

Abstract
Aberrant DNA methylation is an epigenetic hallmark of melanoma, known to play important roles in melanoma formation and progression. Recent advances in genome-wide methylation methods have provided the means to identify differentially methylated genes, methylation signatures, and potential biomarkers. However, despite considerable effort and advances in cataloging methylation changes in melanoma, many questions remain unanswered.
The aim of this review is to summarize recent developments, emerging trends, and important unresolved questions in the field of aberrant DNA methylation in melanoma. In addition to reviewing recent developments, we carefully synthesize the findings in an effort to provide a framework for understanding the current state and direction of the field. To facilitate clarity, we divided the review into DNA methylation changes in melanoma, biomarker opportunities, and therapeutic developments. We hope this review contributes to accelerating the utilization of the diagnostic, prognostic, and therapeutic potential of DNA methylation for the benefit of melanoma patients.
Keywords: Epigenetics, Melanoma, DNA methylation, DNMT, Azacitidine, Biomarker, DNMT1, DNMT3B
Background
Aberrant DNA methylation is an epigenetic hallmark of melanoma, and many studies suggest it plays an important in both melanoma formation and progression [1]. Recent advances in genome-wide methylation methods have provided the means to rapidly identify differentially methylated genes, methylation signatures, and potential biomarkers in melanoma. However, despite considerable effort and advances in cataloging methylation changes in melanoma, many questions remain unanswered. Here, we discuss the role of DNA methylation in gene silencing, its potential for biomarker development and as a therapeutic target, including the potential to enhance responses to immune therapies in melanoma.
Approaches to identifying differentially methylated DNA
New discoveries in melanoma epigenetics have been driven by a continually improving set of tools useful for interrogating the epigenome at multiple levels. In the early 1980s, the gold standard technique developed for identifying differential DNA methylation took advantage of sodium bisulfite’s ability to selectively convert only unmethylated cytosine bases to uracil. After bisulfite conversion, both methylated and unmethylated DNA in regions of interest could be PCR amplified using flanking primers, cloned and Sanger sequenced [2]. Unfortunately, this technique carried a number of limitations, including the need for a significant fraction of an allele to be methylated, as well as significant technical expertise and resources for correct utilization [3]. Though bisulfite conversion and sequencing at individual loci allowed for many early discoveries in epigenetics, the significant labor and cost inherent to DNA sequencing led Herman and Baylin to later develop methylation-specific PCR (MSP) as a way to determine the methylation status of individual sites rapidly and efficiently. MSP utilizes primers specific only to methylated (or unmethylated) DNA, thus producing a PCR product after template bisulfite conversion only when bases of the correct methylation status are present [4]. The use of PCR as the step that distinguishes methylated vs. unmethylated DNA bases increases the sensitivity of the assay, allowing for measurement of significantly smaller amounts of DNA methylation than previous sequencing-based methods. More recently, by combining MSP with quantitative PCR and melting curve analysis, even more sensitive assays based on comparing ratios of methylated vs. unmethylated PCR products have been developed for measuring very low levels of methylation at specific loci [5]. Though MSP and its derivatives allowed for cataloging of methylation patterns in CpG islands of known tumor suppressors and oncogenes, with the increasing accessibility of genome sequencing, investigators have concomitantly become able to undertake epigenetic studies of broader scope. Whole genome bisulfite sequencing (WGBS) using massively parallel next generation sequencing technology has allowed for genome-wide examination of methylation patterns with single-base resolution. Nevertheless, a number of challenges continue to limit the utility of this technique. These include lower fragment complexity due to unmethylated cytosine to thymine conversion, PCR amplification bias for certain methylation states, significant coverage minimums, and difficulties with interpretation of methylation data in the context of other epigenetic changes, such as chromatin states [6, 7], as well as sequencing costs. As a result, multiple approaches have been developed to yield limited whole genome-level information while circumventing some of these limitations. Limited sequencing approaches attempt to reduce the cost and labor of WGBS by focusing on specific subsets within the larger genome. One widely used approach to genome-level methylation analysis is reduced representation bisulfite sequencing (RRBS). Rather than attempting to sequence the entire genome, RRBS begins with restriction enzymatic digestion of whole genomic DNA with a methylation-insensitive enzyme, frequently MspI, followed by size-based fragment purification. Fragments of between 40 and 220 base pairs have been shown to contain representative coverage and enrichment of most promoter and CpG island regions, which make up less than 1% of the genome [8, 9]. After bisulfite treatment, fragments are sequenced using traditional high-throughput sequencing methods. In addition to decreased cost and processing complexity, significantly less input DNA is required for RRBS vs. WGBS, allowing for RRBS use on paraffin-embedded or other limited tissue samples. RRBS also provides greater coverage in highly repetitive regions of the genome, including CpG islands, than WGBS, though RRBS ultimately only measures about 80% of CpG islands and 60% of promoters in the whole genome under optimal conditions [9]. For cancer genomics in particular, however, these advantages allow for the rapid comparison of large numbers of paired somatic and tumor epigenomes for a fraction of the price of whole genome sequencing. Recently, many additional methyl-sensitive endonucleases have been described, such as BisI, GlaI, and PcsI, which have different specificities and generate short fragments which can subsequently be sequenced [10]. Other limited sequencing approaches use microarray or other fixed probe-based capture techniques. BeadChip arrays, such as Illumina’s Infinium HumanMethylation450 system, have emerged as a widely used option for automated, low-cost, and rapid screening of pre-selected methylation sites for large numbers of samples [11]. Advantages include low starting material requirement (<1 μg DNA), relatively low cost, established analysis tools, and comprehensive promoter coverage. Some of the disadvantages include relatively high DNA purity requirement, lack of non-human genomes, and sparsity of non-promoter and non-CpG methylation probes [12]. Alternative approaches of focused differential methylation analysis include enriching for a subpopulation of DNA recognized by antibodies against 5-methylcytosine (MeDIP) or “pulling down” DNA using methyl-binding proteins (MBD-Seq) before sequencing. Comprehensive comparisons of currently available techniques and considerations regarding their use are available in literature [10, 12, 13].
Aberrant methylation changes in melanoma
Silencing of tumor suppressor genes
Focal DNA hypermethylation of tumor suppressor gene promoters is well described in many cancers, including melanoma. Silencing of PTEN, p16/14, and RASSF1A (Ras association domain family 1, isoform A) [14–19] in melanoma has been heavily investigated and is summarized in Table 1 [20]. The PTEN phosphatase converts PIP3 (phosphatidylinositol phosphate) to PIP2, antagonizing PI3K function, and suppressing activation of the PI3K/AKT pathway [21]. Mirmohammadsadegh et al. reported PTEN promoter methylation in 62% of melanoma serum samples examined [15]. Similar results were reported in a study using methylation-specific PCR, where 60% (120/200) exhibited methylation of the PTEN promoter [14]. PTEN methylation correlated with decreased immunohistological PTEN expression and was found to be associated with increased risk of death by Cox regression analysis. Recently, others have confirmed that methylation of PTEN is a significant prognostic factor of poor survival in melanoma [22]. In addition to epigenetic silencing, deletion/mutation occurs commonly and functional loss of PTEN occurs in approximately 40–60% of sporadic melanomas [23–25].
Table 1.
Summary of well described tumor suppressor genes methylation in melanoma
| Gene name | Function | Methylation prevalence (%) | Methylation context | Associated changes | Reference |
|---|---|---|---|---|---|
| PTEN | Inhibitor of PI3K signaling | 6–62 | Promoter hypermethylation | Gene transcription silenced | [14, 15, 22] |
| p16 | Inhibitor of CDK4/6 | 5–27 | Promoter hypermethylation | NRAS mutation associated | [16, 27, 29] |
| p14 | Inhibitor of MDM2 | 41–57 | Promoter hypermethylation | Gene transcription decreased | [17, 31] |
| RASSF1A | Cell cycle regulator | 15–57 | Promoter hypermethylation | Loss of expression | [19, 150] |
| MGMT | DNA repair | 35 | Promoter hypermethylation | No correlation | [150] |
p16 is one of the proteins encoded by the CDKN2A locus and plays an important role in arresting the cell cycle at the G1/S checkpoint, by inhibiting CDK4 and CDK6 and activating Rb [26]. p16 promoter methylation was reported to occur in 25% of analyzed cutaneous melanoma metastases (15/59), with a significant overrepresentation in NRAS-mutated samples [16]. Additional studies reported similar frequencies of p16 methylation of 27% (16/60), albeit others have reported lower frequencies of approximately 5–10% [27–29].
p14, also encoded by the CDKN2A locus, binds and inhibits MDM2 from triggering p53 ubiquitination and targeting it for proteosomal degradation [30]. p14 methylation has been comparatively less studied but has been shown to be hypermethylated in approximately 57% (34/60 samples) of melanoma samples, independently of the p16 promoter (88% of the time), but also in conjunction with p16 promoter methylation (12% of the time) [31], findings that were later corroborated by additional studies [17]. Interestingly, CDKN2A methylation has not been reported in human melanocytic nevi (0/43 samples) [27, 32], in contrast to genetic alterations of this locus, which are well documented.
RASSF1A is a tumor suppressor gene, encoding a microtubule-associated protein that regulates mitotic arrest, cell cycle arrest, and apoptosis [33]. Methylation of two regions of the RASSF1A CpG island was reported in 44 metastatic melanoma tumors and 11 melanoma cell lines using methylation-specific PCR [19]. Overall, RASSF1A was hypermethylated in 55% of melanoma tumors, which correlated with loss of expression of the RASSF1A gene. The expression could be restored with 5-aza-2′-deoxycytidine treatment [19]. A follow-up study investigated the methylation of RARB2, MGMT, DAPK, and RASSF1A in 20 primary melanomas, 86 metastatic melanoma, and 15 cell lines [18]. RASSF1A was methylated in 15% (3/20) of primary tumors and 57% (49/86) of metastatic samples, suggesting that methylation level may correlate with advanced stage. Similarly, Tanemura et al. reported low methylation frequency of RASSF1A in early stage melanoma but approximately 50% in stage III and IV melanoma samples [34], suggesting that RASSF1A methylation may potentially be an indicator of tumor progression. Treatment with 5-aza-deoxycytidine causes demethylation of the RASSF1A promoter and re-expression [18]. A microarray-based screen identified 17 novel genes undergoing methylation-mediated silencing. Three genes have potential tumor-suppressive function, including HOXB13, SYK, and LXN [35, 36]. Methylation of genes classically thought of as oncogenes, such as KIT, has also been described in melanoma, with currently unclear functional roles [37].
Methylation differences between melanocytes, nevi, and melanoma
Apart from known tumor suppressors, over a hundred other genes have been identified as differentially methylated in melanoma relative to melanocytes. Many differentially methylated genes belong to pathways critical for cancer cell survival and growth, including cell cycle, apoptosis, metabolism, DNA repair, PI3K/mTOR signaling, metastasis, and immune response, as summarized in Table 2. Many genome-wide studies have investigated the methylome of melanoma cell lines and clinical samples [35, 38–41]. The first genome-wide integrated analysis of promoter methylation and gene expression compared methylation and expression in eight melanoma cell lines relative to newborn and adult benign melanocytes [39]. The study used an elegant pipeline of linear mixed-effect modeling and manual promoter region filtering. It identified 76 differentially methylated markers, most of which—89% (68/76), were hypermethylated, and only a minority was previously reported in melanoma at the time (COL1A2, RAB33A, DDIT4L, and HOXB13). The results were validated by bisulfite sequencing of COL1A2, NPM2, HSPB6, DDIT4L, and MT1G promoters, which exhibited increasing incidence with advanced melanoma stage, suggesting potential use as predictors of melanoma progression [39]. A subsequent study used a candidate gene approach and investigated the methylation status of 15 cancer-related genes in 16 melanoma cell lines. Melanoma hypermethylation prevalence for ERα (50%), MGMT (50%), RARB2 (44%), RIL (88%), RASSF1A (69%), PAX7 (31%), PGRB (56%), PAX2 (38%), NKX2-3 (63%), OLIG2 (63%), HAND1 (63%), ECAD (88%), CDH13 (44%), MLH1 (0%), and p16 (6%) was reported [40]. Similarly, Bonazzi et al. reported methylation of COL1A2 (24%), THBS1 (31%), TNFRSD10D (66%), and UCHL1 (42%) genes in 12 melanoma cell lines, relative to a reference pool of melanocytes [42]. More recent studies have integrated DNA methylation changes with transcriptional changes (RNA-Seq) and histone modifications (ChIP-Seq). An integrated RNA-Seq and CpG island demethylation profiling of melanocytes and melanomas found that melanocytes undergo progressive hypomethylation, followed by extensive hypermethylation as melanoma progresses [43]. However, these results are based on one stage I melanoma cell line and melanocyte line, one stage III melanoma cell line, and two stage IV melanoma cell lines. Despite these limitations, the study was able to correctly ascertain the methylation status of PCSK1, CYP1B1, QPCT, c-Kit, and TERC genes, which were previously identified by candidate gene approaches. Overall, 821 differentially methylated genes were identified, mostly belonging to a network of developmental pathways [43]. Notably, repetitive DNA elements exhibited extensive hypomethylation in stage III/IV melanoma cell lines, possibly contributing to increased transposition and mutagenesis [43]. The study provided a comprehensive catalog of CpG methylation and expression during melanoma progression, which could be utilized as a resource to better understand how methylation changes evolve in melanoma.
Table 2.
Pathway classification of differentially methylated genes in melanoma
| Pathway/function | Genes (methylation frequencya) | References |
|---|---|---|
| Tumor suppressors | CDH1 (88%), CDKN2A (76%), PTEN (23%), APC (15%), SOCS1 (75%) | [15, 40, 151, 152] |
| Oncogenes | PAX7 (31%), OLIG2 (63%), SYK (2%) | [35, 40] |
| Protein kinases | DAPK1 (19%), HSPB8 (69%) | [152, 153] |
| Differentiation | TNFRSF10A/C/D,CCR7, THBD (20%), BST2 (50%), DPP4 (80%), ENC1 (6%), GDF15 (75%), WIF1 | [35, 154–159] |
| Homeodomain proteins | HOXB13 (20%), PAX2 (38%), PAX7 (31%), NKX2-3 (63%) | [35, 40, 160] |
| Hypoxia | CDKN1B, CDKN1C (35%), CXCR4, LXN (95%) | [35, 36, 151, 154] |
| Transcription factors | PGR (56%), HAND1/2 (15%), PAX7 (31%), ESR1, RUNX3 (23%) | [40, 151, 161] |
| Interferon gamma response | SOCS1 (75%), HLA-A, PTGS2 (20%), SOCS3 (60%), BST2 (50%), XAF1, SOCS2 (44%) | [35, 152, 162–164] |
| Epithelial-to-mesenchymal transition | TFPI2, TIMP3 (13%), COL1A2 (63%), TPM1 (8%), PDLIM4 | [39, 42, 152, 165, 166] |
| PI3K/mTOR | CXCR4, CDKN1B, PTEN (23%) | [151, 154] |
| Metastasis | CDH8 (10%), CDH13 (44%), EPB41L3 (5%), SERPINB5 (100%), TFPI2, SYK (3%), CCR7 | [35, 40, 152, 154, 155] |
| Immune recognition | MAGE-A1,2,3,4, BAGE (83%), HLA-A | [167, 168] |
| Apoptosis | RASSF1A, HSPB6 (69%), TRAILR1 (80%) | [39, 169, 170] |
| DNA repair | MGMT (0–50%) | [151] |
| Metabolism | CYP1B1 (100%), DNAJC15 (50%), CD98 | [35, 43, 171] |
| Other/unknown | FAM78A, LRRC2, PCSK1, PPP1R3C, PTPRG, QPCT, SLC27A3, DERL3, MFAP2, MT1G, WFDC1 | [35, 39, 42, 151] |
aWhere applicable
Significant methylation differences have also been reported between melanocytic nevi and melanoma. A study using microarrays to survey 1505 CpG sites in 27 benign nevi and 22 primary melanomas identified significantly differentially methylated genes that distinguished melanoma from nevi [38]. Seven genes were reported as hypermethylated in melanoma (COL1A2, FRZB, GSTM2, KCNK4, NPR2, TRIP6) and 19 genes were reported as hypomethylated, (CD2, EMR3, CARD15, EV12A, HLA-DP1, IFNG, IL2, ITK, KLK10, LAT, MPO, PSCA, PTHLH, PTHR1, RUNX3, and TNFSF8). Approximately half of the genes had immune-related functions, including T cell regulation (ITK, LAT, CD2, TNFSF8, IFNG, and IL2). However, the study did not differentiate between expression in melanoma lesional cells or infiltrating lymphocytes [38]. Another group studied global DNA methylation in melanoma cell lines and benign nevi using Infinium BeadChip arrays and identified 106 genes hypermethylated in melanoma [41]. The most frequently hypermethylated genes identified were HOXA9, C1orf106, HIST1H3E, MAPK13, and LEP, with MAPK13 methylation in 67% of primary and 85% of metastatic melanomas. Interestingly, transfection of ectopic MAPK13 into cell lines with promoter methylation of MAPK13 decreased proliferation, but did not affect proliferation of cells without endogenous MAPK13 silencing. Gene set enrichment revealed significant overrepresentation of homeobox genes (HOX) and G protein-coupled receptor genes [41]. While numerous DNA methylation differences were identified among melanocytes, nevi, and melanoma, no methylation-based clinically validated assays have been reported to date.
Methylation clusters in melanoma are associated with different biologic behavior
Several studies have suggested that distinct methylation subgroups exist within melanoma and are associated with distinct biologic behavior and survival outcomes. Unsupervised clustering and principal component analysis of genome-wide DNA methylation patterns in melanoma revealed three methylation clusters based on total methylation: MS1, MS2, and MS3 [44]. The MS1 group had the highest methylation level, mainly elevated at promoter islands and poised (“bivalent”) promoters, which are Polycomb repressive complex (PRC2) target genes. The MS3 group had the lowest methylation levels, resembling peripheral blood leukocytes, and MS2 was intermediate. Neither BRAF nor NRAS mutation was clearly associated with the methylation clusters, but homozygous deletions of CDKN2A were more frequent in MS1. There was no significant association of the methylation clusters with histopathology or primary tumor characteristics. Furthermore, the subgroups did not differ by the total number of mutations, but MS1 was enriched for IDH1 R132 hotspot mutations. Metastatic melanoma samples from TCGA with the MS1 signature had significantly inferior survival (~20 months for MS1 vs. 60 months for MS3) [44]. Analyzing differential gene expression between the clusters revealed upregulation of TP53, MDM2, CDK4, CDK6, CCND1, CCNE1, and E2F3, as well as epigenetic modifiers TET1, JARID1B, SWI/SNF chromatin remodelers (SMARCA1/2/4, ARID2, PBRM1), and DNMT3A in the MS1 cluster, and enrichment of an immune signature in the MS3 cluster. The clusters were correlated with different phenotypic behavior based on gene expression analysis. The MS1 cluster was termed “proliferative,” while the MS3 cluster was dominated by an “immune high” signature, possibly explaining the better survival of MS3 cluster patients [44]. However, it remains unclear which immune cells infiltrate and how they interact with melanoma cells. In contrast to these findings, Ecsedi et al. suggest that hypermethylation is an early event in melanoma, that is, methylation gradually decreases with progression (inversely correlated with Breslow thickness), and is associated with less favorable overall survival [45]. They also found that BRAF V600E is associated with specific methylation changes, as opposed to wild-type samples. This finding is consistent with studies suggesting that oncogenic BRAF drives methylation of specific target genes [46, 47]. It should be noted that Ecsedi et al. used an assay method that surveys only 1505 CpGs, as opposed to Lauss et al. who used the HumanMethylation450K platform (~480,000 CpGs). The different findings could possibly be explained by differences in metastatic and primary tumors, use of different methodologies with different scope (Methyl450 vs. GoldenGate Methylation assay), or underlying differences in the sampled populations. Both studies support the existence of at least two distinct methylation clusters in melanoma, which are associated with different gene expression and prognosis. Others have reported genome-wide methylation correlating with Breslow thickness, mutated BRAF, ulceration, and a negative association with mitotic rate [48]. A potential role for DNA methylation in regulating metastasis to the brain was linked to aberrant methylation of the homeobox D cluster [49]. Identifying differentially methylated CpG islands during melanoma progression suggested that genome-wide overall DNA methylation (mostly intragenic) significantly decreased with progression but focal (promoter associated) CpG methylation progressively increased. Analyzing methylation differences between brain metastases and lymph node metastases identified ~1500 differentially methylated CpG sites associated with genes. Enrichment analysis revealed overrepresentation of genes involved in cell cycle regulation, morphology, and assembly, and DNA repair, recombination, and replication [49]. Integrating methylation changes with gene expression changes prompted focus on the homeobox (HOX) gene family, with 10 members of the family undergoing differential methylation [49]. Specifically, HOXD9 was hypermethylated in brain metastases and associated with a 2.7-fold higher risk of death. HOX genes play important roles during development but are silenced in somatic cells by Polycomb repressive complexes. Aberrant expression of HOX genes was reported in different human tumors, including melanoma [50]. SOX9 expression has also been shown to be regulated by DNA methylation in melanoma, where it can regulate invasion, and predict poor survival [51]. In addition to observational studies describing the methylome of melanoma, several studies suggest a BRAF-driven mechanism of aberrant methylation of specific genomic loci in melanoma [46].
DNA methylation of miRNA genes in melanoma
Methylation of microRNA (miRNAs) genes has also been shown to play important roles in melanoma cell survival, proliferation, and migration by affecting epithelial-to-mesenchymal transition (EMT) signaling, cytoskeletal components, p53, and PI3K pathways (Fig. 1). miR-34a methylation and expression silencing was reported in 43% (19/44) of melanoma cell lines and 60% (20/32) of primary melanomas [52]. miR-34a, which is also a p53 transcriptional target, is thought to play a tumor-suppressive role in cancer by inducing cell cycle arrest or apoptosis, at least partly by regulating CDK6 [52]. A liposomal miR-34a mimic recently showed potential activity in acral melanoma in a phase I clinical trial [53]; however, the expansion study (NCT01829971) was subsequently halted due to multiple grade 4 immune-related adverse events. Other members of the miR-34 family, miR-34b and miR-34c, were reported to regulate the expression of natural killer cell receptor ligand ULBP2 [54], a prognostic marker in human melanoma [55], as well as MET, cyclin-dependent kinases, and N-myc expression [56, 57]. Forced expression of miR-34b decreases melanoma cell invasion and adhesion, presumably through affecting cytoskeletal remodeling and expression of cell adhesion network genes, such as THBS2 and DKK1 [58]. miR-211 expression is regulated by DNA methylation (DNMT1) in melanoma and can act as a metabolic switch through regulating HIF-1a protein stability [59] and inhibiting EMT by targeting RAB22A [60]. Ectopic expression of miR-211 in melanoma is associated with decreased cell growth during hypoxia [59]. DNA methylation is also thought to regulate the expression of miR-203 [61]. miR-203 ectopic expression was reported to decrease cell invasion by targeting the Polycomb group gene BMI1 in melanoma [62]. Additionally, Mazar et al. reported that miR-34b/c, miR-489, miR-375, miR-132, miR-519b, miR-654, miR-let-7e, miR-142-3p, miR-200a, miR-145, miR-21, miR-496, and miR-452 expressions can be induced by hypomethylating agents in melanoma cell lines [58, 63]. miR-375-associated CpG island methylation and suppression was reported in melanoma tissue samples from primary and metastatic lesions, but not in melanocytes or melanocytic nevi. Expression of miR-375 could be de-repressed with hypomethylating agents, and ectopic expression inhibited cell proliferation, invasion, and motility [64]. Expression of miR-18b also was reported to be silenced by DNA methylation of a proximal CpG island in melanoma cell lines. Forced re-expression decreased MDM2 and increased p53 expression level, associated with suppression of colony formation [65], suggesting a tumor-suppressive role for miR-18b. Methylation and silencing of miR-196b expression was reported to potentiate mTOR signaling in melanoma [66] and other cancers [67]. miR-205 was reported to be silenced in melanoma compared to melanocytic nevi. Ectopic expression of miR-205 decreased melanoma cell migration in vitro, associated with increased decreased expression of ZEB2 and increased expression of E-cadherin [68]. Silencing of a large miRNA cluster on chromosome 14q32 is reported to be mediated by DNA methylation in melanoma. Within this cluster are miR-376a and miR-376c, which can downregulate IGF1R levels and decrease cell migration and growth [69]. miR-31 is also frequently silenced by DNA methylation in melanoma cells, and forced expression of miR-31 is reported to decrease cell migration and invasion, possibly through targeting SRC, MET, RAB27a, and MAP3K14 [70].
Fig. 1.

Abnormal DNA methylation in melanoma disrupts microRNA expression. In melanocytes and nevi (left), normal DNA methylation patterns contribute to balanced pro-tumorigenic (shown in red) and antitumorigenic (shown in green) microRNA expression. In melanoma (right), aberrant DNA methylation causes expression changes leading to microRNA imbalance and abundance of pro-tumorigenic microRNAs. See text for specific microRNAs disrupted and their targets
Most studies described above suggest that hypermethylation and silencing of miRNAs plays a pro-tumorigenic role in melanoma and that ectopic expression of specific miRNAs could attenuate melanoma proliferation and metastatic potential by targeting EMT signaling, PI3K/AKT pathway, or p53 signaling in vitro and in animal models. Despite solid pre-clinical evidence, very few miRNA liposomal therapies have been investigated in clinical trials in melanoma thus far.
Source of aberrant DNA methylation
The source of aberrant DNA methylation in melanoma, i.e., global hypomethylation and focal hypermethylation, remains elusive [22]. In general, these changes could be mediated by (1) an active process, either through increased activity or overexpression of DNMT enzymes; (2) aberrant targeting of DNMT enzymes, due to altered splicing, altered binding, or altered expression of scaffolding proteins; and (3) a passive process due to changes in other epigenetic modifications that regulate targeting of DNA methylation. The notion that DNA methylation is regulated by, and regulates, proliferative signaling will be discussed here, due to emerging evidence supporting this hypothesis.
PI3K signaling, which is activated in most cancers, can regulate methylation of imprinted regions [71], and inactivation of PI3K signaling decreases DNMT3B level in HCC [72], melanoma, and prostate [73] and other cancers. Aberrant DNA methylation may thus act as a licensor that enables chronic activation of growth-signaling pathways, i.e., PI3K/mTOR signaling through various mechanisms, which are probably cell type specific, but may include PTEN silencing, miRNA-mediated regulation of RICTOR [66], or other components of the PI3K/AKT pathway. Transient activation of proliferative pathways may serve to instruct proliferative DNA methylation patterns by upregulating DNMT expression, altering DNMT splicing, or altering DNMT targeting, by affecting expression of targeting proteins (such as MAFG) [47]. Once aberrantly activated, misguided DNA methylation leads to establishment of abnormal patterns that support chronic activation of growth signaling. BRAF activation and MAPK signaling has been shown to drive tumor-specific methylation programs in colon cancer and possibly melanoma [47]. Our in vivo reports in melanoma and others in specific CNS tumor support [74, 75] this notion, but it remains to be thoroughly investigated in vivo in additional cancers. In vitro, many cancers require DNMT3B for survival and proliferation [76], implying that DNMT3B can regulate proliferative pathways in a wide array of cancers. Apart from this instructive (“driver”) role targeting particular pathways, it is also plausible, but less likely, that aberrant DNA methylation leads to stochastic expression changes that promote tumor progression. CpG microarray analysis of UACC62 melanoma cell lines with and without BRAF knockdown surprisingly identified many hypermethylated and hypomethylated genes, suggesting that mutant BRAF dictates a specific methylation program. Knockdown of BRAF decreased the expression of DNMT1 and EZH2 in the cell line, suggesting that DNMT1 may be the mediating DNMT. However, expression of other DNMTs was not investigated, and rescue of demethylated sites by DNMT1 overexpression was not reported. In addition to proliferative pathways regulating DNA methylation, the converse is also described, establishing a potential possible feedback loop: methylation has been shown to regulate MITF expression, SOX9 expression, MAPK, and PI3K signaling [51, 77, 78]. Recently, loss of LKB1, a tumor suppressor somatically inactivated in ~10% of melanomas, has been shown to funnel glucose derivatives toward serine metabolism, increased S-adenosyl methionine production, DNA hypermethylation, and increased proliferation in the context of Ras/Raf activation [79].
DNA demethylation in melanoma
Shortly following the discovery of 5-hydroxymethylcytosine (5-hmc), active demethylation, and the role of TET family hydroxylases in converting 5-mc to 5-hmc [80–82], an important role for 5-hmc was shown in melanoma [83]. Lian et al. investigated the global level of 5-hmc in human melanocytes, nevi, and melanoma and reported a significant decrease of 5-hmc in melanoma relative to nevi and melanocytes. The loss of 5-hmc correlated with melanoma progression in patient sample tissue microarrays. Importantly, overexpressing IDH2 or TET2 restored 5-hmc levels in melanoma cell lines, suggesting that lack/loss of function of these enzymes plays an essential role in 5-hmc depletion during melanoma progression. Restoring 5-hmc levels by IDH2 and TET2 overexpression was associated with suppressed tumor invasion and growth in a zebrafish melanoma model and mouse xenograft model of melanoma [83], suggesting a potential functional role of this epigenetic mark in melanoma growth and invasion. Several subsequent studies confirmed that 5-hmc is decreased in metastatic melanoma, associated with loss of TET activity [84, 85], and suggest its use as an adjunct marker to distinguish between benign nodal nevi and melanoma in lymph node biopsies [86–88]. While conclusive functional studies in melanoma have not been reported yet, 5-hmc may be enriched in exons and transcription factor binding sites to promote gene expression [89, 90], through distinct 5-hmc readers and transcription factors [91, 92], according to studies in stem cells and CNS. Mutations in IDH1/2 enzymes have been reported in ~6–10% of melanomas [93, 94] and correlate with the CpG island methylator phenotype (CIMP) cluster-defined DNA methylation profile in melanoma samples in TCGA. TET enzyme family expression has been reported as decreased in melanoma, albeit by an unknown mechanism [83, 85]. Certain IDH1 mutations result in gain of function production of oncometabolite 2-HG, a competitive inhibitor of α-KG, which is a necessary cofactor for TET enzymatic activity. An SNP associated with melanoma risk was discovered in an intron of TET2, and somatic mutations of TET2 were reported in approximately 4% of melanoma cases in one study [95]; however, other sequencing studies have not reported TET mutations [93, 96].
Our current understanding of the role of DNA demethylation and oxidative derivatives of 5-mc in melanoma is in its infancy. Currently, there is a large discrepancy between the frequency of loss of 5-hmc in melanoma (nearly universal, according to several reports) and mechanisms potentially explaining loss of 5-hmc (IDH gain of function, TET enzyme loss of function). Additionally, studies investigating 5-fc (5-formylcytosine) and 5-cac (5-carboxycytosine) prevalence and distribution in melanoma have not been reported yet. Early evidence suggests that DNA demethylation plays important roles in melanoma biology and will undoubtedly be a field of active investigation in years to come.
Differential DNA methylation as a biomarker in melanoma
DNA methylation has the potential to be used as a melanoma biomarker in several clinically important settings: (1) differentiating benign lesions (nevi) from melanoma; (2) determining melanoma burden in sentinel lymph nodes; (3) aiding prognosis of stage III patients, which currently exhibit great variability in survival; (4) predicting response to therapies; and (5) monitoring for recurrence using peripheral blood.
Conway et al. identified a 12 CpG locus signature highly predictive of melanoma in a small independent sample set of 29 nevi and 25 melanomas (ROC = 0.95) [38]. Gao et al. reported a diagnostic algorithm consisting of CLDN11, CDH11, PPP1R3C, MAPL13, and GNMT which could help distinguish between melanoma and dysplastic nevi (ROC = 0.81) [97]. Detection of aberrantly methylated SOCS1/2, RASSF1A, MGMT, and CDKN2A in serum has been reported to have diagnostic value in patients with malignant melanoma [98]. Despite reasonable success with the small validation cohorts in the original reports, these signatures have not been further validated by other, larger studies.
Melanoma patients currently exhibit great variability in survival, even within AJCC subgroups [99], and there is great unmet need for a molecular biomarker to further stratify patient risk. Identification of a CpG island methylation signature has been useful in predicting prognosis, diagnosis, and response to treatment in a variety of tumor types [100, 101]. Genome-wide methylation studies have uncovered that tumors with enriched methylation in CpG islands have a particularly aggressive phenotype, termed the CpG island methylator phenotype (CIMP) [102]. CIMP was found to predict prognosis and, in some cases, response to therapy in various cancers [103–109]. Tanemura et al. investigated promoter methylation of tumor-related genes (TRGs): WIF1, TFPI2, RASSF1A, SOCS1, GATA4, RARB2, and a family of MINT (methylated in tumor) genes in an attempt to define a CIMP in melanoma [34]. They reported an association of WIF1, TFPI2, RASSF1A, and SOCS1 hypermethylation with advanced clinical stage in a small cohort of patients (n = 122). Apart from this proposed CIMP signature, several studies reported that DNA methylation of single genes can predict survival of melanoma patients. Lahtz et al. reported that methylation silencing of PTEN, an inhibitor of PI3K signaling, exhibited significant association with poorer survival in melanoma [14].
Sigalotti et al. reported that methylation level of LINE-1 is associated with shorter overall survival in stage III cutaneous melanoma [110]. The 5-year overall survival with hypomethylated LINE-1 was 48%, compared to 7% for hypermethylated sequences [110]. In contrast, Ecsedi et al. reported that hypomethylation of LINE-1 is associated with shorter survival [111] and additionally identified hypermethylation of six genes (DSP, EPHB6, HCK, IL18, IRAK3, and KIT) as a poor prognostic factor associated with decreased overall survival. You et al. reported that increased TSLC1 (tumor suppressor in lung cancer 1) promoter methylation is associated with advanced melanoma stage and shorter progression-free survival in a cohort of 120 melanoma samples [112].
Methylation of the DNA repair protein MGMT was reported to predict response to temozolomide treatment in stage IV melanoma patients [113]. Of 64 patient samples in the study, methylation of MGMT was detected in 25% (16/64) of the samples. In the hypermethylated group, the response rate to temozolomide was 62%, compared to 15% in the unmethylated group. MGMT promoter hypermethylation was also associated with significantly higher response rate and longer progression-free survival in other studies [113, 114]. It should be noted that MGMT methylation status predicts response to temozolomide in glioblastoma [115, 116] and is used clinically for identifying tumors that may respond to temozolomide. Presence of circulating methylated RASSF1A DNA in the blood of melanoma patients was reported as a predictor of therapy response and disease outcome [117]. Circulating methylated RASSF1A DNA was associated with lack of response to therapy, while detection of at least one methylated gene out of RASSF1A, RAR-beta2, and MGMT signatures was associated with shorter overall survival [117]. A study investigating differentially methylated CpG sites between melanoma brain metastases (poor prognosis) and lymph node metastasis from patients with good prognosis reported that HOXD9 methylation was associated with overall survival [49]. In a validation cohort of 145 melanoma patients, HOXD9 hypermethylation in lymph node metastases was associated with shorter progression-free and overall survival [49]. The function of HOXD9 in melanoma is incompletely understood, but studies from HCC suggest that it significantly enhances migration, invasion, and metastasis [118]. Methylation and silencing of ESRP1 has been also associated with altered CD44v6 expression, which could predict brain metastasis-free survival [119].
Sigalotti et al. used genome-wide Methylation 27 beadchip arrays to study 45 stage IIIc melanoma patients [120]. Using K-means clustering, the cohort was classified into a favorable and an unfavorable group, based on global methylation. Median survival in the favorable group was 31.5 months, compared to 10.3 months in the unfavorable group, translating to 5-year overall survival of 41.2 and 0%, respectively [120]. Samples could also be correctly assigned to groups using a 17-gene nearest shrunken centroid methylation signature (FGF4, WNT10B, FLJ33860, PCDHAC2, SLC6A11, GJB5, S100A9, CRHR1, MGC35206, IGLL1, TRIM40, SLC18A2, TUB, GRM4, SLC6A18, and ALOX12B). Methylation of the miR-196b promoter has been associated with a poorer 5-year overall survival in melanoma patients in TCGA by Cox multivariate analysis ([66, 93]).
Bell et al. studied enhancer methylation in a panel of 23 cancers, including melanoma. They reported that differentially methylated enhancer regions were metastatic site specific and determined by microenviromental cues. A significant correlation was found between specific enhancer methylation patterns and patient survival. Specifically, differential methylation of KIT-, CYTL1-, and KIF14-associated enhancers played a role in regulating proliferation [121].
While important and informative, the main limitation of the above studies is relatively small sample cohorts, lack of subsequent validation studies, and reliance on melanoma cell lines (in some studies), which can have an altered DNA methylation landscape. With the availability of large, well-annotated melanoma sample databases, such as TCGA and the International Cancer Genome Consortium, it will now be important to validate the status of the biomarkers discussed above and develop the most promising markers with prospective studies into a methylation signature which can be of clinical utility. Studies investigating DNA methylation as a diagnostic and prognostic tool in melanoma are summarized in Table 3.
Table 3.
Summary of DNA methylation biomarkers in melanoma
| Methylated gene(s) | Methylation association | Hazard ratioa | Reporting study |
|---|---|---|---|
| WIF1, TFPI2, RASSF1A and SOCS1 | Advanced clinical stage | – | [34] |
| 17-gene (see text) | Overall survival | 2.41 (1.02–5.7) | [120] |
| RASSF1A, RAR-beta2, MGMT | Shorter overall survival | 2.38 (1.1–4.8) | [117] |
| KIT, DSP, HCK, IL18 | Shorter overall survival | – | [45] |
| HOXD9 | Shorter overall survival | 2.7 (1.1–6.5) | [49] |
| LINE-1 | Shorter overall survival | 2.63 (1.2–5.6) | [110] |
| MINT31 | Longer overall survival | 0.237 (not reported) | [34] |
| TFPI2 | Presence of metastases | – | [172] |
| PTEN | Shorter overall survival | 3.76 (1.2–11.1) | [14, 22] |
| MGMT | Response to therapy | - | [113] |
| Longer progression-free survival | 2.17 (1.3–3.5) | ||
| TSLC1 | Progression-free survival | - | [112] |
| RASSF1A | Resistance to therapy | 0.21 (0.1–0.9) | [117] |
| ESR1 | Shorter overall survival | 2.31 (1.4–5.5) | [161] |
| Shorter progression free survival | 2.64 (1.3–5.1) | ||
| p73 | Sensitivity to therapy | – | [173] |
aWhere applicable, with 95% confidence interval in parenthesis
DNA methylation as a therapeutic target in melanoma
Nonspecific inhibition of DNA methylation
The reversible nature of DNA methylation makes it a desirable drug target, as opposed to mutations and deletions, which are more challenging to rectify [122]. Two major demethylating agents, 5-aza-2-deoxycytidine (decitabine, DEC) and 5-azacitidine (azacitidine, AZA), are FDA approved for treating myelodysplastic syndrome (MDS) and certain cases of AML [123]. It is still debated whether the clinical efficacy of high-dose AZA is due to demethylating activity [124] or DNA methylation-independent roles [125], notably induction of cytotoxic DNA damage, cell cycle arrest, and induction of tumor suppressors. FDA-approved use of demethylating agents currently remains limited to few hematologic malignancies. The role of decitabine in hematologic malignancies is well recognized with ~75 open clinical trials registered (most for myelodysplastic syndrome and AML). However, there are comparatively few active trials in solid tumors. A phase I/II ongoing clinical trial (NCT01876641) is investigating decitabine in combination with vemurafenib as a treatment for treatment-resistant BRAF V600-mutated metastatic melanoma. Preliminary results, presented at the ASCO 2015 annual meeting, suggest a 21% complete response rate. Complete response rates of <1% seen with vemurafenib alone were reported in the BRIM-3 trial [126], suggesting that DNA methylation may possibly play a role in regulating MAPK signaling and/or resistance to vemurafenib (J Clin Oncol 33, 2015 suppl; abstr 9056). Based on recent results, targeting DNA methylation may also be a synergistic strategy in those melanomas harboring somatic inactivation of the LKB1 tumor suppressor [79].
Inhibition of individual DNMT enzymes
DNA methylation in mammals is established and maintained by the complex interplay of at least three enzymatically active, independently encoded DNA methyltransferases: DNMT1, DNMT3A, and DNMT3B [127–129], summarized in Fig. 2. An additional DNMT3-like protein, DNMT3L, lacks essential components of the MTase (methyltransferase) motif but can bind and regulate the activity of DNMT3A [130] and DNMT3B [131, 132]. The non-redundant roles and specific targeting preferences of DNMTs are just becoming appreciated, owing to elegant enzyme-specific disruption studies [133, 134]. Consistent with this idea, a division of labor among methyltransferases [133] has been proposed and DNMT3B in particular may play specific, non-redundant roles in gene body methylation [135]. While there are currently no DNMT-specific inhibitors, there is extensive evidence suggesting that inhibition of DNMTs may have an antiproliferative effect on melanoma cells.
Fig. 2.
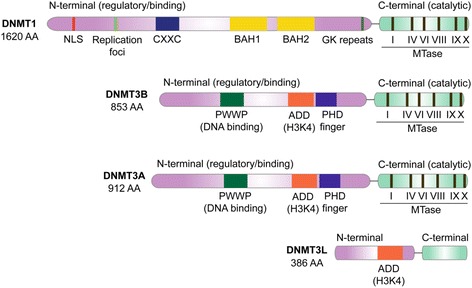
Structural domains of mammalian DNA methyltransferases. NLS nuclear localization signal, BAH bromo-adjacent homology, GK Gly-Lys linker, CXXC cysteine-rich motif, MTase methyltransferase, PWWP Pro-Trp-Trp-Pro motif, ADD ATRX/DNMT3/DNMT3L, PHD plant homeodomain
Antisense depletion of DNMT1 in the MZ2-MEL cell lines was shown to lead to hypomethylation and re-expression of the germ line-specific MAGE-A1 transgene, which is commonly silenced in melanoma [136]. Cancer-germ line gene activation upon DNMT1 transient depletion was observed in 45 human melanoma cell lines using a microarray approach [137]. Ras/Rac1/ERK activation induced increased DNMT1 protein expression during anchorage blockade and in response to oxidative stress. Interestingly, PI3K/AKT activity did not affect DNMT1 protein levels [138]. This study further proposed an increase in DNMT1 as a mediator of global DNA methylation changes and contributor to malignant transformation, although these claims have not been functionally investigated.
Stable RNAi depletion of DNMT3A in B16 melanoma cells decreased tumor growth and metastasis in a xenograft mouse model [139], which was associated with dysregulation of class I and class II MHC genes. The authors hypothesized that loss of DNMT3A increased MHC-dependent antigen presentation, prevented immune escape, and led to a T cell-mediated immune response, but this remains to be functionally investigated.
DNMT3B expression increases with melanoma progression [140], and DNMT3B has been associated with p16INK4A methylation in melanoma [17], as well as regulating mTORC2 signaling in a melanoma model in vivo [66]. Despite many pre-clinical studies suggesting a pro-tumorigenic role for individual DNMT enzymes, there are currently no enzyme-specific inhibitors. This is partly due to incomplete understanding of allosteric control, lack of crystal structures, and complex interactions, i.e., heteropolymer formation of DNMT enzymes [141].
DNA methylation and immunotherapy
The most promising novel therapy for melanoma, and other cancers with many neoantigens such as NSCLC, is immune-checkpoint inhibition [142]. DNA methylation has been long recognized to regulate expression of antigen presentation genes, MHC class I genes, tumor antigens such as MAGE and NY-ESO1, viral response, and interferon pathway genes [143]. DNA methylation was also recently shown to regulate expression of PD-1, PD-L1, PD-L2, and CTLA-4 [144]. Furthermore, many known methylation targets have been identified as differentially expressed in response to anti-CTLA-4, anti-PD-1 combination, or sequential therapy [145]. Perturbing DNA methylation could thus be a potential pathway to augment antitumor immune responses, and pre-clinical evidence supporting this notion has recently emerged. Inhibition of DNA methylation using AZA was shown to increase effectiveness of anti-CTLA4 therapy in a B16 graft model of melanoma in vivo [146] and reactivate expression of endogenous viruses in colon cancer cell lines [147]. Others have identified a distinct melanoma methylation cluster with improved survival rates and overexpression of an immune signature [44]. However, it is difficult to delineate whether the increased sensitivity to anti-CTLA4 is the result of specifically inhibiting DNA methylation and tumor antigen re-expression, triggering interferon signaling, DNA damage, DNA methylation-independent antiproliferative effects of AZA, or some other mechanism. Clinically, lung cancer patients who received AZA were subsequently more likely to respond to immune-checkpoint inhibitors [148]. Sporadic microsatellite instable colorectal cancer, which is associated with promoter methylation of Wnt target ITF2 and hMLH1, has been shown to respond to PD-1 inhibitor immunotherapy in a small trial [149]. Clinical trials combining 5-azacitidine with nivolumab and ipilimumab are currently ongoing in MDS, AML, and NSCLC. In melanoma, a phase I clinical trial (NCT02608437) is currently evaluating combination treatment with guadecitabine (hypomethylating agent) and ipilimumab in patients with unresectable or metastatic disease (J Clin Oncol 34, 2016 suppl; abstr TPS9595). Similarly, combination of oral azacitidine with pembrolizumab is being evaluated as part of a phase II trial in metastatic melanoma (NCT02816021). Results of these clinical trials (Table 4) will provide important information regarding the potential synergistic effects of hypomethylating agents and checkpoint inhibitors in melanoma (Fig. 3).
Table 4.
Summary of current clinical trials targeting DNA methylation in melanoma
| Approach | Phase | Status | Clinical trial identifier |
|---|---|---|---|
| Guadecitabine + ipilimumab for unresectable disease | I | Recruiting | NCT02608437 |
| Oral azacitidine + pembrolizumab for metastatic melanoma | II | Recruiting soon | NCT02816021 |
| Azacitidine + rInterferon alfa2b for stage III/IV unresectable | I/II | Completed, results not reported | NCT00217542 |
| Oral azacitidine bioequivalence study | I | Recruiting | NCT02223052 |
| Decitabine + vemurafenib + cobimetinib for resistant disease | I/II | Recruiting | NCT01876641 |
Fig. 3.
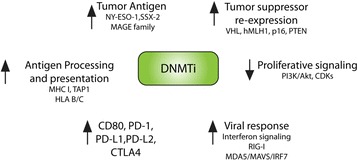
Potential mechanisms of synergy between DNA methylation inhibitors with targeted therapies and immune-checkpoint therapy. Inhibition of DNMTs has been shown to increase expression of melanoma antigens such as NY-ESO-1, MAGE family genes, and SSX-2. Inhibition of methylation can lead to tumor suppressor (VHL, hMLH1, p16, PTEN) re-expression, decreased proliferative signaling, increased viral response through activation of interferon signaling, altered expression of immunocheckpoint receptors, and increased antigen processing
Conclusions
Identification and cataloging of aberrant DNA methylation changes in melanoma, as reported by numerous studies discussed above, is an important first step toward understanding the methylation landscape of human melanoma and utilizing it for clinical benefit. Methylation of immunomodulatory genes, existence of distinct DNA methylation clusters with separate molecular phenotypes, and aberrant methylation of Polycomb targets are some of the general trends emerging in recent literature. However, many challenges remain to be resolved before the potential of targeting DNA methylation can be optimally utilized. Functional evaluation of differentially methylated genes in vivo, i.e., whether the aberrant methylation is directly driving proliferation or is a by-product of malfunctioned methylation machinery (“passenger”), is generally lacking. The origin of aberrant DNA methylation, i.e., altered expression and/or targeting of DNMT enzymes, remains incompletely understood, and identification of DNMT-specific inhibitors has proven to be challenging. Finally, understanding the molecular correlates of melanoma response to immunotherapy is a tremendously important question and one in which DNA methylation may play a crucial role, according to recent studies. Clinical trials investigating potential synergistic effects of immune-checkpoint inhibitors and hypomethylating agents are currently ongoing and may identify methylation-regulated neoantigens. In the interim, DNA methylation signatures may prove useful for identifying patients likely to respond to therapy and may aid decision-making in treatment of melanoma patients.
Acknowledgements
The authors would like to acknowledge Dr. Sherman Weissman, Dr. Qin Yan, and Dr. Narendra Wajapeyee (all of Yale University) for constructive discussion and feedback on the manuscript.
Funding
This work as been supported by the Yale MSTP (NIH T32GM007205) and Ruth L. Kirschstein National Research Service Award (NIH F30 CA196089) to GM.
Availability of data and materials
The MEDLINE database of biomedical and life science journal citations was queried using the search term “methylation” AND “melanoma.” The search returned a total of 738 citations in English between 1977 and 2017. Of the 738 citations, 711 had an abstract available for review. The Web of Science (ISI Web of Knowledge) citation database was queried with the search term “methylation” AND “melanoma.” A total of 1123 citations between 1985 and 2017 were identified, and 778 citations had available abstracts in English. All available unique abstracts with ≥1 citation, a total of 1020 abstracts, were reviewed by GM.
Authors’ contributions
GM conceived the study, reviewed the literature, and wrote the manuscript. NT reviewed the literature and wrote a chapter of the manuscript. GM, MWB, and NT edited the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- AJCC
American Joint Committee on Cancer
- AKT
RAC-alpha serine/threonine-protein kinase
- AML
Acute myeloid leukemia
- ASCO
American Society for Clinical Oncology
- AZA
Azacitidine
- BRAF
v-Raf murine sarcoma viral oncogene homolog B
- CIMP
CpG island methylator phenotype
- CNS
Central nervous system
- CTLA-4
Cytotoxic T lymphocyte-associated protein 4
- DAPK
Death-associated protein kinase 1
- DEC
Decitabine
- DNA
Deoxyribonucleic acid
- DNMT
DNA methyltransferase
- DSP
Desmoplakin
- ECAD
E-cadherin
- EMT
Epithelial-to-mesenchymal transition
- ERKs
Extracellular-regulated kinases
- FRZB
Frizzled-related protein
- GNMT
Glycine N-methyltransferase
- HCC
Hepatocellular carcinoma
- HCK
Hematopoietic cell kinase
- HG
Hydroxyglutarate
- HIF
Hypoxia-induced factor
- HLA
Human leukocyte antigen
- HOX
Hoemeobox
- IDH
Isocitrate dehydrogenase
- IFNG
Interferon gamma
- IL
Interleukin
- ITK
Interleukin 2-inducible T cell kinase
- KG
Ketoglutarate
- LATs
Linker for activation of T cells
- LEP
Leptin
- LINEs
Long interspersed nuclear elements
- LXN
Latexin
- MAFG
v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog G
- MAGE
Melanoma antigen/melanoma-associated antigen
- MAPKs
Mitogen-activated protein kinases
- MDS
Myelodysplastic syndrome
- MET
Mesenchymal-epithelial transition factor
- MGMT
O-6-Methylguanine-DNA methyltransferase
- MHC
Major histocompatibility complex
- MINT
Msx2-interacting nuclear target
- MITF
Microphthalmia-associated transcription factor
- MPO
Myeloperoxidase
- MSP
Methyl-specific PCR
- NRAS
Neuroblastoma RAS viral oncogene homolog
- NSCLC
Non-small cell lung cancer
- PGRB
Promoter regions B
- PSCA
Prostate stem cell antigen
- PTEN
Phosphatase and tensin homolog
- PTHLH
Parathyroid hormone-like hormone
- QPCT
Glutaminyl-peptide cyclotransferase
- RAR
Retinoic acid receptor
- RICTOR
Raptor independent component of mTOR complex 2
- ROC
Receiver operator characteristic
- RRBS
Reduced representation bisulfite sequencing
- SNP
Single nucleotide polymorphism
- SWI/SNF
SWItch/Sucrose Non-Fermentable
- SYK
Spleen tyrosine kinase
- TCGA
The Cancer Genome Atlas
- TERC
Telomerase RNA component
- TET
Ten-eleven translocation methylcytosine dioxygenase
- TUB
Tubby bipartite transcription factor
- WGBS
Whole genome bisulfite sequencing
References
- 1.Schinke C, et al. Aberrant DNA methylation in malignant melanoma. Melanoma Res. 2010;20(4):253–65. doi: 10.1097/CMR.0b013e328338a35a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frommer M, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89(5):1827–31. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herman JG, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93(18):9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herman JG, Baylin SB. Methylation-specific PCR. Curr Protoc Hum Genet. 2001;Chapter 10:Unit 10 6. doi: 10.1002/0471142905.hg1006s16. [DOI] [PubMed] [Google Scholar]
- 5.Kristensen LS, et al. Sensitive Melting Analysis after Real Time- Methylation Specific PCR (SMART-MSP): high-throughput and probe-free quantitative DNA methylation detection. Nucleic Acids Res. 2008;36(7):e42. doi: 10.1093/nar/gkn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adusumalli S, et al. Methodological aspects of whole-genome bisulfite sequencing analysis. Brief Bioinform. 2015;16(3):369–79. doi: 10.1093/bib/bbu016. [DOI] [PubMed] [Google Scholar]
- 7.Crary-Dooley FK, et al. A comparison of existing global DNA methylation assays to low-coverage whole-genome bisulfite sequencing for epidemiological studies. Epigenetics. 2017:0:1-9. [DOI] [PMC free article] [PubMed]
- 8.Meissner A, et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33(18):5868–77. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu H, et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6(4):468–81. doi: 10.1038/nprot.2010.190. [DOI] [PubMed] [Google Scholar]
- 10.Kurdyukov S, Bullock M. DNA methylation analysis: choosing the right method. Biology (Basel) 2016;5:1. doi: 10.3390/biology5010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teh AL, et al. Comparison of methyl-capture sequencing vs. Infinium 450K methylation array for methylome analysis in clinical samples. Epigenetics. 2016;11(1):36–48. doi: 10.1080/15592294.2015.1132136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yong WS, Hsu FM, Chen PY. Profiling genome-wide DNA methylation. Epigenetics Chromatin. 2016;9:26. doi: 10.1186/s13072-016-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrison A, Parle-McDermott A. DNA methylation: a timeline of methods and applications. Front Genet. 2011;2:74. doi: 10.3389/fgene.2011.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lahtz C, et al. Methylation of PTEN as a prognostic factor in malignant melanoma of the skin. J Invest Dermatol. 2010;130(2):620–2. doi: 10.1038/jid.2009.226. [DOI] [PubMed] [Google Scholar]
- 15.Mirmohammadsadegh A, et al. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006;66(13):6546–52. doi: 10.1158/0008-5472.CAN-06-0384. [DOI] [PubMed] [Google Scholar]
- 16.Jonsson A, et al. High frequency of p16(INK4A) promoter methylation in NRAS-mutated cutaneous melanoma. J Invest Dermatol. 2010;130(12):2809–17. doi: 10.1038/jid.2010.216. [DOI] [PubMed] [Google Scholar]
- 17.Venza M, et al. Epigenetic regulation of p14ARF and p16INK4A expression in cutaneous and uveal melanoma. Biochim Biophys Acta. 2015;1849(3):247–56. doi: 10.1016/j.bbagrm.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Hoon DS, et al. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene. 2004;23(22):4014–22. doi: 10.1038/sj.onc.1207505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spugnardi M, et al. Epigenetic inactivation of RAS association domain family protein 1 (RASSF1A) in malignant cutaneous melanoma. Cancer Res. 2003;63(7):1639–43. [PubMed] [Google Scholar]
- 20.Roh MR, et al. Promoter methylation of PTEN is a significant prognostic factor in melanoma survival. J Invest Dermatol. 2016;136(5):1002–11. [DOI] [PubMed]
- 21.Stahl JM, et al. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003;63(11):2881–90. [PubMed] [Google Scholar]
- 22.Micevic G, et al. Attenuation of genome-wide 5-methylcytosine level is an epigenetic feature of cutaneous malignant melanomas. Melanoma research. 2017;27(2):85–96. [DOI] [PMC free article] [PubMed]
- 23.Zhou XP, et al. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am J Pathol. 2000;157(4):1123–8. doi: 10.1016/S0002-9440(10)64627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Birck A, et al. Mutation and allelic loss of the PTEN/MMAC1 gene in primary and metastatic melanoma biopsies. J Invest Dermatol. 2000;114(2):277–80. doi: 10.1046/j.1523-1747.2000.00877.x. [DOI] [PubMed] [Google Scholar]
- 25.Tsao H, et al. Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene. 1998;16(26):3397–402. doi: 10.1038/sj.onc.1201881. [DOI] [PubMed] [Google Scholar]
- 26.Serrano M, et al. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85(1):27–37. doi: 10.1016/S0092-8674(00)81079-X. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalgo ML, et al. Low frequency of p16/CDKN2A methylation in sporadic melanoma: comparative approaches for methylation analysis of primary tumors. Cancer Res. 1997;57(23):5336–47. [PubMed] [Google Scholar]
- 28.Fujimoto A, et al. p16INK4a inactivation is not frequent in uncultured sporadic primary cutaneous melanoma. Oncogene. 1999;18(15):2527–32. doi: 10.1038/sj.onc.1202803. [DOI] [PubMed] [Google Scholar]
- 29.Straume O, et al. Significant impact of promoter hypermethylation and the 540 C>T polymorphism of CDKN2A in cutaneous melanoma of the vertical growth phase. Am J Pathol. 2002;161(1):229–37. doi: 10.1016/S0002-9440(10)64174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pomerantz J, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92(6):713–23. doi: 10.1016/S0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 31.Freedberg DE, et al. Frequent p16-independent inactivation of p14ARF in human melanoma. J Natl Cancer Inst. 2008;100(11):784–95. doi: 10.1093/jnci/djn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welch J, et al. Lack of genetic and epigenetic changes in CDKN2A in melanocytic nevi. J Invest Dermatol. 2001;117(2):383–4. doi: 10.1046/j.0022-202x.2001.01391.x. [DOI] [PubMed] [Google Scholar]
- 33.Dammann R, et al. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet. 2000;25(3):315–9. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 34.Tanemura A, et al. CpG island methylator phenotype predicts progression of malignant melanoma. Clin Cancer Res. 2009;15(5):1801–7. doi: 10.1158/1078-0432.CCR-08-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muthusamy V, et al. Epigenetic silencing of novel tumor suppressors in malignant melanoma. Cancer Res. 2006;66(23):11187–93. doi: 10.1158/0008-5472.CAN-06-1274. [DOI] [PubMed] [Google Scholar]
- 36.Muthusamy V, et al. The hematopoietic stem cell regulatory gene latexin has tumor-suppressive properties in malignant melanoma. J Invest Dermatol. 2013;133(7):1827–33. doi: 10.1038/jid.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dahl C, et al. KIT is a frequent target for epigenetic silencing in cutaneous melanoma. J Invest Dermatol. 2015;135(2):516–24. doi: 10.1038/jid.2014.372. [DOI] [PubMed] [Google Scholar]
- 38.Conway K, et al. DNA-methylation profiling distinguishes malignant melanomas from benign nevi. Pigment Cell Melanoma Res. 2011;24(2):352–60. doi: 10.1111/j.1755-148X.2011.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koga Y, et al. Genome-wide screen of promoter methylation identifies novel markers in melanoma. Genome Res. 2009;19(8):1462–70. doi: 10.1101/gr.091447.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tellez CS, et al. CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009;19(3):146–55. doi: 10.1097/CMR.0b013e32832b274e. [DOI] [PubMed] [Google Scholar]
- 41.Gao L, et al. Genome-wide promoter methylation analysis identifies epigenetic silencing of MAPK13 in primary cutaneous melanoma. Pigment Cell Melanoma Res. 2013;26(4):542–54. doi: 10.1111/pcmr.12096. [DOI] [PubMed] [Google Scholar]
- 42.Bonazzi VF, et al. Cross-platform array screening identifies COL1A2, THBS1, TNFRSF10D and UCHL1 as genes frequently silenced by methylation in melanoma. PLoS One. 2011;6(10):e26121. doi: 10.1371/journal.pone.0026121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li JL, et al. Genome-wide methylated CpG island profiles of melanoma cells reveal a melanoma coregulation network. Sci Rep. 2013;3:2962. doi: 10.1038/srep02962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lauss M, et al. DNA methylation subgroups in melanoma are associated with proliferative and immunological processes. BMC Med Genomics. 2015;8:73. doi: 10.1186/s12920-015-0147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ecsedi S, et al. DNA methylation characteristics of primary melanomas with distinct biological behaviour. PLoS One. 2014;9(5):e96612. doi: 10.1371/journal.pone.0096612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hou P, et al. The BRAF(V600E) causes widespread alterations in gene methylation in the genome of melanoma cells. Cell Cycle. 2012;11(2):286–95. doi: 10.4161/cc.11.2.18707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang M, et al. Common BRAF(V600E)-directed pathway mediates widespread epigenetic silencing in colorectal cancer and melanoma. Proc Natl Acad Sci U S A. 2016;113(5):1250–5. doi: 10.1073/pnas.1525619113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas NE, et al. DNA methylation profiles in primary cutaneous melanomas are associated with clinically significant pathologic features. Pigment Cell Melanoma Res. 2014;27(6):1097–105. doi: 10.1111/pcmr.12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marzese DM, et al. Epigenome-wide DNA methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. Hum Mol Genet. 2014;23(1):226–38. doi: 10.1093/hmg/ddt420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maeda K, et al. Altered expressions of HOX genes in human cutaneous malignant melanoma. Int J Cancer. 2005;114(3):436–41. doi: 10.1002/ijc.20706. [DOI] [PubMed] [Google Scholar]
- 51.Cheng PF, et al. Methylation-dependent SOX9 expression mediates invasion in human melanoma cells and is a negative prognostic factor in advanced melanoma. Genome Biol. 2015;16:42. doi: 10.1186/s13059-015-0594-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lodygin D, et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7(16):2591–600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 53.Beg MS, et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs. 2016:1–9. [DOI] [PMC free article] [PubMed]
- 54.Heinemann A, et al. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012;72(2):460–71. doi: 10.1158/0008-5472.CAN-11-1977. [DOI] [PubMed] [Google Scholar]
- 55.Paschen A, et al. Differential clinical significance of individual NKG2D ligands in melanoma: soluble ULBP2 as an indicator of poor prognosis superior to S100B. Clin Cancer Res. 2009;15(16):5208–15. doi: 10.1158/1078-0432.CCR-09-0886. [DOI] [PubMed] [Google Scholar]
- 56.Lujambio A, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A. 2008;105(36):13556–61. doi: 10.1073/pnas.0803055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong F, Lou D. MicroRNA-34b/c suppresses uveal melanoma cell proliferation and migration through multiple targets. Mol Vis. 2012;18:537–46. [PMC free article] [PubMed] [Google Scholar]
- 58.Mazar J, et al. Epigenetic regulation of microRNA genes and the role of miR-34b in cell invasion and motility in human melanoma. PLoS One. 2011;6(9):e24922. doi: 10.1371/journal.pone.0024922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mazar J, et al. MicroRNA 211 functions as a metabolic switch in human melanoma cells. Mol Cell Biol. 2016;36(7):1090–108. doi: 10.1128/MCB.00762-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu H, Yang W. MiR-211 is epigenetically regulated by DNMT1 mediated methylation and inhibits EMT of melanoma cells by targeting RAB22A. Biochem Biophys Res Commun. 2016;476(4):400–5. doi: 10.1016/j.bbrc.2016.05.133. [DOI] [PubMed] [Google Scholar]
- 61.Noguchi S, et al. DNA methylation contributes toward silencing of antioncogenic microRNA-203 in human and canine melanoma cells. Melanoma Res. 2015;25(5):390–8. doi: 10.1097/CMR.0000000000000183. [DOI] [PubMed] [Google Scholar]
- 62.Chang X, et al. MiR-203 inhibits melanoma invasive and proliferative abilities by targeting the polycomb group gene BMI1. Biochem Biophys Res Commun. 2015;456(1):361–6. doi: 10.1016/j.bbrc.2014.11.087. [DOI] [PubMed] [Google Scholar]
- 63.Piletic K, Kunej T. MicroRNA epigenetic signatures in human disease. Arch Toxicol. 2016;90(10):2405–19. doi: 10.1007/s00204-016-1815-7. [DOI] [PubMed] [Google Scholar]
- 64.Mazar J, et al. Epigenetic regulation of microRNA-375 and its role in melanoma development in humans. FEBS Lett. 2011;585(15):2467–76. doi: 10.1016/j.febslet.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 65.Dar AA, et al. The role of miR-18b in MDM2-p53 pathway signaling and melanoma progression. J Natl Cancer Inst. 2013;105(6):433–42. doi: 10.1093/jnci/djt003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Micevic G, et al. DNMT3b modulates melanoma growth by controlling levels of mTORC2 component RICTOR. Cell Rep. 2016;14(9):2180–92. doi: 10.1016/j.celrep.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li NA, et al. miR-196b regulates gastric cancer cell proliferation and invasion via PI3K/AKT/mTOR signaling pathway. Oncol Lett. 2016;11(3):1745–9. doi: 10.3892/ol.2016.4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu S, et al. Loss of microRNA-205 expression is associated with melanoma progression. Lab Invest. 2012;92(7):1084–96. doi: 10.1038/labinvest.2012.62. [DOI] [PubMed] [Google Scholar]
- 69.Zehavi L, et al. Silencing of a large microRNA cluster on human chromosome 14q32 in melanoma: biological effects of mir-376a and mir-376c on insulin growth factor 1 receptor. Mol Cancer. 2012;11:44. doi: 10.1186/1476-4598-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Asangani IA, et al. Genetic and epigenetic loss of microRNA-31 leads to feed-forward expression of EZH2 in melanoma. Oncotarget. 2012;3(9):1011–25. doi: 10.18632/oncotarget.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Popkie AP, et al. Phosphatidylinositol 3-kinase (PI3K) signaling via glycogen synthase kinase-3 (Gsk-3) regulates DNA methylation of imprinted loci. J Biol Chem. 2010;285(53):41337–47. doi: 10.1074/jbc.M110.170704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mei C, et al. Transcriptional and post-transcriptional control of DNA methyltransferase 3B is regulated by phosphatidylinositol 3 kinase/Akt pathway in human hepatocellular carcinoma cell lines. J Cell Biochem. 2010;111(1):158–67. doi: 10.1002/jcb.22684. [DOI] [PubMed] [Google Scholar]
- 73.Agarwal S, et al. Mahanine restores RASSF1A expression by down-regulating DNMT1 and DNMT3B in prostate cancer cells. Mol Cancer. 2013;12(1):99. doi: 10.1186/1476-4598-12-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spence T, et al. CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol. 2014;128(2):291–303. doi: 10.1007/s00401-014-1291-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kleinman CL, et al. Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet. 2014;46(1):39–44. doi: 10.1038/ng.2849. [DOI] [PubMed] [Google Scholar]
- 76.Beaulieu N, et al. An essential role for DNA methyltransferase DNMT3B in cancer cell survival. J Biol Chem. 2002;277(31):28176–81. doi: 10.1074/jbc.M204734200. [DOI] [PubMed] [Google Scholar]
- 77.Lauss M, et al. Genome-wide DNA methylation analysis in melanoma reveals the importance of CpG methylation in MITF regulation. J Invest Dermatol. 2015;135(7):1820–8. doi: 10.1038/jid.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Micevic, G, et al. DNMT3b modulates melanoma growth by controlling levels of mTORC2 component RICTOR. Cell Rep. 2016. [DOI] [PMC free article] [PubMed]
- 79.Kottakis F, et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016;539(7629):390–5. doi: 10.1038/nature20132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ito S, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310):1129–33. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146(6):866–72. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lian CG, et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150(6):1135–46. doi: 10.1016/j.cell.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gustafson CB, et al. Epigenetic reprogramming of melanoma cells by vitamin C treatment. Clin Epigenetics. 2015;7(1):51. doi: 10.1186/s13148-015-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gambichler T, Sand M, Skrygan M. Loss of 5-hydroxymethylcytosine and ten-eleven translocation 2 protein expression in malignant melanoma. Melanoma Res. 2013;23(3):218–20. doi: 10.1097/CMR.0b013e32835f9bd4. [DOI] [PubMed] [Google Scholar]
- 86.Lee JJ, et al. 5-Hydroxymethylcytosine expression in metastatic melanoma versus nodal nevus in sentinel lymph node biopsies. Mod Pathol. 2015;28(2):218–29. doi: 10.1038/modpathol.2014.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Uchiyama R, et al. 5-Hydroxymethylcytosine as a useful marker to differentiate between malignant melanomas and benign melanocytic nevi. J Dermatol Sci. 2014;73(2):161–3. doi: 10.1016/j.jdermsci.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 88.Rodic N, et al. Diagnostic utility of 5-hydroxymethylcytosine immunohistochemistry in melanocytic proliferations. J Cutan Pathol. 2015;42(11):807–14. doi: 10.1111/cup.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pastor WA, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473(7347):394–7. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu M, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149(6):1368–80. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mellen M, et al. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151(7):1417–30. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Spruijt CG, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152(5):1146–59. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 93.Cancer Genome Atlas, N Genomic classification of cutaneous melanoma. Cell. 2015;161(7):1681–96. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shibata T, et al. Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am J Pathol. 2011;178(3):1395–402. doi: 10.1016/j.ajpath.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Song F, et al. Identification of a melanoma susceptibility locus and somatic mutation in TET2. Carcinogenesis. 2014;35(9):2097–101. doi: 10.1093/carcin/bgu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krauthammer M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44(9):1006–14. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gao L, et al. Promoter CpG island hypermethylation in dysplastic nevus and melanoma: CLDN11 as an epigenetic biomarker for malignancy. J Invest Dermatol. 2014;134(12):2957–66. doi: 10.1038/jid.2014.270. [DOI] [PubMed] [Google Scholar]
- 98.Marini A, et al. Epigenetic inactivation of tumor suppressor genes in serum of patients with cutaneous melanoma. J Invest Dermatol. 2006;126(2):422–31. doi: 10.1038/sj.jid.5700073. [DOI] [PubMed] [Google Scholar]
- 99.Balch CM, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199–206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3(4):253–66. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 101.Mikeska T, Craig JM. DNA methylation biomarkers: cancer and beyond. Genes (Basel) 2014;5(3):821–64. doi: 10.3390/genes5030821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4(12):988–93. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 103.Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96(15):8681–6. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Van Rijnsoever M, et al. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin Cancer Res. 2003;9(8):2898–903. [PubMed] [Google Scholar]
- 105.Bae JM, et al. Prognostic implication of the CpG island methylator phenotype in colorectal cancers depends on tumour location. Br J Cancer. 2013;109(4):1004–12. doi: 10.1038/bjc.2013.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Juo YY, et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Ann Oncol. 2014;25(12):2314–27. doi: 10.1093/annonc/mdu149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Abe M, et al. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Res. 2005;65(3):828–34. [PubMed] [Google Scholar]
- 108.An C, et al. Prognostic significance of CpG island methylator phenotype and microsatellite instability in gastric carcinoma. Clin Cancer Res. 2005;11(2 Pt 1):656–63. [PubMed] [Google Scholar]
- 109.Baysan M, et al. G-cimp status prediction of glioblastoma samples using mRNA expression data. PLoS One. 2012;7(11):e47839. doi: 10.1371/journal.pone.0047839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sigalotti L, et al. Methylation levels of the “long interspersed nucleotide element-1” repetitive sequences predict survival of melanoma patients. J Transl Med. 2011;9:78. doi: 10.1186/1479-5876-9-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ecsedi SI, et al. Transposable hypomethylation is associated with metastatic capacity of primary melanomas. Int J Clin Exp Pathol. 2013;6(12):2943–8. [PMC free article] [PubMed] [Google Scholar]
- 112.You Y, et al. TSLC1 gene silencing in cutaneous melanoma. Melanoma Res. 2010;20(3):179–83. doi: 10.1097/CMR.0b013e32833413c0. [DOI] [PubMed] [Google Scholar]
- 113.Tuominen R, et al. MGMT promoter methylation is associated with temozolomide response and prolonged progression-free survival in disseminated cutaneous melanoma. Int J Cancer. 2015;136(12):2844–53. doi: 10.1002/ijc.29332. [DOI] [PubMed] [Google Scholar]
- 114.Cesinaro AM, et al. Prognostic significance of MGMT gene promoter methylation in differently treated metastatic melanomas. Pathology. 2012;44(4):313–7. doi: 10.1097/PAT.0b013e328353a0ff. [DOI] [PubMed] [Google Scholar]
- 115.Hegi ME, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 116.Weller M, et al. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol. 2010;6(1):39–51. doi: 10.1038/nrneurol.2009.197. [DOI] [PubMed] [Google Scholar]
- 117.Mori T, et al. Predictive utility of circulating methylated DNA in serum of melanoma patients receiving biochemotherapy. J Clin Oncol. 2005;23(36):9351–8. doi: 10.1200/JCO.2005.02.9876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lv X, et al. HOXD9 promotes epithelial-mesenchymal transition and cancer metastasis by ZEB1 regulation in hepatocellular carcinoma. J Exp Clin Cancer Res. 2015;34:133. doi: 10.1186/s13046-015-0245-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 119.Marzese DM, et al. Brain metastasis is predetermined in early stages of cutaneous melanoma by CD44v6 expression through epigenetic regulation of the spliceosome. Pigment Cell Melanoma Res. 2015;28(1):82–93. doi: 10.1111/pcmr.12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sigalotti L, et al. Whole genome methylation profiles as independent markers of survival in stage IIIC melanoma patients. J Transl Med. 2012;10:185. doi: 10.1186/1479-5876-10-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bell RE, et al. Enhancer methylation dynamics contribute to cancer plasticity and patient mortality. Genome Res. 2016;26(5):601–11. doi: 10.1101/gr.197194.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4(2):143–53. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 123.Saunthararajah Y. Key clinical observations after 5-azacytidine and decitabine treatment of myelodysplastic syndromes suggest practical solutions for better outcomes. Hematology Am Soc Hematol Educ Program. 2013;2013:511–21. doi: 10.1182/asheducation-2013.1.511. [DOI] [PubMed] [Google Scholar]
- 124.Navada SC, et al. Clinical development of demethylating agents in hematology. J Clin Invest. 2014;124(1):40–6. doi: 10.1172/JCI69739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res. 2009;15(12):3938–46. doi: 10.1158/1078-0432.CCR-08-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6(8):597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 128.Okano M, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–57. doi: 10.1016/S0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 129.Bestor TH. Cloning of a mammalian DNA methyltransferase. Gene. 1988;74(1):9–12. doi: 10.1016/0378-1119(88)90238-7. [DOI] [PubMed] [Google Scholar]
- 130.Chedin F, Lieber MR, Hsieh CL. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci U S A. 2002;99(26):16916–21. doi: 10.1073/pnas.262443999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gowher H, et al. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J Biol Chem. 2005;280(14):13341–8. doi: 10.1074/jbc.M413412200. [DOI] [PubMed] [Google Scholar]
- 132.Chen ZX, et al. Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J Cell Biochem. 2005;95(5):902–17. doi: 10.1002/jcb.20447. [DOI] [PubMed] [Google Scholar]
- 133.Tiedemann RL, et al. Acute depletion redefines the division of labor among DNA methyltransferases in methylating the human genome. Cell Rep. 2014;9(4):1554–66. doi: 10.1016/j.celrep.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liao J, et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet. 2015;47(5):469–78. doi: 10.1038/ng.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Baubec T, et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 2015. [DOI] [PubMed]
- 136.Loriot A, et al. Transient down-regulation of DNMT1 methyltransferase leads to activation and stable hypomethylation of MAGE-A1 in melanoma cells. J Biol Chem. 2006;281(15):10118–26. doi: 10.1074/jbc.M510469200. [DOI] [PubMed] [Google Scholar]
- 137.Cannuyer J, et al. A gene expression signature identifying transient DNMT1 depletion as a causal factor of cancer-germline gene activation in melanoma. Clin Epigenetics. 2015;7:114. doi: 10.1186/s13148-015-0147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Molognoni F, et al. Ras and Rac1, frequently mutated in melanomas, are activated by superoxide anion, modulate Dnmt1 level and are causally related to melanocyte malignant transformation. PLoS One. 2013;8(12):e81937. doi: 10.1371/journal.pone.0081937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Deng T, et al. An essential role for DNA methyltransferase 3a in melanoma tumorigenesis. Biochem Biophys Res Commun. 2009;387(3):611–6. doi: 10.1016/j.bbrc.2009.07.093. [DOI] [PubMed] [Google Scholar]
- 140.Nguyen T, et al. Downregulation of microRNA-29c is associated with hypermethylation of tumor-related genes and disease outcome in cutaneous melanoma. Epigenetics. 2011;6(3):388–94. doi: 10.4161/epi.6.3.14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jeltsch A, Jurkowska RZ. Allosteric control of mammalian DNA methyltransferases—a new regulatory paradigm. Nucleic Acids Res. 2016. [DOI] [PMC free article] [PubMed]
- 142.Valpione S, Campana LG. Immunotherapy for advanced melanoma: future directions. Immunotherapy. 2016;8(2):199–209. doi: 10.2217/imt.15.111. [DOI] [PubMed] [Google Scholar]
- 143.Licht JD. DNA methylation inhibitors in cancer therapy: the immunity dimension. Cell. 2015;162(5):938–9. doi: 10.1016/j.cell.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 144.Yang H, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28(6):1280–8. doi: 10.1038/leu.2013.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Das R, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194(3):950–9. doi: 10.4049/jimmunol.1401686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chiappinelli KB, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–86. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Roulois D, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162(5):961–73. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wrangle J, et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget. 2013;4(11):2067–79. doi: 10.18632/oncotarget.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Le DT, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hoon DS. Are circulating tumor cells an independent prognostic factor in patients with high-risk melanoma? Nat Clin Pract Oncol. 2004;1(2):74–5. doi: 10.1038/ncponc0041. [DOI] [PubMed] [Google Scholar]
- 151.Furuta J, et al. Promoter methylation profiling of 30 genes in human malignant melanoma. Cancer Sci. 2004;95(12):962–8. doi: 10.1111/j.1349-7006.2004.tb03184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu S, et al. Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment Cell Melanoma Res. 2008;21(5):545–58. doi: 10.1111/j.1755-148X.2008.00484.x. [DOI] [PubMed] [Google Scholar]
- 153.Li B, et al. Overload of the heat-shock protein H11/HspB8 triggers melanoma cell apoptosis through activation of transforming growth factor-beta-activated kinase 1. Oncogene. 2007;26(24):3521–31. doi: 10.1038/sj.onc.1210145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mori T, et al. Epigenetic up-regulation of C-C chemokine receptor 7 and C-X-C chemokine receptor 4 expression in melanoma cells. Cancer Res. 2005;65(5):1800–7. doi: 10.1158/0008-5472.CAN-04-3531. [DOI] [PubMed] [Google Scholar]
- 155.Sigalotti L, et al. Epigenetics of human cutaneous melanoma: setting the stage for new therapeutic strategies. J Transl Med. 2010;8:56. doi: 10.1186/1479-5876-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gallagher WM, et al. Multiple markers for melanoma progression regulated by DNA methylation: insights from transcriptomic studies. Carcinogenesis. 2005;26(11):1856–67. doi: 10.1093/carcin/bgi152. [DOI] [PubMed] [Google Scholar]
- 157.Zhang F, Dai X, Wang Y. 5-Aza-2′-deoxycytidine induced growth inhibition of leukemia cells through modulating endogenous cholesterol biosynthesis. Mol Cell Proteomics. 2012;11(7):M111 016915. doi: 10.1074/mcp.M111.016915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bonazzi VF, Irwin D, Hayward NK. Identification of candidate tumor suppressor genes inactivated by promoter methylation in melanoma. Genes Chromosomes Cancer. 2009;48(1):10–21. doi: 10.1002/gcc.20615. [DOI] [PubMed] [Google Scholar]
- 159.Lin YC, et al. Wnt inhibitory factor-1 gene transfer inhibits melanoma cell growth. Hum Gene Ther. 2007;18(4):379–86. doi: 10.1089/hum.2006.005. [DOI] [PubMed] [Google Scholar]
- 160.Besaratinia A, Tommasi S. Epigenetics of human melanoma: promises and challenges. J Mol Cell Biol. 2014;6(5):356–67. doi: 10.1093/jmcb/mju027. [DOI] [PubMed] [Google Scholar]
- 161.Mori T, et al. Estrogen receptor-alpha methylation predicts melanoma progression. Cancer Res. 2006;66(13):6692–8. doi: 10.1158/0008-5472.CAN-06-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fonsatti E, et al. Methylation-regulated expression of HLA class I antigens in melanoma. Int J Cancer. 2003;105(3):430–1. doi: 10.1002/ijc.11077. [DOI] [PubMed] [Google Scholar]
- 163.Tokita T, et al. Methylation status of the SOCS3 gene in human malignant melanomas. Int J Oncol. 2007;30(3):689–94. [PubMed] [Google Scholar]
- 164.Ng KC, et al. XAF1 expression is significantly reduced in human melanoma. J Invest Dermatol. 2004;123(6):1127–34. doi: 10.1111/j.0022-202X.2004.23467.x. [DOI] [PubMed] [Google Scholar]
- 165.Nazarian R, Jazirehi AR. TFPI2 methylation can serve as an epigenetic biomarker of metastatic melanoma. Epigenomics. 2013;5(3):252–3. [PubMed] [Google Scholar]
- 166.Das AM, et al. Tissue inhibitor of metalloproteinase-3 (TIMP3) expression decreases during melanoma progression and inhibits melanoma cell migration. Eur J Cancer. 2016;66:34–46. doi: 10.1016/j.ejca.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 167.De Smet C, et al. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc Natl Acad Sci U S A. 1996;93(14):7149–53. doi: 10.1073/pnas.93.14.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Grunau C, et al. Frequent DNA hypomethylation of human juxtacentromeric BAGE loci in cancer. Genes Chromosomes Cancer. 2005;43(1):11–24. doi: 10.1002/gcc.20155. [DOI] [PubMed] [Google Scholar]
- 169.Maat W, et al. Epigenetic inactivation of RASSF1a in uveal melanoma. Invest Ophthalmol Vis Sci. 2007;48(2):486–90. doi: 10.1167/iovs.06-0781. [DOI] [PubMed] [Google Scholar]
- 170.Bae SI, et al. Reversal of methylation silencing of Apo2L/TRAIL receptor 1 (DR4) expression overcomes resistance of SK-MEL-3 and SK-MEL-28 melanoma cells to interferons (IFNs) or Apo2L/TRAIL. Oncogene. 2008;27(4):490–8. doi: 10.1038/sj.onc.1210655. [DOI] [PubMed] [Google Scholar]
- 171.Theodosakis N, et al. Integrative discovery of CD98 as a melanoma biomarker. Pigment Cell Melanoma Res. 2016;29(3):385–7. doi: 10.1111/pcmr.12464. [DOI] [PubMed] [Google Scholar]
- 172.Lo Nigro C, et al. Methylated tissue factor pathway inhibitor 2 (TFPI2) DNA in serum is a biomarker of metastatic melanoma. J Invest Dermatol. 2013;133(5):1278–85. doi: 10.1038/jid.2012.493. [DOI] [PubMed] [Google Scholar]
- 173.Shen L, et al. Drug sensitivity prediction by CpG island methylation profile in the NCI-60 cancer cell line panel. Cancer Res. 2007;67(23):11335–43. doi: 10.1158/0008-5472.CAN-07-1502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The MEDLINE database of biomedical and life science journal citations was queried using the search term “methylation” AND “melanoma.” The search returned a total of 738 citations in English between 1977 and 2017. Of the 738 citations, 711 had an abstract available for review. The Web of Science (ISI Web of Knowledge) citation database was queried with the search term “methylation” AND “melanoma.” A total of 1123 citations between 1985 and 2017 were identified, and 778 citations had available abstracts in English. All available unique abstracts with ≥1 citation, a total of 1020 abstracts, were reviewed by GM.