN microparticles using a single W/O emulsion technique to form CS-ZN-NIMs (Figure 2). The resulting CS-ZN-NIMs were characterized for size using SEM microscopy and image processing techniques. The single W/O emulsion resulted in solid, spherical particles ranging in size from 2 μm to 30 μm (Table 1, Figure 3). Increasing the initial ZN solution concentration from 12 to 25% led to a slight increase in average particle diameter from 8.76 μm to 10.63 μm (Table 1, Figure 3).
Figure 2.

Formation of CS-ZN-NIMs using a single water-in-oil emulsion (W/O). Briefly, CS/DNA NPs were formed via ionic gelation as described. The formed NPs were added to a 90% aqueous ethanol solution of ZN at varying concentrations (5–25% w/v). The ZN-CS/DNA NPs suspension was then added to the continuous phase (corn oil) under constant stirring to form the single W/O emulsion. Solvent evaporation results in ZN microsphere solidification, encapsulating the CS/DNA NPs to form CS-ZN-NIMs. The resulting particles were separated from the continuous phase, washed to remove oil, and collected.
Table 1.
CS-ZN-NIM Size and CS/DNA NP encapsulation and loading in ZN microparticles
| ZN Solution Concentration (%) | Average Particle Size (μm) | CS/DNA NP Encapsulation in ZN Microparticles (%)
|
CS/DNA NP Loading in ZN Microparticles (μg DNA/g ZN )
|
||||
|---|---|---|---|---|---|---|---|
| CS:DNA = 5 | CS:DNA = 10 | CS:DNA = 25 | CS:DNA = 5 | CS:DNA = 10 | CS:DNA = 25 | ||
| 12 | 8.76 ± 2.12 | 72.26 ± 4.0 | 74.27 ± 3.7 | 44.46 ± 1.29 | 185.63 ± 9.48 | 174.46 ± 22.9 | 62.60 ± 3.77 |
| 15 | 8.35 ± 0.51 | 52.15 ± 12.1 | 79.52 ± 8.9 | 54.98 ± 14.4 | 100.07 ± 19.6 | 157.67 ± 13.8 | 62.44 ± 10.9 |
| 25 | 10.63 ± 0.24 | 48.70 ± 19.4 | 81.17 ± 0.79 | 44.01 ± 3.22 | 83.41 ± 9.46 | 110.25 ± 11.7 | 29.28 ± 1.75 |
Figure 3.

SEM micrographs of CS-ZN-NIMs formed with varying initial ZN concentrations. (A) 12% zein (530x magnification). (B) 15% zein (570x magnification). (C) 25% (680x magnification).
3.4 CS/DNA loading and encapsulation into ZN microparticles
Loading of CS/DNA NPs into ZN microparticles using the single W/O emulsion was found to be dependent upon both the initial ZN concentration used in the emulsion as well as the properties of the CS/DNA NPs being loaded, specifically the weight ratio of CS to DNA (Table 1). Loading generally decreased as the initial ZN concentration increased, and the highest overall loadings of 185.63 and 174.46 μg DNA/g ZN were observed at a ZN concentration of 12% with CS/DNA NPs formed at CS:DNA ratios of 5 and 10, respectively (Table 1). CS/DNA NPs formed at a CS:DNA ratio of 25 resulted in the lowest loading for all ZN concentrations used. Encapsulation efficiency was found to be dependent on the CS:DNA ratio of the CS:DNA NPs loaded into the ZN microparticles (Table 1). Particles formed with CS/DNA NPs at a CS:DNA ratio of 10 exhibited the highest encapsulation efficiencies of 74, 79 and 81% for CS-ZN-NIMs formed with 12, 15 and 25% ZN solutions, respectively. CS-ZN-NIMs formed with 15 and 25% ZN and loaded with CS/DNA NPs at a CS:DNA ratio of 5 and 25 showed a decrease in encapsulation efficiency to below 60% (Table 1).
3.5 Biocompatibility of CS-ZN-NIMs: cellular viability assay
ZN has been shown to positively influence cell viability and proliferation, possibly due to internalization of its degradation products, which has been shown to have antioxidant properties [25]. To determine the effect of CS-ZN-NIMs on cell viability and overall biocompatibility, a series of cellular viability studies were performed (Figure 4). HEK293T cells cultured with CS-ZN-NIMs showed significantly increased (p ≤ 0.001) viability relative to control cells (Figure 4A). The viability of Caco-2 and RAW 264.7 cells cultured with CS-ZN-NIMs was not statistically different compared to vehicle control cells that received no treatment, with the exception of CS-ZN-NIMs formed with 12 and 15% ZN, which resulted in slightly increased viability of Caco-2 cells (p ≤ 0.05 and p ≤ 0.01, respectively) (Figure 4B and C).
Figure 4.

Biocompatibility of CS-ZN-NIMs incubated with HEK293T (A), Caco-2 (B), and RAW 264.7 (C) cells. Cellular proliferation was measured using the WST-1 assay and absorbance values were normalized to cells that received no particle treatment. ***, **, and * indicate p ≤ 0.001, 0.01, 0.05, respectively for treatments compared to cell control condition. All data represent mean ± SEM (n = 6).
3.6 Simulated gastric fluid release
Protection of the encapsulated DNA payload from degradation, in addition to minimal DNA release in the gastric compartment, is essential to achieving successful gene delivery via the oral route. To investigate the release profile of CS/DNA NPs from CS-ZN-NIMs, a series of SGF release studies were performed (Figure 5). The CS-ZN-NIMs exhibited sustained DNA release for up to 240 minutes, with particles formed with 15 and 25% ZN showing a slower rate of release when compared to particles formed with 12% ZN for the first 60 minutes of incubation (see Figure 5 legend for statistics). After 45 minutes in SGF, a time frame similar to the average gastric residence time (from 10 to 120 minutes) for fasted human stomachs [49]. CS-ZN-NIMs formed with either 15% and 25% ZN still retained 56.9 ± 3.05 and 61.0 ± 10.2% of the encapsulated DNA, respectively, while particles formed with 12% ZN and naked CS NPs retained 35.5 ± 0.8% and 50.2 ± 1.0% of encapsulated DNA, respectively (Figure 5).
Figure 5.

CS/DNA NP release from CS-ZN-NIMs formed with 12% ZN (●), 15% ZN (◆), and 25% ZN (■), and DNA release from naked CS/DNA NPs (▼) incubated in SGF. The shaded portion of the figure represents the mean gastric residence time for solid oral dosage forms in fasted human stomachs. All data represent mean ± SEM (n = 3). *, p ≤ 0.05 for CS NPs and 12% ZN. +++, p ≤ 0.001 for CS NPs and 15 and 25% ZN. xx, p ≤ 0.01 for 12 and 25% ZN. ◆, p ≤ 0.05 for 12 and 15% ZN. +, p ≤ 0.05 for CS NPs and 15 and 25% ZN. x, p ≤ 0.05 for 12 and 25% ZN.
3.7 SIF Treatment of CS-ZN-NIMs: Enzyme-mediated release of CS/DNA NPs and Transfection Profiles
To investigate the ability of CS-ZN-NIMs to release transfection competent CS/DNA NPs in the intestinal compartment, a series of SIF incubations were performed. The CS/DNA NPs released from CS-ZN-NIMs during incubation were collected and subsequently delivered to cells, and the amount of transgene expression was quantified after a 24-hour incubation. For SIF studies, the properties of the inner core of CS/DNA NPs loaded into ZN microparticles were held constant at 1.0 mg/mL CS, a CS:TPP ratio of 8, and a CS:DNA ratio of 10 (Figure 6A). While the overall trend in transgene expression was found to increase with increasing ZN concentration, there were no statistical differences in transgene expression with increasing ZN concentrations, with the exception of the 5 and 25% ZN conditions (p ≤ 0.05) (Figure 6A). However, when incubated in the absence of enzyme, the ability of all CS-ZN-NIM formulations (formed with any percentage of ZN) to mediate transgene expression was dramatically reduced (p ≤ 0.05 for 10, 12, 15% ZN particles and p ≤ 0.001 for 25% ZN particles), demonstrating an intestinal enzyme-mediated degradation of the ZN microparticles and release of CS/DNA NPs. Next, to ensure that the W/O emulsion encapsulation process did not affect the transfection properties of the CS/DNA NPs, a series of transfections following SIF treatment were performed with CS-ZN-NIMs loaded with CS/DNA NPs formed at CS:DNA ratios of 5, 10 and 25 because low CS:DNA ratios have been implicated in improved transfection when using low molecular weight CS [50] (Figure 6B). CS-ZN-NIMs loaded with CS/DNA NPs at a CS:DNA ratio of 5 and then subjected to SIF conditioning resulted in the highest transgene expression, followed by particles loaded with CS/DNA NPs formed at CS:DNA ratios of 10 and 25 (Figure 6B, see figure legend for a description of statistics). Finally, CS-ZN-NIMs were incubated in SIF for varying periods of time including 0, 15, 30, and 60 minutes. Transgene expression increased with increasing incubation time in SIF for CS-ZN-NIMs formed from 12, 15, and 25% ZN (Figure 6C), further indicating ZN degradation and CS/DNA NP release is an enzyme-mediated process.
Figure 6.

Transgene expression mediated by CS-ZN-NIMs following incubation in simulated intestinal fluid (SIF). (A) The effect of intestinal enzyme-mediated degradation of CS-ZN-NIMs on transgene expression. Particles formed with varying zein solutions (5, 7, 10, 12, 15, and 25%) were incubated in either SIF containing pancreatin (+ SIF) or PBS (− SIF) ***, *, and ns indicate p ≤ 0.001, 0.05, and no significance, respectively for +SIF conditions compared to their respective −SIF condition (n = 6). While the overall trend in transgene expression was found to increase with increasing ZN concentration, there were no statistical differences in transgene expression with increasing ZN concentrations, with the exception of the 5 and 25% ZN conditions (p ≤ 0.05). (B) Transgene expression of CS-ZN-NIMs formed with varying zein solutions and varying CS/DNA NPs (n = 3). Asterisks (*) indicate significance between CS:DNA = 5 and CS:DNA = 10 conditions, ***, and ** p ≤ 0.001 and 0.01, respectively. Plus signs (+) indicate significance between CS:DNA = 5 and CS:DNA = 25 conditions, +++, + p ≤ 0.001 and 0.05, respectively. Diamonds (◆) indicate significance between CS:DNA = 10 and CS:DNA = 25 conditions ◆◆, ◆ p ≤ 0.01 and 0.05, respectively. (C) Transgene expression mediated by CS-ZN-NIMs after varying incubation times in SIF containing pancreatin.
3.8 In vitro simulation of oral delivery
To test the ability of the outer ZN microparticle to protect the CS/DNA NPs throughout the whole GI tract, particles formed from 12 and 15% ZN were subjected to in vitro simulated GI transit (15 mins in SGF containing pepsin followed by 60 minutes in SIF containing pancreatin). Due to the discrepancy observed in duplicating transfection (note differences in transfection observed in independent experiments in Fig. 6A and 6B) with the 25% ZN particles, and the low CS/DNA NP loading, we chose to exclude these particles from further testing. The resulting NPs were then delivered to HEK293T cells (Figure 7). CS-ZN-NIMs formed with both ZN concentrations were able to protect CS/DNA NPs from SGF and release transfection-competent CS/DNA NPs upon SIF treatment (Figure 7). CS-ZN-NIMs formed with 12 and 15% ZN induced significantly higher transgene expression when compared to naked CS/DNA NPs subjected to the same simulated GI treatment (p ≤ 0.05), resulting in 22 and 20-fold increases in transgene expression, respectively (Figure 7).
Figure 7.

Transgene expression mediated by CS-ZN-NIMs after complete simulated GI tract transit. CS-ZN-NIMs formed with varying initial zein solutions were subjected to 15 minutes incubation in SGF containing pepsin, followed immediately by 60 minutes incubation in SIF containing pancreatin. ** signifies p ≤ 0.01 when compared to naked CS NPs subjected to the same treatment. Fold change increase in transgene expression over naked CS NPs is shown in parentheses. All data represent mean ±SEM (n = 6).
3.8 In vivo oral delivery of CS-ZN-NIMs
Orally delivered DNA has great promise for use in a wide variety of preventative and curative therapeutic applications, including DNA vaccination against infectious pathogens. To investigate the ability of CS-ZN-NIMs to successfully protect and deliver DNA cargo through the GI tract and induce adaptive immune responses in vivo, mice were orally dosed with CS-ZN-NIMs encapsulating either 35 or 100 μg of the pEGFP-LUC plasmid. For these preliminary in vivo studies we chose 12% ZN particles for their high degree of CS/DNA NP loading, as shown in Table 1. Three weeks after a single oral delivery, fecal samples from each mouse were collected and examined for the presence of IgA antibodies against the GFP protein (Figure 8). Mice orally administered CS-ZN-NIMs developed fecal anti-GFP IgA titers comparable to those from control mice immunized intraperitoneally with GFP in Addavax (Figure 8). These IgA titers were also significantly greater that those observed in mice given an irrelevant antigen. Moreover, the IgA titers from mice dosed with CS-ZN-NIMs were significantly greater than those from mice receiving naked CS/DNA NPs (P ≤ 0.001 for 35 μg dose and P ≤ 0.01 for 100 μg dose). These data demonstrate that CS-ZN-NIMs are capable of protecting DNA cargo during intestinal transit such that it can ultimately mediate in vivo gene expression and induce mucosal immune responses.
Figure 8.
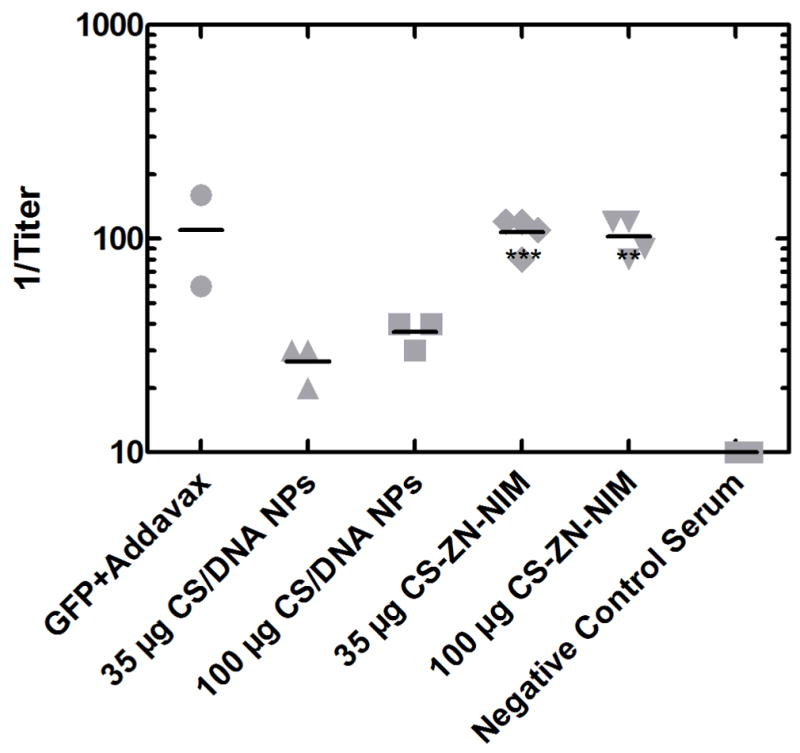
Anti-GFP IgA titers following a single oral delivery of CS-ZN-NIMs. CS-ZN-NIM amounts equivalent to 35 and 100 μg of pEGFP-LUC were delivered to 6-week old male BALB/c mice via oral gavage. Three weeks after gavage, fecal samples were collected, diluted 1:20 w/v in PBS and anti-GFP IgA antibody titers were evaluated using an ELISA assay. Fecal samples from mice immunized with non-GFP antigen was used as a negative control. Endpoint titers were determined using the optical densities measured for the negative control as a cutoff for endpoint dilutions. ( *** p ≤ 0.001, ** p ≤ 0.01)
4. Discussion
Oral gene delivery has the potential to be an effective delivery strategy for both gene therapy and DNA vaccination applications. However, gene delivery via the oral route is complicated by the harsh and variable conditions of the GI tract. In this study, we describe the development of CS-ZN nano-in-microparticles (CS-ZN-NIMs) consisting of inner CS/DNA NP cores encapsulated in a protective ZN microparticle, and we demonstrate their potential for mediating oral nonviral gene delivery. These materials were selected for their individual properties that fulfill the requirements of protection in the gastric environment, as well as subsequent delivery to target cells in the intestine. Moreover, the natural abundance of both materials, coupled with the simple and mild processing conditions used in particle formation, allows for simple scale-up, which is crucial for future clinical applications.
In the development of the CS-ZN-NIMs oral delivery system, full characterization of the CS/DNA core NPs was essential to understand particle formation variables that affect the transfection efficiency of the NPs, and therefore the system in its entirety. The use of CS as a natural polymer for the development of gene delivery vectors has been widely documented [3, 51–53]. The presence of primary amine groups, with a pKa of approximately 6.5, causes protonation at physiological conditions and leads to a net positive charge. This property allows CS to electrostatically combine and condense with negatively charged plasmid DNA to form nano-sized complexes. In this study, we investigated the use of oligosaccharide CS/DNA NPs crosslinked with TPP as the nonviral carrier component of our dual particle oral delivery system. Previous reports have found that several particle formation parameters, including the molecular weight and degree of deacetylation of CS, CS concentration, CS:DNA ratio, and the CS:TPP ratio are important in determining the size, charge and transfection efficiency of CS delivery systems [50, 53, 54]. Here, the size and net positive charge of CS complexes were found to increase as the ratio of CS to TPP increased, which agrees with previous reports [54, 55]. A similar dependence on CS:TPP ratio was observed for the encapsulation efficiency of DNA within CS complexes, with decreased encapsulation observed for increasing CS:TPP ratios. Finally, the in vitro transfection efficiency of the CS complexes was found to be dependent upon both the CS:TPP ratio and the CS concentration used in CS formation (Figure 1). The general trend of decreasing transgene expression with increasing CS:TPP ratio is hypothesized to be directly related to the decrease in encapsulation efficiency and the increasing size of the complexes observed with increasing CS:TPP ratio, as complex size is a critical factor for efficient endocytosis and cellular uptake [56, 57]. The results of these studies highlight the tunable nature these delivery systems to achieve characteristics that are critical to efficient gene delivery, specifically size, charge, encapsulation, and transfection efficiency.
In order to achieve successful gene delivery via the oral route, it is critical that the DNA cargo be protected from the highly acidic pH of the gastric environment and from degradation by endogenous nucleases, as well as gastric and intestinal enzymes. Oral gene delivery systems based on CS have been reported. However, these systems achieve only modest success, presumably due to incomplete protection in the stomach because of the solubility of CS in acidic aqueous conditions [58, 59]. To overcome the limitations of CS delivery systems, we report the use of ZN to further encapsulate the CS/DNA NPs. ZN has recently received interest as a natural biomaterial for a variety of applications including tissue engineering [60–62] and drug and gene delivery [26, 63] due to its inherent biocompatibility and biodegradability. Moreover, ZN, being composed of over 50% hydrophobic amino acids, is amphiphilic in nature, which allows it to self-assemble into nanoparticles [27] and microparticles [29] as well as uniform films that can be used as protective coatings [64]. Taking advantages of these properties, ZN has already been used in tableting and coating applications in both the food and pharmaceutical industries [34, 65].
The use of ZN as a natural material for the encapsulation of therapeutic compounds, including DNA, requires the selection of a suitable solvent that is compatible with the compound to be encapsulated. ZN’s amino acid composition renders it insoluble in aqueous conditions, and therefore requires a binary solvent for complete solubilization. The most commonly used solvent for ZN is aqueous ethanolic solutions between 55 and 90% (v/v) ethanol [66]. To this end, various ethanolic ZN solutions were investigated for use in the W/O emulsion method developed here (data not shown), which involved a single emulsion of ethanolic ZN into the continuous phase of 100% corn oil. It was found that 90% ethanolic solutions of ZN resulted in uniform spherical microparticles containing CS/DNA NPs at the ZN concentrations investigated in this study. This phenomenon was likely due to the increased rate of solvent evaporation relative to lower concentration ethanolic solutions, an important factor implicated in the formation of microparticles using emulsion type encapsulation processes (data not shown). The addition of the non-ionic surfactant span-80 resulted in smaller, more uniform particles due to increased stability of the interface between the continuous (corn oil) and dispersed phases (ethanolic ZN-CS/DNA NP suspension) of the emulsion system (data not shown).
Encapsulation of the CS/DNA NPs within ZN microparticles was determined to be dependent upon the concentration of ZN solution used in the W/O emulsion. To increase the encapsulation of the positively charged CS/DNA NPs within the ZN microparticles, the initial pH of the ZN solutions was raised to approximately 9 to impart an overall negative charge to the ZN (isoelectric point of ZN is 6.8) [67], leading to an increased association of CS/DNA NPs with ZN. Increasing the concentration of the ZN solution from 12 to 25% tended to increase the encapsulation efficiency of CS/DNA nanoparticles within the ZN microparticles for CS/DNA NPs formed at a CS:DNA ratio of 10 (Table 1). This increase could be attributed to the increase in the viscosity of the dispersed phase, leading to decreased diffusion of the CS/DNA NPs from the dispersed phase into the continuous phase during the emulsion process [68, 69]. Increasing the viscosity of the dispersed phase (through increasing ZN concentration) also resulted in larger particles (Table 1 and Figure 2).
Loading of CS/DNA particles into ZN microparticles reached a maximum of ~180 μg DNA/g ZN, which is lower than what has been reported for the loading of hydrophobic molecules into ZN microparticles [30]. It has been shown previously that hydrophilic molecules are more difficult to load into ZN nano- and microparticles than more hydrophobic molecules, presumably due to decreased hydrophobic interactions between the hydrophilic molecules and hydrophobic amino acids of the ZN molecule [30, 45]. However, the maximum encapsulation efficiency of DNA within the ZN microparticles (~80%) was found to be higher than similar multi-particulate systems for DNA delivery [1].
In previous works, ZN has been shown to have degradation products that are beneficial to cellular proliferation, and cells cultured on ZN films have enhanced viability. Further characterization of the CS-ZN-NIMs revealed no cytotoxicity in several cell types relevant to oral delivery, including intestinal epithelial cells and macrophages (Figure 4). These results agree with the extensive field of evidence supporting ZN’s biocompatibility in biomedical applications [25, 26, 37, 70, 71]. We expect the positive effect of ZN degradation products on cell health to be an advantage of the proposed delivery system. The rapid degradation of the ZN microparticle in the intestine could lead to an elevated concentration of degradation products in the intestinal environment, which in turn has the potential to improve cell health and potentially decrease transfection-associated cellular toxicity [25, 62].
Encapsulation of various compounds in ZN, as well as coating particles with ZN, have been shown to decrease the release profile under simulated gastric conditions [72]. To investigate the ability of CS-ZN-NIMs to protect the CS/DNA NPs from degradation and reduce the release of DNA with the gastric compartment, a series of simulated gastric release studies were performed (Figure 5). CS-ZN-NIMs formed from 15 and 25% ZN both exhibited a slower release of DNA in gastric conditions as compared to un-encapsulated CS/DNA NPs during a period of time that is representative of the average gastric residence time in humans. The gastric release profile for particles formed with 15 and 25% ZN indicated slower release kinetics versus that of particles formed with 12% ZN for the first 60 minutes of the release. These data support other studies investigating ZN microparticles for oral delivery or evaluating ZN as an enteric coating, both of which found that increasing the amount of ZN, either to form the coating or during the microparticle formation, led to decreased release rates [72–74]. Those studies, in addition to the data presented here, suggest that the relative amount of ZN used in the preparation of CS-ZN-NIMs influences the protective capabilities of the particles, with increasing amounts of ZN resulting in improved protection. It is worth noting that CS/DNA NPs exhibited slower release kinetics than CS-ZN-NIMs formed using 12% ZN, an unexpected result considering the pKa of the amine groups of CS (~6.5). At the low pH of the SGF dissolution media used during the SGF release, the amine groups should have been completely protonated, resulting in electrostatic repulsion between CS polymer chains and dissolution of CS NPs at pH 1.2. Although our result was surprising and will be the focus of future studies, we would like to highlight that the ZN coating not only serves to slow DNA release, but also to protect the structure and properties of the CS/DNA NPs to allow efficient cellular transfection. This protective effect was evident in our in vitro simulated GI transit studies (Figure 7) as well as in our in vivo oral delivery study (Figure 8). Further, data from our release studies indicate that CS-ZN-NIMs formed with 12, 15 and 25% ZN retain 49, 71 and 77%, respectively, of the encapsulated DNA after a 30-minute incubation in SGF (Figure 5). Although gastric residence and emptying time can vary widely between individuals, for most solid uniform oral dosage forms the gastric residence time in fasted human stomachs is on the order of 10–120 minutes, with an average residence time of ~45 minutes [49]. Therefore, given the appropriate dosing schedule and regimen, CS-ZN-NIMs have the potential to reach the small intestine with nearly 50–70% of the CS/DNA NP cargo remaining intact with the potential to achieve successful transfection.
To better understand the DNA encapsulation characteristics, as well as assess the ability of CS-ZN-NIMs to release CS/DNA NPs in a site-specific manner for DNA delivery in the small intestine, a series of in vitro SIF incubations and transfection experiments were performed. CS-ZN-NIMs incubated in SIF containing pancreatin for 60 minutes were found to mediate transfection in HEK293T cells, with an increase in transgene expression observed for particles formed with increasing concentration of the ZN solutions used in the W/O emulsion. Moreover, the ability of the CS-ZN-NIMs to mediate transfection was found to be dependent on the intestinal enzyme-mediated degradation of the ZN microparticles, as transgene expression was negligible when particles were incubated in PBS (i.e., no degradation of the outer ZN microparticle) (Figure 6). These data support previous work from us [26] and others and indicate that non-enzymatic hydrolytic degradation of ZN microparticles occurs very slowly, further supporting the use of ZN in oral applications [29]. The lack of transgene expression from particles incubated in PBS also indicates complete encapsulation of the CS/DNA NPs within the ZN microparticles as opposed to surface adsorption, further confirming the high encapsulation efficiencies observed. Furthermore, CS-ZN-NIMs incubated in SIF for varying amounts of time showed increasing transgene expression as incubation time increased (Figure 6C). The time-dependence of transgene expression levels observed after SIF incubation of CS-ZN-NIMs further indicates that the degradation of ZN is an enzyme-mediated process occurring over a period of approximately one hour, leading to sustained release of CS/DNA NPs.
In addition to simulated gastric release and intestinal delivery, in vitro simulations of complete GI transit were performed to demonstrate the feasibility of the CS-ZN-NIMs as an oral gene delivery system. Both CS-ZN-NIMs and un-encapsulated, bare CS/DNA NPs were subjected to sequential simulated gastric and simulated intestinal fluid and then assessed for their ability to mediate transgene expression in HEK293T cells. Only cells treated with GI-conditioned CS-ZN-NIMs resulted in luciferase expression, whereas naked CS/DNA NPs subjected to the same treatment did not produce any transgene expression. These results indicate that encapsulation of the CS/DNA NPs within the ZN microparticles not only delays the release of CS/DNA NPs in the gastric environment and allows for DNA release in the small intestine, but it also protects the structure of the CS/DNA NPs essential for achieving successful gene delivery.
The protection of gene delivery vectors and their release in the intestinal compartment has important implications for oral delivery applications including gene therapy, which requires the delivery of the therapeutic gene to the local site of inflammation, and DNA vaccines, which have the potential to transfect immune cells of the gut-associated lymphoid tissues to produce mucosal and systemic immunity. To this end, CS-ZN-NIMs were evaluated for their ability to protect CS/DNA NPs and mediate effective transfection in vivo in mice, and more specifically for their ability to elicit an immune response to the encoded model antigen, GFP (Figure 8). For the purposes of this study, we chose to investigate the ability of CS-ZN-NIMs to induce the production of anti-GFP IgA antibodies as a measure of both transgene expression and immune induction in the small intestine. This indirect measure of transgene expression allowed us to overcome limitations in directly quantifying fluorescent transgene expression due to the autofluorescent signal of degraded zein [26]. Future studies will aim to directly evaluate transgene expression through fluorescence microscopy and measuring in vivo luminescence using a luciferase reporter gene, to confirm the preliminary evidence of GFP expression in the intestinal mucosa presented here.
The presence of anti-GFP IgA antibodies in feces following oral delivery of CS-ZN-NIMs indicates adequate protection and release of CS/DNA NPs and the subsequent successful transfection and production of the GFP protein by cells in vivo. Anti-GFP IgA titers in some mice given CS-ZN-NIMs were as high as titers in mice given GFP protein in Addavax as a positive control, again indicating that sufficient transgene expression and subsequent protein production occurred. IgA antibodies are predominately associated with mucosal immune responses in the intestinal tract [75]. To induce such responses, antigen-activated B cells migrate to the periphery of B-cell follicles where they undergo maturation and expansion into primarily short-lived, IgM secreting cells that contribute to the first wave of antigen-specific antibody production by the adaptive immune response [76]. Further interaction of antigen-specific B cells with activated CD4+ T cells through CD40-CD40L receptor-ligand binding induces class-switch recombination in B cells, allowing for the production of other antibody isotypes, including IgA. Our data provide evidence of GFP production following oral delivery of CS-ZN-NIMs, and suggest that subsequent sampling by APCs, either DCs or B cells themselves, resulted in the induction of an adaptive humoral immune response. Even with these results, further investigation into the ability of this delivery system to induce high affinity, neutralizing IgG antibodies and antigen-specific cytotoxic T cell responses will be required should this delivery system be used for DNA vaccination applications. Nonetheless, these data combined highlight the potential of CS-ZN-NIMs as an oral delivery system for improving gene delivery and DNA vaccination.
5. Conclusions
In this study, we have described the development and characterization of a multi-component particulate delivery system for oral gene delivery. We have successfully fabricated CS/DNA NPs of tunable size, charge, and transfection efficiency and successfully encapsulated them into ZN microparticles using a simple W/O emulsion technique. The resulting CS-ZN-NIMs exhibited tunable CS/DNA NP loading, high encapsulation efficiencies, protection within gastric conditions, and intestinal enzyme-mediated release of transfection-competent CS/DNA NPs that were capable of mediating transfection in vitro. We also demonstrated the ability of CS-ZN-NIMs to successfully mediate transfection in vivo, with evidence of the induction of a primary immune response against the encoded model antigen. Future studies will further investigate the potential of CS-ZN-NIMs for oral DNA vaccination in vivo, improve CS/DNA NP loading, and fully characterize the induction of the immune response generated by CS-ZN-NIMs.
Acknowledgments
Nebraska Research Initiative, UNL IANR ARD, National Science Foundation (CBET-1254415), Center for Nanohybrid Functional Materials (NSF EPS-1004094), American Heart Association (#10SDG2640217), the University of Nebraska Foundation (Layman Funds), National Institute of General Medical Sciences of the National Institutes of Health (P20GM104320), the Crohn’s and Colitis Foundation of America (#3578), the Nebraska Corn Board, UNL Research Council Interdisciplinary Seed Grant, UNL Research Council-Tobacco Settlement Funds Biomedical Seed Grant and USDA CSREES-Nebraska [NEB-21-146 and NEB-26-211] are acknowledged for funding. We wish to thank Dr. Abby Geis, Hatem Kittana, and Carlos Gomes Neto from Dr. Amanda Ramer-Tait’s laboratory for their assistance with oral gavage. We also want to thank Anna Lampe from Dr. Deborah Brown’s laboratory for her assistance with oral gavage and antibody analysis. The authors have no conflict of interest to declare.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bhavsar MD, Amiji MM. Gastrointestinal distribution and in vivo gene transfection studies with nanoparticles-in-microsphere oral system (NiMOS) J Control Release. 2007;119(3):339–48. doi: 10.1016/j.jconrel.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 2.des Rieux A, Fievez V, Garinot M, Schneider YJ, Preat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release. 2006;116(1):1–27. doi: 10.1016/j.jconrel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 3.Guliyeva U, Oner F, Ozsoy S, Haziroglu R. Chitosan microparticles containing plasmid DNA as potential oral gene delivery system. Eur J Pharm Biopharm. 2006;62(1):17–25. doi: 10.1016/j.ejpb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Chang AG, Wu GY. Gene therapy: applications to the treatment of gastrointestinal and liver diseases. Gastroenterology. 1994;106(4):1076–84. doi: 10.1016/0016-5085(94)90771-4. [DOI] [PubMed] [Google Scholar]
- 5.Page DT, Cudmore S. Innovations in oral gene delivery: challenges and potentials. Drug Discov Today. 2001;6(2):92–101. doi: 10.1016/s1359-6446(00)01600-7. [DOI] [PubMed] [Google Scholar]
- 6.Kriegel C, Amiji M. Oral TNF-alpha gene silencing using a polymeric microsphere-based delivery system for the treatment of inflammatory bowel disease. J Control Release. 2011;150(1):77–86. doi: 10.1016/j.jconrel.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friend DR. New oral delivery systems for treatment of inflammatory bowel disease. Adv Drug Deliv Rev. 2005;57(2):247–265. doi: 10.1016/j.addr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Rothman S, Tseng H, Goldfine I. Oral gene therapy: a novel method for the manufacture and delivery of protein drugs. Diabetes Technol Ther. 2005;7(3):549–57. doi: 10.1089/dia.2005.7.549. [DOI] [PubMed] [Google Scholar]
- 9.Dhadwar SS, Kiernan J, Wen J, Hortelano G. Repeated oral administration of chitosan/DNA nanoparticles delivers functional FVIII with the absence of antibodies in hemophilia A mice. J Thromb Haemost. 2010;8(12):2743–2750. doi: 10.1111/j.1538-7836.2010.04116.x. [DOI] [PubMed] [Google Scholar]
- 10.Bowman K, Sarkar R, Raut S, Leong KW. Gene transfer to hemophilia A mice via oral delivery of FVIII–chitosan nanoparticles. J Control Release. 2008;132(3):252–259. doi: 10.1016/j.jconrel.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quade-Lyssy P, Milanov P, Abriss D, Ungerer C, Konigs C, Seifried E, et al. Oral gene therapy for hemophilia B using chitosan-formulated FIX mutants. J Thromb Haemost. 2014;12(6):932–42. doi: 10.1111/jth.12572. [DOI] [PubMed] [Google Scholar]
- 12.Chou FF, Huang SC, Chang SF, Liaw J, Hung PH. Oral gene therapy for hypoparathyroidism: a rat model. Hum Gene Ther. 2009;20(11):1344–50. doi: 10.1089/hum.2009.015. [DOI] [PubMed] [Google Scholar]
- 13.Goldmann K, Ensminger SM, Spriewald BM. Oral gene application using chitosan-DNA nanoparticles induces transferable tolerance. Clin Vaccine Immunol. 2012;19(11):1758–64. doi: 10.1128/CVI.00186-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He C, Yin L, Tang C, Yin C. Multifunctional polymeric nanoparticles for oral delivery of TNF-alpha siRNA to macrophages. Biomaterials. 2013;34(11):2843–54. doi: 10.1016/j.biomaterials.2013.01.033. [DOI] [PubMed] [Google Scholar]
- 15.Shedlock DJ, Weiner DB. DNA vaccination: antigen presentation and the induction of immunity. J Leukoc Biol. 2000;68(6):793–806. [PubMed] [Google Scholar]
- 16.Donnelly JJ, Ulmer JB, Shiver JW, Liu MA. DNA VACCINES. Ann Rev Immunol. 1997;15(1):617–648. doi: 10.1146/annurev.immunol.15.1.617. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen DN, Green JJ, Chan JM, Langer R, Anderson DG. Polymeric Materials for Gene Delivery and DNA Vaccination. Adv Mater. 2009;21(8):847–867. doi: 10.1002/adma.200801478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen SC, Jones DH, Fynan EF, Farrar GH, Clegg JCS, Greenberg HB, et al. Protective Immunity Induced by Oral Immunization with a Rotavirus DNA Vaccine Encapsulated in Microparticles. J Virol. 1998;72(7):5757–5761. doi: 10.1128/jvi.72.7.5757-5761.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaneko H, Bednarek I, Wierzbicki A, Kiszka I, Dmochowski M, Wasik TJ, et al. Oral DNA vaccination promotes mucosal and systemic immune responses to HIV envelope glycoprotein. Virology. 2000;267(1):8–16. doi: 10.1006/viro.1999.0093. [DOI] [PubMed] [Google Scholar]
- 20.Jones DH, Corris S, McDonald S, Clegg JCS, Farrar GH. Poly(dl-lactide-co-glycolide)-encapsulated plasmid DNA elicits systemic and mucosal antibody responses to encoded protein after oral administration. Vaccine. 1997;15(8):814–817. doi: 10.1016/s0264-410x(96)00266-6. [DOI] [PubMed] [Google Scholar]
- 21.Roy K, Mao HQ, Huang SK, Leong KW. Oral gene delivery with chitosan--DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat Med. 1999;5(4):387–91. doi: 10.1038/7385. [DOI] [PubMed] [Google Scholar]
- 22.Jain S, Singh P, Mishra V, Vyas SP. Mannosylated niosomes as adjuvant-carrier system for oral genetic immunization against hepatitis B. Immunol Lett. 2005;101(1):41–9. doi: 10.1016/j.imlet.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Bhavsar MD, Amiji MM. Development of novel biodegradable polymeric nanoparticles-in-microsphere formulation for local plasmid DNA delivery in the gastrointestinal tract. AAPS PharmSciTech. 2008;9(1):288–94. doi: 10.1208/s12249-007-9021-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howard KA, Li XW, Somavarapu S, Singh J, Green N, Atuah KN, et al. Formulation of a microparticle carrier for oral polyplex-based DNA vaccines. BBA-Gen Subjects. 2004;1674(2):149–157. doi: 10.1016/j.bbagen.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 25.Sun QS, Dong J, Lin ZX, Yang B, Wang JY. Comparison of cytocompatibility of zein film with other biomaterials and its degradability in vitro. Biopolymers. 2005;78(5):268–74. doi: 10.1002/bip.20298. [DOI] [PubMed] [Google Scholar]
- 26.Regier MC, Taylor JD, Borcyk T, Yang Y, Pannier AK. Fabrication and characterization of DNA-loaded zein nanospheres. J Nanobiotechnology. 2012;10:44. doi: 10.1186/1477-3155-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong Q, Jin M. Zein nanoparticles produced by liquid–liquid dispersion. Food Hydrocolloid. 2009;23(8):2380–2387. [Google Scholar]
- 28.Zhang Y, Niu Y, Luo Y, Ge M, Yang T, Yu L, et al. Fabrication, characterization and antimicrobial activities of thymol-loaded zein nanoparticles stabilized by sodium caseinate–chitosan hydrochloride double layers. Food Chem. 2014;142(0):269–275. doi: 10.1016/j.foodchem.2013.07.058. [DOI] [PubMed] [Google Scholar]
- 29.Hurtado-Lopez P, Murdan S. Formulation and characterisation of zein microspheres as delivery vehicles. J Drug Deliv Sci Technol. 2005;15(4):267–272. [Google Scholar]
- 30.Karthikeyan K, Vijayalakshmi E, Korrapati PS. Selective interactions of zein microspheres with different class of drugs: an in vitro and in silico analysis. AAPS PharmSciTech. 2014;15(5):1172–80. doi: 10.1208/s12249-014-0151-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Q, Reddy N, Yang Y. Cytocompatible cross-linking of electrospun zein fibers for the development of water-stable tissue engineering scaffolds. Acta Biomater. 2010;6(10):4042–51. doi: 10.1016/j.actbio.2010.04.024. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Lim LT, Kakuda Y. Electrospun zein fibers as carriers to stabilize (−)-epigallocatechin gallate. J Food Sci. 2009;74(3):C233–40. doi: 10.1111/j.1750-3841.2009.01093.x. [DOI] [PubMed] [Google Scholar]
- 33.Khalil AA, Deraz SF, Elrahman SA, El-Fawal G. Enhancement of mechanical properties, microstructure, and antimicrobial activities of zein films cross-linked using succinic anhydride, eugenol, and citric Acid. Prep Biochem Biotechnol. 2015;45(6):551–67. doi: 10.1080/10826068.2014.940967. [DOI] [PubMed] [Google Scholar]
- 34.Yamada K, Takahashi H, Noguchi A. Improved water resistance in edible zein films and composites for biodegradable food packaging. Int J Food Sci Tech. 1995;30(5):599–608. [Google Scholar]
- 35.Hurtado-López P, Murdan S. An investigation into the adjuvanticity and immunogenicity of zein microspheres being researched as drug and vaccine carriers. J Pharm Pharmacol. 2006;58(6):769–774. doi: 10.1211/jpp.58.6.0007. [DOI] [PubMed] [Google Scholar]
- 36.Lai LF, Guo HX. Preparation of new 5-fluorouracil-loaded zein nanoparticles for liver targeting. Int J Pharm. 2011;404(1–2):317–323. doi: 10.1016/j.ijpharm.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 37.Karthikeyan K, Lakra R, Rajaram R, Korrapati PS. Development and characterization of zein-based microcarrier system for sustained delivery of aceclofenac sodium. AAPS PharmSciTech. 2012;13(1):143–9. doi: 10.1208/s12249-011-9731-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aydin R, Pulat M. 5-Fluorouracil Encapsulated Chitosan Nanoparticles for pH-Stimulated Drug Delivery: Evaluation of Controlled Release Kinetics. J Nanomater. 2012;2012:10. [Google Scholar]
- 39.Chen H, Zhong Q. Processes improving the dispersibility of spray-dried zein nanoparticles using sodium caseinate. Food Hydrocolloid. 2014;35:358–366. [Google Scholar]
- 40.Luo Y, Teng Z, Wang Q. Development of zein nanoparticles coated with carboxymethyl chitosan for encapsulation and controlled release of vitamin D3. J Agric Food Chem. 2012;60(3):836–43. doi: 10.1021/jf204194z. [DOI] [PubMed] [Google Scholar]
- 41.Fu JX, Wang HJ, Zhou YQ, Wang JY. Antibacterial activity of ciprofloxacin-loaded zein microsphere films. Mater Sci Eng, C. 2009;29(4):1161–1166. [Google Scholar]
- 42.Parris N, Cooke PH, Hicks KB. Encapsulation of essential oils in zein nanospherical particles. J Agric Food Chem. 2005;53(12):4788–92. doi: 10.1021/jf040492p. [DOI] [PubMed] [Google Scholar]
- 43.Lee S, Alwahab NSA, Moazzam ZM. Zein-based oral drug delivery system targeting activated macrophages. Int J Pharm. 2013;454(1):388–393. doi: 10.1016/j.ijpharm.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 44.Berstein H, Morrel E, Mathiowitz E, Schwaller K, Beck TR. Protein microspheres and methods of using them. Google Patents. 1991 [Google Scholar]
- 45.Lau E, Giddings S, Mohammed S, Dubois P, Johnson S, Stanley R, et al. Encapsulation of Hydrocortisone and Mesalazine in Zein Microparticles. Pharmaceutics. 2013;5(2):277. doi: 10.3390/pharmaceutics5020277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zou T, Gu L. TPGS Emulsified Zein Nanoparticles Enhanced Oral Bioavailability of Daidzin: In Vitro Characteristics and In Vivo Performance. Mol Pharm. 2013;10(5):2062–2070. doi: 10.1021/mp400086n. [DOI] [PubMed] [Google Scholar]
- 47.Qin C, Li H, Xiao Q, Liu Y, Zhu J, Du Y. Water-solubility of chitosan and its antimicrobial activity. Carbohydr Polym. 2006;63(3):367–374. [Google Scholar]
- 48.Calvo P, Remuñán-López C, Vila-Jato JL, Alonso MJ. Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J Appl Polym Sci. 1997;63(1):125–132. [Google Scholar]
- 49.Weitschies W, Wedemeyer RS, Kosch O, Fach K, Nagel S, Söderlind E, et al. Impact of the intragastric location of extended release tablets on food interactions. J Control Release. 2005;108(2–3):375–385. doi: 10.1016/j.jconrel.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 50.Csaba N, Koping-Hoggard M, Alonso MJ. Ionically crosslinked chitosan/tripolyphosphate nanoparticles for oligonucleotide and plasmid DNA delivery. Int J Pharm. 2009;382(1–2):205–14. doi: 10.1016/j.ijpharm.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 51.Mao HQ, Roy K, Troung-Le VL, Janes KA, Lin KY, Wang Y, et al. Chitosan-DNA nanoparticles as gene carriers: synthesis, characterization and transfection efficiency. J Control Release. 2001;70(3):399–421. doi: 10.1016/s0168-3659(00)00361-8. [DOI] [PubMed] [Google Scholar]
- 52.Kim TH, Park IK, Nah JW, Choi YJ, Cho CS. Galactosylated chitosan/DNA nanoparticles prepared using water-soluble chitosan as a gene carrier. Biomaterials. 2004;25(17):3783–3792. doi: 10.1016/j.biomaterials.2003.10.063. [DOI] [PubMed] [Google Scholar]
- 53.Lavertu M, Méthot S, Tran-Khanh N, Buschmann MD. High efficiency gene transfer using chitosan/DNA nanoparticles with specific combinations of molecular weight and degree of deacetylation. Biomaterials. 2006;27(27):4815–4824. doi: 10.1016/j.biomaterials.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 54.Fan W, Yan W, Xu Z, Ni H. Formation mechanism of monodisperse, low molecular weight chitosan nanoparticles by ionic gelation technique. Colloids Surf B Biointerfaces. 2012;90:21–7. doi: 10.1016/j.colsurfb.2011.09.042. [DOI] [PubMed] [Google Scholar]
- 55.Dudhani AR, Kosaraju SL. Bioadhesive chitosan nanoparticles: Preparation and characterization. Carbohydr Polym. 2010;81(2):243–251. [Google Scholar]
- 56.Kunath K, von Harpe A, Fischer D, Petersen H, Bickel U, Voigt K, et al. Low-molecular-weight polyethylenimine as a non-viral vector for DNA delivery: comparison of physicochemical properties, transfection efficiency and in vivo distribution with high-molecular-weight polyethylenimine. Journal of Controlled Release. 2003;89(1):113–125. doi: 10.1016/s0168-3659(03)00076-2. [DOI] [PubMed] [Google Scholar]
- 57.Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Size-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticles. International Journal of Pharmaceutics. 2002;244(1–2):105–115. doi: 10.1016/s0378-5173(02)00315-0. [DOI] [PubMed] [Google Scholar]
- 58.Rinaudc M, Pavlov G, Desbrières J. Solubilization of Chitosan in Strong Acid Medium. Int J Polym Anal Ch. 1999;5(3):267–276. [Google Scholar]
- 59.Rinaudo M, Pavlov G, Desbrières J. Influence of acetic acid concentration on the solubilization of chitosan. Polymer. 1999;40(25):7029–7032. [Google Scholar]
- 60.Dong J, Sun Q, Wang JY. Basic study of corn protein, zein, as a biomaterial in tissue engineering, surface morphology and biocompatibility. Biomaterials. 2004;25(19):4691–4697. doi: 10.1016/j.biomaterials.2003.10.084. [DOI] [PubMed] [Google Scholar]
- 61.Wang HJ, Gong SJ, Lin ZX, Fu JX, Xue ST, Huang JC, et al. In vivo biocompatibility and mechanical properties of porous zein scaffolds. Biomaterials. 2007;28(27):3952–3964. doi: 10.1016/j.biomaterials.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 62.Tu J, Wang H, Li H, Dai K, Wang J, Zhang X. The in vivo bone formation by mesenchymal stem cells in zein scaffolds. Biomaterials. 2009;30(26):4369–4376. doi: 10.1016/j.biomaterials.2009.04.054. [DOI] [PubMed] [Google Scholar]
- 63.Matsuda Y, Suzuki T, Sato E, Sato M, Koizumi S, Unno K, et al. Novel preparation of Zein microspheres conjugated with PS-K available for cancer immunotherapy. Chem Pharm Bull. 1989;37(3):757–759. doi: 10.1248/cpb.37.757. [DOI] [PubMed] [Google Scholar]
- 64.Luo Y, Zhang B, Cheng WH, Wang Q. Preparation, characterization and evaluation of selenite-loaded chitosan/TPP nanoparticles with or without zein coating. Carbohydr Polym. 2010;82(3):942–951. [Google Scholar]
- 65.Winters EP, Deardorff DL. Zein as a film-type coating for medicinal tablets. J Am Pharm Assoc. 1958;47(8):608–612. doi: 10.1002/jps.3030470823. [DOI] [PubMed] [Google Scholar]
- 66.Manley RH, Evans CD. Binary Solvents for Zein. Ind Eng Chem. 1943;35(6):661–665. [Google Scholar]
- 67.Cabra V, Arreguin R, Galvez A, Quirasco M, Vazquez-Duhalt R, Farres A. Characterization of a 19 kDa alpha-zein of high purity. J Agric Food Chem. 2005;53(3):725–9. doi: 10.1021/jf048530s. [DOI] [PubMed] [Google Scholar]
- 68.Yang CY, Tsay SY, Tsiang RC. An enhanced process for encapsulating aspirin in ethyl cellulose microcapsules by solvent evaporation in an O/W emulsion. J Microencapsul. 2000;17(3):269–77. doi: 10.1080/026520400288256. [DOI] [PubMed] [Google Scholar]
- 69.Mehta RC, Thanoo BC, Deluca PP. Peptide containing microspheres from low molecular weight and hydrophilic poly(d,l-lactide-co-glycolide) J Control Release. 1996;41(3):249–257. [Google Scholar]
- 70.Wang HJ, Lin ZX, Liu XM, Sheng SY, Wang JY. Heparin-loaded zein microsphere film and hemocompatibility. J Control Release. 2005;105(1–2):120–131. doi: 10.1016/j.jconrel.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 71.Luo Y, Teng Z, Wang TT, Wang Q. Cellular uptake and transport of zein nanoparticles: effects of sodium caseinate. J Agric Food Chem. 2013;61(31):7621–9. doi: 10.1021/jf402198r. [DOI] [PubMed] [Google Scholar]
- 72.Liu X, Sun Q, Wang H, Zhang L, Wang JY. Microspheres of corn protein, zein, for an ivermectin drug delivery system. Biomaterials. 2005;26(1):109–115. doi: 10.1016/j.biomaterials.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 73.Georget DM, Barker SA, Belton PS. A study on maize proteins as a potential new tablet excipient. Eur J Pharm Biopharm. 2008;69(2):718–26. doi: 10.1016/j.ejpb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 74.Dudlin W. Oral Targeted Drug Delivery Systems: Enteric Coating. In: Hong Wen KP, editor. Oral Controlled Release Formulation Design and Drug Delivery: Theory to Practice. John Wiley & Sons; 2010. pp. 205–223. [Google Scholar]
- 75.Cerutti A. The regulation of IgA class switching. Nature reviews Immunology. 2008;8(6):421–434. doi: 10.1038/nri2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seifert M, Przekopowitz M, Taudien S, Lollies A, Ronge V, Drees B, et al. Functional capacities of human IgM memory B cells in early inflammatory responses and secondary germinal center reactions. Proc Natl Acad Sci USA. 2015;112(6):E546–E555. doi: 10.1073/pnas.1416276112. [DOI] [PMC free article] [PubMed] [Google Scholar]