

Abstract
Background
It has been known for more than 100 years that aneuploidy is an essence of cancer. The question is what keeps the genome stable, thereby preventing aneuploidy. For the past 25 years, it has been proposed that p53 is the “guardian of the genome.” However, it has been shown that inactivation of p53 does not cause aneuploidy. Another essence of cancer is global DNA hypomethylation, which causes destabilization of the genome and subsequent aneupoloidy. Yet, another essence of cancer is excessive use of methionine, resulting in methionine dependence. Methionine dependence is due to possible “metabolic reprogramming” due to carcinogens, including chemical agents and infectious organisms, such as Helicobacter pylori, that result in altered and excessive transmethylation in cancer cells. Cancer cells appear to have a “methyl-sink” whereby methyl groups are diverted from DNA.
Conclusion
DNA hypomethylation destabilizes the genome, leading to aneuploidy and subsequent selection and speciation into autonomous cancers, leading to the conclusion that DNA methylation is the “guardian of the genome.”
Electronic supplementary material
The online version of this article (doi:10.1186/s13039-017-0314-8) contains supplementary material, which is available to authorized users.
Keywords: Methionine, Methionine dependence, Unbalanced transmethylation, Global DNA hypomethylation, Chromosome instability, Aneuploidy, Cancer speciation
“E pur si muove” Galileo
Is p53 “the guardian of the genome”?
Since Lane designated p53 as the “guardian of the genome” [1], this has become the dominant paradigm [2–4].
DNA hypomethylation in cancer
It has been long known that global DNA hypomethylation is a general characteristic of cancer, as originally discovered by our laboratory [5, 6]. DNA hypomethylation is an early event in the formation of cancer as it appears in pre-malignant tissue [7]. The progression of DNA hypomethylation may be driven by “oncogenes” [8].
Hints regarding the effect of DNA hypomethylation on genome integrity come from Perucho et al. [9] on the relationship of Helicobacter pylori to gastric cancer. Helicobacter pylori infection is a very important risk factor for gastric cancer. Perucho et al. [9] observed that H. pylori appears to cause a field defect of DNA hypomethylation in a region of the gastric mucosa prior to cancer development that is not reversed by H. pylori eradication. Perucho et al. [9] also observed that enhanced DNA hypomethylation, was associated with a more invasive and advanced stage type of gastric cancer.
Other studies have indicated that the greater extent of DNA hypomethylation, the more malignant is the cancer. The total levels of DNA 5-methylcytosine were determined in a series of highly metastatic cell lines which had been isolated from a poorly metastatic human melanoma tumor line, MeWo, by Kerbel’s group [10]. Similar to what was observed in gastric cancer described above, the more metastatic the melanoma sublines were, the more extensively they were hypomethylated [10].
It has also been shown that global DNA hypomethylation increases as cervical dysplasia progresses to cervical carcinoma [11].
Compared to benign tumors, atypical and malignant meningiomas were observed to have increased global DNA hypomethylation [12].
DNA hypomethylation leads to aneuploidy
Jaenish et al. generated mice with a mutant DNA methyltransferase 1 (DNMT1) allele. The mutation reduces Dnmt1 expression to 10% of normal levels and results in substantial genome-wide hypomethylation in all tested tissues. The mutant mice developed T cell lymphomas which had a high frequency of chromosome 15 trisomy. DNMT-deficient HCT-116 colon cancer cells had a high degree of genome instability leading to aneuploidy, including many novel chromosomal translocations [13]. These results indicate that DNA hypomethylation plays a causal role in chromosomal instability, aneuploidy, and subsequent cancer [14].
Methionine
Methionine [15] is an essential amonio acid; however, it can be synthesized from homocysteine [16] and methyl-tetrahydrofolate [17, 18] in a vitamin B12-dependent reaction. Methionine is a component of proteins. Methionine is also the universal methyl donor via its activated form S-adenosylmethionine [19].
Deprivation of methionine leads to cancer
Copeland and Salmon observed the development of liver, lung, and other cancers in a significant percentage of rats on a diet deficient in choline, which is a precursor of methionine [20]. This was the first solid evidence that methionine is involved in cancer [21].
Subsequently, Ghoshal and Farber observed that Fischer 344 male rats fed a choline-methionine deficient diet for 1–2 years during which the rats developed a 100% incidence of preneoplastic hepatocyte nodules. Subsequently, 51% of the rats developed hepatocellular carcinoma. Supplement of the diet with 0.8% choline chloride prevented the development of both the precancerous nodules and subsequent cancer [22].
The cancers resulting from methionine/choline-deprived diets in the rats were probably due to methyl shortage and subsequent DNA hypomethylation, since methionine is the source of methyl groups for DNA methylation. DNA hypomethylation appears to be an early event in choline/methionine-deficient diets, irreversible [23–26] and mediated by a deficiency in S-adenosylmethionine [27].
Methionine dependence and altered transmethylation in cancer
The appearance of cancer in rats on choline- and methionine-deficient diets gave an early hint that perturbed methionine metabolism is involved in cancer. Sugimura noted almost 60 years ago that rat tumor growth was slowed by giving the rats a defined diet depleted in methionine for a short period of time [28]. Approximately 45 years ago, it was observed that mouse leukemia cells in culture required very high levels of methionine in order to proliferate [29]. Subsequently, most cancer cell lines were found to be methionine dependent [30, 31]. These cell lines were derived from various cancer types including liver, pancreatic ovarian, submaxillary, brain, lung, bladder, prostate, breast, kidney, cervical, colon, fibrosarcoma, osteosarcoma, rhabdomyosarcoma, leiomyosarcoma, neuroblastoma, glioblastoma and melanoma [30, 31]. Normal unestablished cell strains, thus far characterized, grow well in methionine-depleted medium [30, 32]. Many of the cancer cell lines tested have little else in common other than the fact that they are methionine-dependent and have altered methionine metabolism [33]. Human patient tumors, including tumors of the colon, breast, ovary, prostate, and a melanoma, were also found to be methionine dependent in Gelfoam® histoculture [34]. The high frequency of occurrence of methionine dependence in diverse types of human cancer cells indicated that methionine dependence could be an important step in oncogenic transformation.
Three lines of evidence indicated that methionine-dependent cancer cells synthesize large amounts of methionine endogenously through the reaction catalyzed by N 5-methyltetrahydropteroyl-l-glutamate: l-homocysteine S-methyltransferase (EC 2.1.1.13) [35].
The activity of the methyltransferase involved in methionine biosynthesis was comparable in extracts of malignant and normal cells [35].
The uptake of radioactive label from [5-14C] methyltetrahydropteroyl-l-glutamic acid (N 5-methyl-H4PteGlu), the methyl donor for methionine biosynthesis, was at least as great in the malignant cells as in the normal cells and was nearly totally dependent on the addition of homocysteine, the methyl acceptor in methionine biosynthesis [33, 35].
The majority of the labeled methyl groups incorporated by cancer cells was recovered as methionine [35].
We subsequently observed that although methionine-dependent cancer cells synthesized a normal amount of methionine, the level of free methionine and S-adenosylmethionine (AdoMET), which is synthesized from methionine and is the universal methyl donor, were very low in cancer cells in methionine (MET)-depleted homocysteine (HCY)-supplemented medium (MET−HCY+). In contrast, exogenously supplied MET resulted in normal levels of AdoMET. The ratio of AdoMET to S-adenosylhomocysteine (AdoHCY) is low in methionine-dependent cancer cells growing in MET−HCY+ medium, but is normal in MET+HCY− medium. We determined that the low AdoMET/AdoHCY ratio probably limits growth of MET-dependent cells in MET−HCY+ medium [36, 37].
We subsequently observed that cancer cells have enhanced overall rates of transmethylation compared to normal human fibroblasts (Fig. 1) [38]. Transmethylation rates were measured by blocking AdoHCY hydrolase and measuring AdoHCY, which accumulates as a result of transmethylation from AdoMET. The excess methionine used for the enhanced transmethylation rates appears to be the basis of the methionine dependence of cancer cells which explains the low levels of free methionine and the low AdoMET/AdoHCY ratio in cancer cells under methionine deprivation [37, 38]. The elevated methionine use in cancer cells has been termed the “Hoffman effect” [39], analogous to the “Warburg effect” for enhanced glucose utilization in cancer [40]. The alteration of such a fundamental process as transmethylation in cancer may be indicative of its importance in the oncogenic process [32].
Fig. 1.
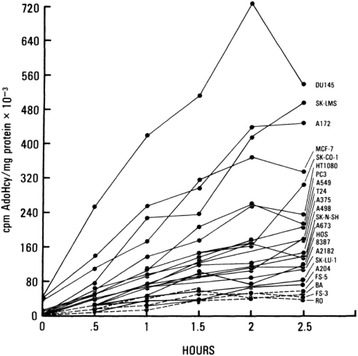
Recombinant methioninase (rMETase) traps cancer cells in S/G2 phase. Time-course imaging of HeLa-FUCCI cells treated with rMETase (1.0 unit/ml). Kinetics of rMETase trapping of cells in S/G2. Images were acquired with the FV1000 confocal microscope (Olympus, Tokyo, Japan). In the FUCCI system, the cells in G0/G1, S, or G2/M phases appear red, yellow, or green, respectively [66]
The abnormal elevated transmethylation and subsequent depletion of AdoMET appears to result in hypomethylation of DNA in cancer cells, since the methionine methyl group may be diverted to a methyl sink in the cancer cells and not sufficiently available for normal DNA methylation. For example, when the W-256 cancer cell line was cultured for 24 h in MET−HCY+ medium, the extent of methylation of nucleic acids and the acid-soluble pool of methionine were decreased. However, there was increased methylation activity of both an endogenous substrate and Escherichia coli tRNA. Methionine deprivation of the W-256 cells resulted in a large increase in the Vmax value for methylation of tRNA, without any change in the Km value for AdoMET [41]. These results indicated that methyl groups originating from methionine may be diverted from DNA in cancer cells.
Rare cells from methionine-dependent cancer cell lines regained the normal ability to grow in MET−HCY+ medium. These cell lines were termed methionine-independent revertants. Two revertants isolated from SV40-transformed cells had regained the ability to grow similar to normal cells in MET−HCY+ medium without substantial changes in methionine biosynthesis activity. Increased methionine biosynthesis thus is not a prerequisite to reversion from methionine dependence to independence [42, 43].
Methionine-independent revertants also had much lower basal transmethylation rates than parental methionine-dependent cell lines. For example, when comparing the methionine-dependent parent SP1 cell line and its methionine-independent revertant, the revertant SP1-R reduced its transmethylation rate. These results further suggested that methionine dependence is due to an increase in the rate of transmethylation reactions [44].
We then demonstrated that methionine-independent revertants of cancer cells concomitantly revert for characteristics associated with cancer. Of the 13 cancer-cell methionine-independent revertants characterized, 5 showed increased anchorage dependence for growth as reflected by reduced cloning efficiencies in methylcellulose, 8 showed an increased serum requirement for optimal growth, 8 showed decreased cell density in medium containing high serum, and 3 altered their cell morphology significantly. Thus, the methionine-independent revertants become more normal-like. Eight of the 13 had increased chromosome numbers, which probably played a role in reversion. Thus by selecting for methionine independence, it is possible to select for heterogeneous malignant-transformation revertants, which became less malignant, indicating further a relationship between altered methionine metabolism and oncogenic transformation [43].
Growth arrest of methionine-dependent cancer cells in MET −HCY+ medium resulted in a reduction in the percentage of mitotic cells in the cell population. Fluorescence-activated cell cycle analysis demonstrated that the cells are arrested in the S/G2 phases of the cell cycle in MET −HCY+ medium. This is in contrast to a G1-phase accumulation of cells, which occurs only in methionine-supplemented medium at very high cell densities and is similar to the G1 block seen in cultures of normal fibroblasts at high density [45]. Depletion of methionine in vivo by recombinant methioninase (rMETase) also resulted in S/G2 cell cycle arrest in tumors (Fig. 2) [46].
Fig. 2.
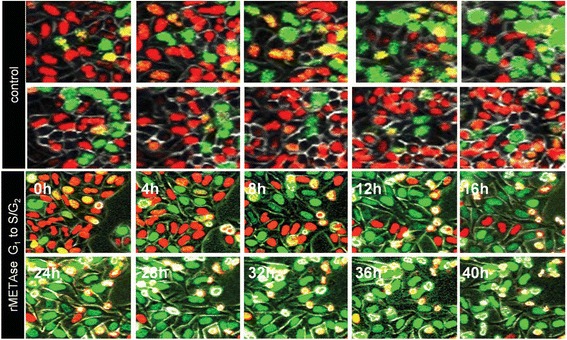
Efficacy of recombinant methioninase (rMETase) on growth of human colon tumors HCT 15 in nude mice. rMETase (5 or 10 units/g every 8 h) was administered by i.p. injection in nude mice with human colon tumor HCT 15, growing s.c. [54]
In summary, the altered methionine metabolism and transmethylation in cancer cells may be the initiating event of DNA hypomethylation and subsequent aneuploidy.
Progression of aneuploid cells to autonomous cancer
Duesberg et al. [47] have proposed that carcinogenesis, such as that caused by SV40 virus, is due to induced aneuploidy, which destabilizes the karyotype which progressively evolves to autonomous clonal cancers.
Duesberg et al. [48] proposed that other carcinogens induce aneuploidy in cancer in a similar way. They induced carcinogenesis with nitrosourea and observed noninvasive mammary tumors after 2 or more months and invasive carcinomas after 6 or more months. As with SV40, these researchers observed that aneuploid karyotypes, with varying non-clonal hyperplasias, formed rare cancer “species” with individual clonal karyotypes, which became autonomous cancers.
From perturbed methionine metabolism to DNA hypomethylation to a destabilized genome to aneuploidy to cancer
We suggest here that the pre-malignant DNA hypomethylation such as that observed by Perucho et al. [9] results from the unbalanced excess transmethylation which diverts methyl groups into where they are not available for normal methylation processes such as DNA methylation. The excess transmethylation rate explains the “methionine dependence” of cancer cells. Hypomethylation of DNA then results in the initiation of aneuploidy and subsequent speciation to autonomous cancer.
Thus, we propose that carcinogenesis is initiated by perturbation of methionine metabolism and transmethylation (“metabolic reprogramming”) resulting in DNA hypomethylation, thereby destabilizing the karyotype, which sets off a chain reaction of aneuploidizations as Duesberg proposes, which generate ever more abnormal karyotypes and eventually cancer-specific combinations [49]. It is possible that the degree of methionine dependence of cancer cells, which may be influenced by their extent of methionine biosynthesis from homocysteine, may affect how much DNA hypomethylation has occurred in cancer cells.
This hypothesis of cancer generation described in this review can accommodate mutated “oncogenes,” “driver genes,” “tumor suppressor genes,” etc., that may influence the behavior of cancers. However, with possible rare exceptions, individual gene alterations do not account for the global changes resulting in methionine dependence, global DNA hypomethylation, and altered karotype evolution, all of which are the essences of cancer.
Every hypothesis should be testable and be able to be negated. The present hypothesis for example predicts that premalignant tissue such as observed in the stomach with H. pylori infection or SV-40 infection, other infectious agents such as human papiloma virus, Epstein-Barr virus [50], or carcinogens such as nitrosourea or cigarette smoke, should have an elevated methionine requirement and excess and altered transmethylation leading to DNA hypomethylation as shown in the stomach after H. pylori infection by Perucho et al. [9]. The hypothesis predicts that chronic perturbation of methionine metabolism should be carcinogenic, such as a chronic methionine-depleted diet. The hypothesis also predicts that short-term deprivation of methionine should be curative to cancer and in mouse models; this has shown to be the case (Fig. 3) [35, 51–55]. However, important aspects of using methionine deprivation to treat cancer still remains to be investigated, including side effects on other metabolic pathways as well as the influence of the extent of methionine-synthesis capacity of cancer cells on the outcome of this therapy.
Fig. 3.
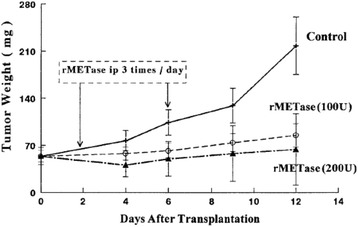
Rates of transmethylation of human tumor cell lines and normal human fibroblast cell strains. All cells were labeled with 100 μM [35S]-methionine-containing medium (25 μCi/ml) for 24 h. Periodate-oxidized 3-deazaadenosine was added to a concentration of 10 pM and the accumulation of [35S] AdoHcy was measured at half- hour intervals. Solid lines are human cancer cell lines. Dashed lines are human normal cell strains [38]
The hypothesis also explains why the use of [11C] methionine is so effective in positron emission tomography (PET) imaging, since the cancers use excessive methionine for their aberrant excess transmethylation and therefore take up excess [11C] methionine. For example, the higher specificity and sensitivity of [11C]-labeled methionine in PET (MET-PET) such as in gliomas has been demonstrated [56]. Because of these properties, MET-PET is the most popular amino acid tracer used in PET imaging of brain tumors and provides a high detection rate of brain tumors and metastasis and accurate tumor delineation [57]. MET-PET can also distinguish between recurrent glioma and necrosis [58]. MET-PET was heterogeneous in areas that MRI showed to be homogeneous, demonstrating the higher sensitivity of MET-PET. Malignant histopathology was detected in the areas of the tumor with the highest MET uptake, suggesting higher MET uptake correlates to a higher degree of malignancy consistent with our hypothesis [59]. MET-PET also could distinguish cures in melanoma [60].
Conclusion
The current paradigm is that p53 is the “guardian of the genome” [1–4, 61]. However, DNA methylation may be a more global “guardian of the genome.” When DNA methylation is disrupted by aberrant (“re-programmed”) methionine metabolism, DNA becomes globally hypomethylated resulting in chromosome instability and aneuploidy leading to possible “cellular speciation” and clonal cancer [62].
It is hypothesized that carcinogens may have their initial effect by “reprogramming” methionine metabolism, in particular transmethylation. Such carcinogens include chemicals as well as infectious agents such as SV-40 virus and H. pylori. It is remarkable that all types of cancers tested are found to be methionine dependent [32], which is perhaps the only known general metabolic abnormality in cancer [63]. This methionine metabolism “reprogramming” results in global [5, 6] and gene-specific [64] DNA hypomethylation, another general defect in cancer, which in turn leads to aneuploidy, another general defect in cancer which in turn leads to cancer “speciation.” Vogelstein et al. [65] reported that inactivation of p53 did not lead to aneuploidy. Thus, it seems that the genome has another “guardian”, DNA methylation is a likely candidate, and when it is perturbed, the genome destabilizes leading to aneuploidy and cancer [14]. Please see (Additional file 1).
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
This paper is a review. All data discussed are published and referenced in the manuscript.
Authors’ contributions
RMH wrote the manuscript.
Competing interests
The author declares that he has no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable since this manuscript is a review.
Dedication
This paper is dedicated to the 80th birthday of Professor Peter H. Duesberg, of the University of California, Berkeley.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Supplemental Material. Hoffman’s Rules for Success in Science. (DOCX 57 kb)
References
- 1.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/S0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 4.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 5.Diala ES, Hoffman RM. Hypomethylation of HeLa cell DNA and the absence of 5-methylcytosine in SV40 and adenovirus (type 2) DNA: analysis by HPLC. Biochem Biophys Res Commun. 1982;107:19–26. doi: 10.1016/0006-291X(82)91663-1. [DOI] [PubMed] [Google Scholar]
- 6.Diala ES, Cheah MSC, Rowitch D, Hoffman RM. Extent of DNA methylation in human tumor cells. J Natl Cancer Inst. 1983;71:755–764. [PubMed] [Google Scholar]
- 7.Goelz SE, Vogelstein B, Hamilton SR, Feinberg AP. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science. 1985;228:187–190. doi: 10.1126/science.2579435. [DOI] [PubMed] [Google Scholar]
- 8.Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI, Chen X, Kojima K, Thung S, Bronson RT, Lachenmayer A, Revill K, Alsinet C, Sachidanandam R, Desai A, Senbanerjee S, Ukomadu C, Llovet JM, Sadler KC. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014;25:196–209. doi: 10.1016/j.ccr.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leodolter A, Alonso S, González B, Ebert MP, Vieth M, Röcken C, Wex T, Peitz U, Malfertheiner P. Somatic DNA Hypomethylation in H. pylori-Associated High-Risk Gastritis and Gastric Cancer: Enhanced Somatic Hypomethylation Associates with Advanced Stage Cancer. Clin Transl Gastroenterol. 2015;6:e85. doi: 10.1038/ctg.2015.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liteplo RG, Kerbel RS. Reduced levels of DNA 5-methylcytosine in metastatic variants of the human melanoma cell line MeWo. Cancer Res. 1987;47:2264–2267. [PubMed] [Google Scholar]
- 11.Kim YI, Giuliano A, Hatch KD, Schneider A, Nour MA, Dallal GE, Selhub J, Mason JB. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer. 1994;74:893–899. doi: 10.1002/1097-0142(19940801)74:3<893::AID-CNCR2820740316>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 12.Gao F, Shi L, Russin J, Zeng L, Chang X, He S, Chen TC, Giannotta SL, Weisenberger DJ, Zada G, Mack WJ, Wang K. DNA methylation in the malignant transformation of meningiomas. Plos One. 2013;8:e54114. doi: 10.1371/journal.pone.0054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karpf AR, Matsui S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005;65:8635–8639. doi: 10.1158/0008-5472.CAN-05-1961. [DOI] [PubMed] [Google Scholar]
- 14.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 15.Barger G, Coyne FP. The amino acid methionine: constitution and synthesis. Biochem J. 1928;22:1417–1425. doi: 10.1042/bj0221417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butz LW, Duvigneaud V. The formation of a homologue of cysteine by the decomposition of methionine with sulfuric acid. J Biol Chem. 1932;99:135–142. [Google Scholar]
- 17.Weissbach H, Taylor RT. Role of vitamin B12 in methionine biosynthesis. Fed Proc. 1966;25:1649–1656. [PubMed] [Google Scholar]
- 18.Finkelstein JD, Kyle WE, Harris BJ. Methionine metabolism in mammals: regulatory effects of S-adenosylhomocysteine. Arch Biochem Biophys. 1974;165:774–779. doi: 10.1016/0003-9861(74)90306-3. [DOI] [PubMed] [Google Scholar]
- 19.Cantoni G. The nature of the active methyl donor formed enzymatically from L-methinine and adenosinetriphosphate. J Am Chem Soc. 1953;74:2942–2943. doi: 10.1021/ja01131a519. [DOI] [Google Scholar]
- 20.Copeland DH, Salmon WD. The occurrence of neoplasms in the liver, lungs, and other tissues of rats as a result of prolonged choline deficiency. Am J Pathol. 1946;22:1059–1079. [PubMed] [Google Scholar]
- 21.McCully K. Homocysteine metabolism, atherosclerosis and diseases of aging. Compr Physiol. 2016;6:471–505. doi: 10.1002/cphy.c150021. [DOI] [PubMed] [Google Scholar]
- 22.Ghoshal AK, Farber E. The induction of liver cancer by dietary deficiency of choline and methionine without added carcinogens. Carcinogenesis. 1984;5:1367–1370. doi: 10.1093/carcin/5.10.1367. [DOI] [PubMed] [Google Scholar]
- 23.Christman JK, Chen M-L, Sheikhnejad G, Dizik M, Abileah S, Wainfan E. Methyl deficiency, DNA methylation, and cancer: Studies on the reversibility of the effects of a lipotrope-deficient diet. J Nutr Biochem. 1993;4:672–680. doi: 10.1016/0955-2863(93)90106-7. [DOI] [Google Scholar]
- 24.Wainfan E, Dizik M, Stender M, Christman JK. Rapid appearance of hypomethylated DNA in livers of rats fed cancer-promoting, methyl-deficient diets. Cancer Res. 1989;49:4094–4097. [PubMed] [Google Scholar]
- 25.Tsujiuchi T, Tsutsumi M, Sasaki Y, Takahama M, Konishi Y. Hypomethylation of CpG sites and c-myc gene overexpression in hepatocellular carcinomas, but not hyperplastic nodules, induced by a choline-deficient L-amino acid-defined diet in rats. Jpn J Cancer Res. 1999;90:909–913. doi: 10.1111/j.1349-7006.1999.tb00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimizu K, Onishi M, Sugata E, Sokuza Y, Mori C, Nishikawa T, Honoki K, Tsujiuchi T. Disturbance of DNA methylation patterns in the early phase of hepatocarcinogenesis induced by a choline-deficient L-amino acid-defined diet in rats. Cancer Sci. 2007;98:1318–1322. doi: 10.1111/j.1349-7006.2007.00564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shivapurkar N, Poirier LA. Tissue levels of S-adenosylmethionine and S-adenosylhomocysteine in rats fed methyl-deficient, amino acid-defined diets for 1 to 5 weeks. Carcinogenesis. 1983;4:1051–1057. doi: 10.1093/carcin/4.8.1051. [DOI] [PubMed] [Google Scholar]
- 28.Sugimura T, Birnbaum SM, Winitz M, Greenstein JP. Quantitative nutritional studies with water-soluble, chemically defined diets. VIII. The forced feeding of diets each lacking in one essential amino acid. Arch Biochem Biophys. 1959;81:448–455. doi: 10.1016/0003-9861(59)90225-5. [DOI] [PubMed] [Google Scholar]
- 29.Chello PL, Bertino JR. Dependence of 5-methyltetrahydrofolate utilization by L5178Y murine leukemia cells in vitro on the presence of hydroxycobalamin and transcobalamin II. Cancer Res. 1973;33:1898–1904. [PubMed] [Google Scholar]
- 30.Mecham JO, Rowitch D, Wallace CD, Stern PH, Hoffman RM. The metabolic defect of methionine dependence occurs frequently in human tumor cell lines. Biochem Biophys Res Commun. 1983;117:429–434. doi: 10.1016/0006-291X(83)91218-4. [DOI] [PubMed] [Google Scholar]
- 31.Tan Y, Xu M, Hoffman RM. Broad selective efficacy of recombinant methioninase and polyethylene glycol-modified recombinant methioninase on cancer cells in vitro. Anticancer Res. 2010;30:1041–1046. [PubMed] [Google Scholar]
- 32.Hoffman RM. Altered methionine metabolism, DNA methylation and oncogene expression in carcinogenesis. A review and synthesis. Biochim Biophys Acta. 1984;738:49–87. doi: 10.1016/0304-419x(84)90019-2. [DOI] [PubMed] [Google Scholar]
- 33.Stern PH, Wallace CD, Hoffman RM. Altered methionine metabolism occurs in all members of a set of diverse human tumor cell lines. J Cell Physiol. 1984;119:29–34. doi: 10.1002/jcp.1041190106. [DOI] [PubMed] [Google Scholar]
- 34.Guo HY, Herrera H, Groce A, Hoffman RM. Expression of the biochemical defect of methionine dependence in fresh patient tumors in primary histoculture. Cancer Res. 1993;53:2479–2483. [PubMed] [Google Scholar]
- 35.Hoffman RM, Erbe RW. High in vivo rates of methionine biosynthesis in transformed human and malignant rat cells auxotrophic for methionine. Proc Natl Acad Sci USA. 1976;73:1523-1527. [DOI] [PMC free article] [PubMed]
- 36.Coalson DW, Mecham JO, Stern PH, Hoffman RM. Reduced availability of endogenously synthesized methionine for S-adenosylmethionine formation in methionine dependent cancer cells. Proc Natl Acad Sci U S A. 1982;79:4248–4251. doi: 10.1073/pnas.79.14.4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borrego SL, Fahrmann J, Datta R, Stringari C, Grapov D, Zeller M, Chen Y, Wang P, Baldi P, Gratton E, Fiehn O, Kaiser P. Metabolic changes associated with methionine stress sensitivity in MDA-MB-468 breast cancer cells. Cancer Metab. 2016;4:9. doi: 10.1186/s40170-016-0148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stern PH, Hoffman RM. Elevated overall rates of transmethylation in cell lines from diverse human tumors. In Vitro. 1984;20:663–670. doi: 10.1007/BF02619617. [DOI] [PubMed] [Google Scholar]
- 39.Murakami T, Li S, Han Q, Tan Y, Kiyuna T, Igarashi K, Kawaguchi K, Hwang HK, Miyaki K, Singh AS, et al. Recombinant methioninase effectively targets a Ewing’s sarcoma in a patient-derived orthotopic xenograft (PDOX) nude-mouse model. Oncotarget, in press. [DOI] [PMC free article] [PubMed]
- 40.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 41.Tisdale MJ. Effect of methionine deprivation on methylation and synthesis of macromolecules. Br J Cancer. 1980;42:121–128. doi: 10.1038/bjc.1980.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffman RM, Jacobsen SJ, Erbe RW. Reversion to methionine independence by malignant rat and SV40-transformed human fibroblasts. Biochem Biophys Res Commun. 1978;82:228–234. doi: 10.1016/0006-291X(78)90600-9. [DOI] [PubMed] [Google Scholar]
- 43.Hoffman RM, Jacobsen SJ, Erbe RW. Reversion to methionine independence in simian virus 40-transformed human and malignant rat fibroblasts is associated with altered ploidy and altered properties of transformation. Proc Natl Acad Sci U S A. 1979;76:1313–1317. doi: 10.1073/pnas.76.3.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Judde JG, Ellis M, Frost P. Biochemical analysis of the role of transmethylation in the methionine dependence of tumor cells. Cancer Res. 1989;49:4859–4865. [PubMed] [Google Scholar]
- 45.Hoffman RM, Jacobsen SJ. Reversible growth arrest in simian virus 40-transformed human fibroblasts. Proc Natl Acad Sci U S A. 1980;77:7306–7310. doi: 10.1073/pnas.77.12.7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yano S, Li S, Han Q, Tan Y, Bouvet M, Fujiwara T, Hoffman RM. Selective methioninase-induced trap of cancer cells in S/G2 phase visualized by FUCCI imaging confers chemosensitivity. Oncotarget. 2014;5:8729–8736. doi: 10.18632/oncotarget.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bloomfield M, Duesberg P. Karyotype alteration generates the neoplastic phenotypes of SV40-infected human and rodent cells. Mol Cytogenet. 2015;8:79. doi: 10.1186/s13039-015-0183-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bloomfield M, McCormack A, Mandrioli D, Fiala C, Aldaz CM, Duesberg P. Karyotypic evolutions of cancer species in rats during the long latent periods after injection of nitrosourea. Mol Cytogenet. 2014;7:71. doi: 10.1186/s13039-014-0071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duesberg P, Li R. Multistep carcinogenesis: a chain reaction of aneuploidizations. Cell Cycle. 2003;2:202–210. doi: 10.4161/cc.2.3.382. [DOI] [PubMed] [Google Scholar]
- 50.Kanai Y, Hirohashi S. Alterations of DNA methylation associated with abnormalities of DNA methyltransferases in human cancers during transition from a precancerous to a malignant state. Carcinogenesis. 2007;28:2434–2442. doi: 10.1093/carcin/bgm206. [DOI] [PubMed] [Google Scholar]
- 51.Kokkinakis DM, Schold SC, Jr, Hori H, Nobori T. Effect of long-term depletion of plasma methionine on the growth and survival of human brain tumor xenografts in athymic mice. Nutr Cancer. 1997;29:195–204. doi: 10.1080/01635589709514624. [DOI] [PubMed] [Google Scholar]
- 52.Kokkinakis DM, von Wronski MA, Vuong TH, Brent TP, Schold SC., Jr Regulation of O6-methylguanine-DNA methyltransferase by methionine in human tumour cells. Br J Cancer. 1997;75:779–788. doi: 10.1038/bjc.1997.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kokkinakis DM, Hoffman RM, Frenkel EP, Wick JB, Han Q, Xu M, Tan Y, Schold SC. Synergy between methionine stress and chemotherapy in the treatment of brain tumor xenografts in athymic mice. Cancer Res. 2001;61:4017–4023. [PubMed] [Google Scholar]
- 54.Tan Y, Sun X, Xu M, Tan X-Z, Sasson A, Rashidi B, Han Q, Tan X-Y, Wang X, An Z, Sun F-X, Hoffman RM. Efficacy of recombinant methioninase in combination with cisplatin on human colon tumors in nude mice. Clin Cancer Res. 1999;5:2157–2163. [PubMed] [Google Scholar]
- 55.Yoshioka T, Wada T, Uchida N, Maki H, Yoshida H, Ide N, Kasai H, Hojo K, Shono K, Maekawa R, et al. Anticancer efficacy in vivo and in vitro, synergy with 5-fluorouracil, and safety of recombinant methioninase. Cancer Res. 1998;58:2583–2587. [PubMed] [Google Scholar]
- 56.Grosu AL, Weber WA, Riedel E, Jeremic B, Nieder C, Franz M, Gumprecht H, Jaeger R, Schwaiger M, Molls M. L-(methyl-11C) methionine positron emission tomography for target delineation in resected high-grade gliomas before radiotherapy. Int J Radiat Oncol Biol Phys. 2005;63:64–74. doi: 10.1016/j.ijrobp.2005.01.045. [DOI] [PubMed] [Google Scholar]
- 57.Glaudemans AW, Enting RH, Heesters MA, Dierckx RA, van Rheenen RW, Walenkamp AM, Slart RH. Value of 11C-methionine PET in imaging brain tumours and metastases. Eur J Nucl Med Mol Imaging. 2013;40:615–635. doi: 10.1007/s00259-012-2295-5. [DOI] [PubMed] [Google Scholar]
- 58.Tsuyuguchi N, Takami T, Sunada I, Iwai Y, Yamanaka K, Tanaka K, Nishikawa M, Ohata K, Torii K, Morino M, Nishio A, Hara M. Methionine positron emission tomography for differentiation of recurrent brain tumor and radiation necrosis after stereotactic radiosurgery—in malignant glioma. Ann Nucl Med. 2004;18:291–296. doi: 10.1007/BF02984466. [DOI] [PubMed] [Google Scholar]
- 59.Nariai T, Tanaka Y, Wakimoto H, Aoyagi M, Tamaki M, Ishiwata K, Senda M, Ishii K, Hirakawa K, Ohno K. Usefulness of L-[methyl-11C] methionine-positron emission tomography as a biological monitoring tool in the treatment of glioma. J Neurosurg. 2005;103:498–507. doi: 10.3171/jns.2005.103.3.0498. [DOI] [PubMed] [Google Scholar]
- 60.Tamura K, Yoshikawa K, Ishikawa H, Hasebe M, Tsuji H, Yanagi T, Suzuki K, Kubo A, Tsujii H. Carbon-11-methionine PET imaging of choroidal melanoma and the time course after carbon ion beam radiotherapy. Anticancer Res. 2009;29:1507–1514. [PubMed] [Google Scholar]
- 61.Vousden K. Guardian of the genome. Cancer. 2015;1:10–11. [Google Scholar]
- 62.Kamiyama H, Suzuki K, Maeda T, Koizumi K, Miyaki Y, Okada S, Kawamura YJ, Samuelsson JK, Alonso S, Konishi F, Perucho M. DNA demethylation in normal colon tissue predicts predisposition to multiple cancers. Oncogene. 2012;31:5029–5037. doi: 10.1038/onc.2011.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoffman RM. Development of recombinant methioninase to target the general cancer-specific metabolic defect of methionine dependence: a 40-year odyssey. Expert Opin Biol Ther. 2015;15:21–31. doi: 10.1517/14712598.2015.963050. [DOI] [PubMed] [Google Scholar]
- 64.Cheah MSC, Wallace CD, Hoffman RM. Hypomethylation of DNA in human cancer cells: a site-specific change in the c-myc oncogene. J Natl Cancer Inst. 1984;73:1057–1065. [PubMed] [Google Scholar]
- 65.Bunz F, Fauth C, Speicher MR, Dutriaux A, Sedivy JM, Kinzler KW, Vogelstein B, Lengauer C. Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer Res. 2002;62:1129–1133. [PubMed] [Google Scholar]
- 66.Yano S, Takehara K, Zhao M, Tan Y, Han Q, Li S, Bouvet M, Fujiwara T, Hoffman RM. Tumor-specific cell-cycle decoy by Salmonella typhimurium A1-R combined with tumor-selective cell-cycle trap by methioninase overcome tumor intrinsic chemoresistance as visualized by FUCCI imaging. Cell Cycle. 2016;15:1715–1723. doi: 10.1080/15384101.2016.1181240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This paper is a review. All data discussed are published and referenced in the manuscript.