

Abstract
With more than 500,000 copies, mammalian-wide interspersed repeats (MIRs), a sub-group of SINEs, represent ∼2.5% of the human genome and one of the most numerous family of potential targets for the RNA polymerase (Pol) III transcription machinery. Since MIR elements ceased to amplify ∼130 myr ago, previous studies primarily focused on their genomic impact, while the issue of their expression has not been extensively addressed. We applied a dedicated bioinformatic pipeline to ENCODE RNA-Seq datasets of seven human cell lines and, for the first time, we were able to define the Pol III-driven MIR transcriptome at single-locus resolution. While the majority of Pol III-transcribed MIR elements are cell-specific, we discovered a small set of ubiquitously transcribed MIRs mapping within Pol II-transcribed genes in antisense orientation that could influence the expression of the overlapping gene. We also identified novel Pol III-transcribed ncRNAs, deriving from transcription of annotated MIR fragments flanked by unique MIR-unrelated sequences, and confirmed the role of Pol III-specific internal promoter elements in MIR transcription. Besides demonstrating widespread transcription at these retrotranspositionally inactive elements in human cells, the ability to profile MIR expression at single-locus resolution will facilitate their study in different cell types and states including pathological alterations.
Keywords: SINE, mammalian-wide interspersed repeats, RNA polymerase III, RNA-Seq, ENCODE
1. Introduction
Mammalian-wide Interspersed Repeats (MIRs) represent an ancient family of tRNA-derived Short INterspersed Elements (SINEs) found in all mammalian genomes, whose amplification seems to have ceased in the ancestors of placental mammals.1–3 This is in contrast with Alu elements, the most abundant SINEs, which are still transpositionally active.4 It is thought that MIRs may have arisen following the fusion of a tRNA molecule with the 3′-end of an existing Long INterspersed Element (LINE).5 The complete MIR element is about 260 bp in length and comprises a tRNA-related 5′ head, a 70-bp conserved central domain containing a 15-bp core sequence, and a LINE-related sequence located at the 3′-end5 (Fig. 1). In an earlier study first describing MIRs, two segments overlapping with the LINE-related region were described as separate interspersed repeats MER24 and DBR.2
Figure 1.

Representation of the structure of a mammalian-wide interspersed repeat (MIR). A tRNA-related region contains A- and B-box promoter elements driving Pol III transcription by being recognized by TFIIIC. Core-SINE indicates a highly conserved central sequence, followed by a LINE-related region. Pol III is expected to terminate at the first encountered termination signal (Tn) which may be located at varying distances from the end of the MIR body.
MIR elements were actively propagating prior to the radiation of mammals and before placental mammals separated. For this reason their age was originally estimated to be ∼130 million years (myr) even if it has been suggested that the CORE-SINE6 may have originated ∼550 myr ago due to the remarkable similarity between Ther-1 (the MIR consensus in placental mammals which corresponds to that revealed earlier in humans) and the OR2 SINE of octopuses.7 Intriguingly, there are observations suggesting that the core region may serve some general function in mammalian genomes, because the level of sequence conservation is higher than the 3′ and 5′-flanking sequences.8
In the human genome, there are more than 500,000 annotated MIRs.9 Based on their sequence similarity, they have been grouped into 4 subfamilies named MIR, MIRb, MIRc, and MIR3. Like all SINEs, MIRs are thought to be transcribed by the RNA Polymerase III (Pol III) machinery, with the assembly factor TFIIIC recognizing the A- and B-box internal control regions within the tRNA-derived portion of the element.2 As well established for tRNA gene promoters, once bound TFIIIC recruits TFIIIB [composed of Brf1, Bdp1 and the TATA box-binding protein (TBP)], which in turn recruits Pol III.10 The first experimental verifications of MIRs as Pol III targets in the human genome have come from the results of genome-wide location analyses of the Pol III machinery in human11,12 and mouse cells.13,14 Interestingly, one of these studies revealed that, in human immortalized fibroblasts, the Pol III machinery is consistently associated with a MIR located in the first intron of the POLR3E gene, coding for a specific subunit of Pol III (RPC5). A significant enrichment of components of the Pol III machinery was also observed at a dozen other MIRs, thus supporting the notion that MIRs, although transpositionally inactive, can undergo autonomous transcription.11,13,14
Unlike Alus that, although non-autonomous in retrotransposition, nevertheless exploit the LINE-encoded machinery to amplify in the genome, MIRs have ceased to amplify by retrotransposition ∼130 Myr ago. This could be due to many reasons, including for example the inactivation of L2 (the partner LINE for MIR elements) or mutations in LINE-related regions of MIRs. Nevertheless, it can be speculated that since part of these elements are still transcriptionally active at least in the human and mouse genome,11 changes in the 3′ tail due to sequence mutations could potentially rescue the retrotransposition potential of some of them. Recent studies have shown that enhancers are a platform from which MIRs can exert a regulatory function in the human genome9 and that MIRs can also serve as insulators acting as chromatin barriers or enhancer-blocking elements.15 Nothing is known, however, about the possible involvement of MIR expression in their role at enhancers/insulators. In general, in spite of some advancement in our knowledge of how the massive presence of MIRs impacts genome biology, their contribution to the human transcriptome is still largely unexplored, as are their transcription mechanism and regulation. In particular, the cis-acting elements controlling MIR transcription have never been systematically studied.
In this study, by applying a recently developed bioinformatics pipeline to ENCODE RNA-seq data, we define for the first time the MIR expression profiles of human cells, and we investigate on the in cis requirements for MIR transcription by biochemical analysis in vitro.
2. Materials and methods
2.1. Bioinformatic pipeline for MIR expression
For MIR RNA identification we used the Cold Spring Harbor Laboratory (CSHL) long RNA-seq data within ENCODE16 of seven human cell lines (GM12878, H1-hESC, HeLa-S3, HepG2, HUVEC, K562, and NHEK). The RNA-Seq reads were aligned to the human reference genome GRCh37/hg19 as previously described.17 PolyA+ and PolyA- datasets (bam files) were merged to streamline the analyses. We were not interested in the differences between these two features because MIR RNAs usually do not have a poly(A) tail.
The bam files from each dataset, containing RNA-Seq reads aligned to the reference genome (GRCh37/hg19) using TopHat aligner, along with the annotated MIRs, were submitted to a Python script that performs the identification of individual MIR transcripts. The script first builds stranded coverage vectors for the whole genome, using the bam file supplied and only considering uniquely mapped reads (tag NH:i:1 in the bam file). Then, for each annotated MIR having an expression coverage value over a calculated background noise threshold, the script calculates the coordinates of the corresponding expected full-length consensus element (see Supplementary Methods), to take into account the fact that many of the annotated MIR elements are truncated. Finally a flanking region filter is applied to the identified expected full-length element, as described in Supplementary Methods, in order to exclude false positives arising from MIR elements embedded in Pol II transcripts. The filter is designed to do this by imposing a significantly lower expression coverage value to the flanking regions immediately upstream and downstream of the expected full-length SINE, thus discriminating between genuine SINE RNAs and those that are ‘passengers’ of longer Pol II transcripts or part of their trailers extending downstream of their annotated 3′UTRs. Figure 2 shows a schematic representation of the pipeline, which is improved with respect to the one previously employed to distinguish genuine from ‘passenger’ Alu RNAs17 allowing us to control each step and to identify both Alu and MIR transcripts of the SINE class of retrotransposons.
Figure 2.

Bioinformatic pipeline flowchart. Shown is a flow diagram of the improved bioinformatic pipeline for the identification of autonomously expressed SINE loci from RNA-seq data sets. See text and Supplementary Methods for details.
Only MIRs which passed the final filter of the pipeline in both ENCODE RNA-Seq replicates were considered to represent autonomously expressed MIR loci and will be referred to as ‘expression-positive’. The complete list of expression-positive MIRs are reported in Supplementary Table S1.
To further support the identification of unique MIR transcripts found in Hela-S3 and K562 cells we intersected the ChIP-seq peak data from ENCODE/Stanford/Yale/USC/Harvard (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeSydhTfbs/) and Transcription Factor Binding Sites (TFBS) from ENCODE data uniformly processed by the ENCODE Analysis Working Group (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wg EncodeRegTfbsClustered/wgEncodeReg TfbsClustered WithCells V3.bed.gz) for some Pol III transcription factors (TFs) binding sites with the coordinates of the expected full-length expression-positive MIRs, extended to 200 bp upstream. To identify other Pol II TFs associated with expression-positive MIR elements, we intersected, for each cell line, the 500 bp upstream of the expected full-length MIRs with the coordinates of the TF binding sites from ENCODE data, also uniformly processed by the ENCODE Analysis Working Group. The lists of Pol III-associated expression-positive MIRs and of those associated with other TFs are reported in Supplementary Table S2.
2.2. Plasmid construction
Four human MIR loci (whose chromosome coordinates are reported in Table 3), together with their 5′- and 3′-flanking regions, were PCR-amplified and cloned into pGEM®-T Easy vector (Promega) using the oligonucleotides listed in Supplementary Table S4. Constructs containing a targeted mutation of the B box internal control element were generated as described previously.17 Upstream deletion constructs employed forward PCR primers generating amplicons truncated at position −25 with respect to MIR A box at the 5′-end. Truncated amplicons were inserted into pGEM®-T Easy and the constructs selected for in vitro transcription contained the 5′-truncated insert in the same orientation as their wild-type MIR counterpart, to minimize the influence of vector sequence on transcription efficiency.
Table 3.
MIRs subjected to in vitro transcription analysis
| MIR | Expression in cell linesa | Predicted length of primary transcript(s)b |
|---|---|---|
| MIR_dup2285 (chr16:22309780-22309939) | GM12878, H1-hESC, HeLa-S3, HepG2, HUVEC, K562, NHEK | 124/131 (T4), 223/230 (T3GT2), 243/250 (T4), 354/361 (T3CT, downstream of annotated MIR but within the cloned sequence) |
| MIR_dup3493 (chr1:34943459-34943727) | GM12878, H1-hESC, K562, NHEK | 177 (TAT3), 213 (TAT3), 277 (T2AT2). Expected transcripts originating from terminators more downstream of the annotated MIR but within the cloned sequence: 305 (T2AT2), 365 (T2AT3), 393 (T4) |
| MIRb_dup5848 (chr2:71762977-71763215) | H1-hESC, HepG2, NHEK | 119 (TCT3), 256 (T3CT), 358 (T5) |
| MIRc_dup2189 (chr14:89445565-89445634) | H1-hESC, K562, NHEK | 137 (T3AT) and 140 (T4), 207 (T2GT3), 250 (T5) |
aThe column lists, for each MIR element, the cell lines in which it was found to be expressed by ENCODE RNA-seq data analysis.
bThe reported transcript lengths were calculated by assuming as TSS the A or G residue closest to the position 12 bp upstream of the A box. To estimate the 3’ end of the transcript, both canonical (Tn with n ≥ 4) and non-canonical T-rich Pol III terminators [17] were considered both within and downstream of MIR body sequence (indicated in parentheses after the transcript length). For canonical terminators, the four Us corresponding to the first four Ts of the termination signal were considered as part of the transcripts; for non-canonical terminators, all the nucleotides of the terminator were considered as incorporated into the RNA. In the case of MIR_dup2285, for which two possible A boxes could drive transcription, the expected lengths of both putative alternative transcripts are indicated (Supplementary Fig. S1).
2.3. In vitro transcription
All recombinant plasmids for in vitro transcription reactions were purified with the Qiagen Plasmid Mini kit (Qiagen). Reaction mixtures (final volume: 25 µl) contained 500 ng of template DNA, 70 mM KCl, 5 mM MgCl2, 1 mM DTT, 2.5% glycerol, 20 mM Tris–HCl pH 8, 5 mM phosphocreatine, 2 µg/ml alpha-amanitin, 0.4 U/µl SUPERase-In (Ambion) as RNase inhibitor, 40 µg of HeLa cell nuclear extract,18 0.5 mM each of ATP, CTP, and GTP, 0.025 mM UTP and 5µCi of [α-32P]UTP (Perkin-Elmer). Reactions were allowed to proceed for 60 min at 30 °C before being stopped by the addition of 75 µl of nuclease-free water and 100 µl of phenol:chloroform (1:1) pH 5.5. Purified labeled RNA products were resolved on a 6% polyacrylamide, 7 M urea gel and visualized, and quantified with a Cyclone Phosphor Imager (PerkinElmer) and the Quantity One software (Bio-Rad).
2.3. Availability
The Python script allowing to identify individual MIR transcripts is available at the following URL: https://github.com/davidecarnevali/SINEsFind.
3. Results
3.1. A bioinformatic pipeline for the identification of transcriptionally active MIR loci from RNA-Seq datasets
To leverage the potential of our previous work,17 we developed a pipeline in order to improve the identification of Pol III SINE transcripts (see Materials and Methods and Supplementary Methods). We focused on the identification of MIR transcripts by applying the improved pipeline to the aligned Long RNA-Seq reads from ENCODE,17 considering for analysis a subset of the annotated MIR elements from the GRCh37/hg19 UCSC RepeatMasker track classified according to their genomic location: (i) MIRs mapping in intergenic regions (i.e. outside RefSeq, Ensembl, and lincRNA genes), (ii) MIRs mapping within RefSeq, Ensembl, and lincRNA genes but in antisense orientation, and (iii) MIRs fully contained within introns of RefSeq, Ensembl, and lincRNA genes in sense orientation and not overlapping with any exon. We refer to the first two groups together as ‘intergenic/antisense’.
Figure 2 provides a schematic representation of our improved pipeline. The main improvements with respect to the previously described pipeline17 consist in the estimation of the ‘background’ coverage signal for each sample (instead using a fixed value for all the samples) and the definition of the flanking regions based on the global alignment of each annotated element with its consensus sequence (See Materials and Methods and Supplementary Methods).
3.2. General features of MIR transcriptomes emerging from ENCODE RNA-seq data analysis
A preliminary survey of the expression coverage of the MIR loci identified as expression-positive showed us both convincing MIR transcription profiles and less clearcut ones with noise background signals upstream and downstream of the MIR element probably deriving from the sequencing of introns or unknown Pol II transcripts. Even the latter cases, however, displayed expression coverage enrichment in the region of the annotated MIR element, suggesting the possibility of autonomous Pol III transcription as a pathway for their biogenesis, perhaps occurring in parallel with Pol II transcription of the host gene with intron-containing MIR RNAs generated by processing from longer Pol II transcripts. Since the tendency to high noise-to-background signals is more frequently observed in MIR element mapping within introns of Pol II genes in sense orientation, which constitute 79% of the expression-positive MIRs (see Supplementary Table S1), we decided to mainly focus on the expression of intergenic/antisense MIRs, while a few examples of gene-hosted sense-oriented MIRs will be addressed later in the Results section.
The full list of MIRs identified as expression-positive by our search strategy is reported in Supplementary Table S1. As summarized in Table 1 each of the cell lines expressed a limited number of MIR elements (ranging from 145 in the case of GM12878 cells to 435 in the case of HUVEC cells). Of the whole set of 1301 expression-positive MIR loci a small percentage (∼21%) were intergenic/antisense, which is in contrast with the global distribution of all the annotated MIRs regardless of their expression where the percentage of the intergenic/antisense MIRs is much higher (∼68%) (see Supplementary Table S1). A significant fraction (∼39%) of intergenic/antisense MIRs mapped in antisense orientation to annotated Pol II-transcribed genes. For comparison, the fraction of intergenic/antisense expression-positive Alus mapping in antisense orientation to the overlapping genes in the same cell lines was only 22%,17 suggesting that MIRs antisense to Pol II genes could have a more specific role, possibly correlated with the regulation of the overlapping gene. A small fraction (∼20%) of all the expression-positive MIRs were found to be expressed in more than one cell line (see Supplementary Table S1), while this percentage decreased to 12% considering only the intergenic/antisense MIRs (Table 1). This suggests a marked cell line specificity in MIR expression (even though few MIRs seem to be ubiquitously expressed as will be further reported below) greater than the one found for Alu expression where the fraction of expression-positive Alus expressed in more than one cell line was higher (∼24%).17
Table 1.
Statistic of expression-positive MIR elements in selected cell lines
| Cell line | Total MIRsa | Intergenic/ antisense | Intergenic/ antisense sharedb | Antisensec |
|---|---|---|---|---|
| GM12878 | 145 | 39 | 12 | 16 |
| H1-hESC | 280 | 52 | 18 | 23 |
| HeLa-S3 | 188 | 32 | 6 | 15 |
| HepG2 | 348 | 46 | 16 | 23 |
| HUVEC | 435 | 59 | 7 | 18 |
| K562 | 161 | 42 | 13 | 18 |
| NHEK | 158 | 55 | 15 | 25 |
| ALLd | 1301 | 271 | 33 | 105 |
aFor each cell line, the column reports the number of MIRs considered as autonomously expressed in both ENCODE RNA-seq replicates.
bFor each cell line, the column reports the number of intergenic/antisense MIRs that are also expressed in one or more different cell lines.
cReported in this column are the numbers of intergenic MIRs mapping with an antisense orientation to either protein-coding, non protein-coding or lincRNA genes.
dThe numbers in this raw refer to individual MIRs expressed in one or more cell lines.
As summarized in Table 2, no significant under- or over-representation of any particular MIR subfamily within the set of expressed MIRs was observed (see Supplementary Table S1).
Table 2.
Subfamily distribution of expression-positive MIRs
| MIR subfamily | Total genomica | Expressed genomica | Total intergenic/ antisensea | Expressed intergenic/ antisensea |
|---|---|---|---|---|
| MIR | 171148 (30%) | 436 (33%) | 115860 (30%) | 101 (37%) |
| MIRb | 219279 (38%) | 487 (37%) | 148567 (38%) | 104 (38%) |
| MIRc | 100252 (17%) | 202 (16%) | 67888 (17%) | 34 (13%) |
| MIR3 | 87763 (15%) | 176 (14%) | 59078 (15%) | 32 (12%) |
aReported are the absolute copy numbers and (in parentheses) the percentages of MIRs of each sub-family considered relative to (from left to right): the total set of genomic MIRs (intergenic/antisense plus intronic sense MIRs); the set of MIRs found to be expression-positive in one or more cell line (intergenic/antisense plus intronic sense ones); the genomic set of intergenic/antisense MIRs; the set of intergenic/antisense MIRs found to be expression-positive in one or more cell lines.
3.3. Survey of expressed MIRs according to location and base-resolution expression profile
The ∼6 × 105 annotated MIRs are not all complete in sequence, and represent only a portion of the canonical full-length MIR element. Among expression-positive MIRs, we found both complete and incomplete elements and, correspondingly, three main types of base-resolution expression profiles: (i) full-length or almost full-length MIRs (Fig. 3A and B), covered by sequence reads along all their extension; (ii) incomplete MIRs representing either the left or the right portion of the canonical full-length MIR but whose transcript coverage tends to correspond to the one of a fully transcribed canonical MIR [more specifically, transcript coverage tends to extend into the downstream MIR-unrelated region for incomplete MIRs lacking the 3′ moiety (Fig. 3C), while it tends to start in an upstream MIR-unrelated region, possessing functional A and B-boxes, for incomplete MIRs lacking the 5′ moiety (Fig. 3E)]; (iii) incomplete MIRs lacking the 3′ moiety (thus containing A and B boxes) and producing transcripts that do not extend outside of the MIR sequence (Fig. 3D).
Figure 3.
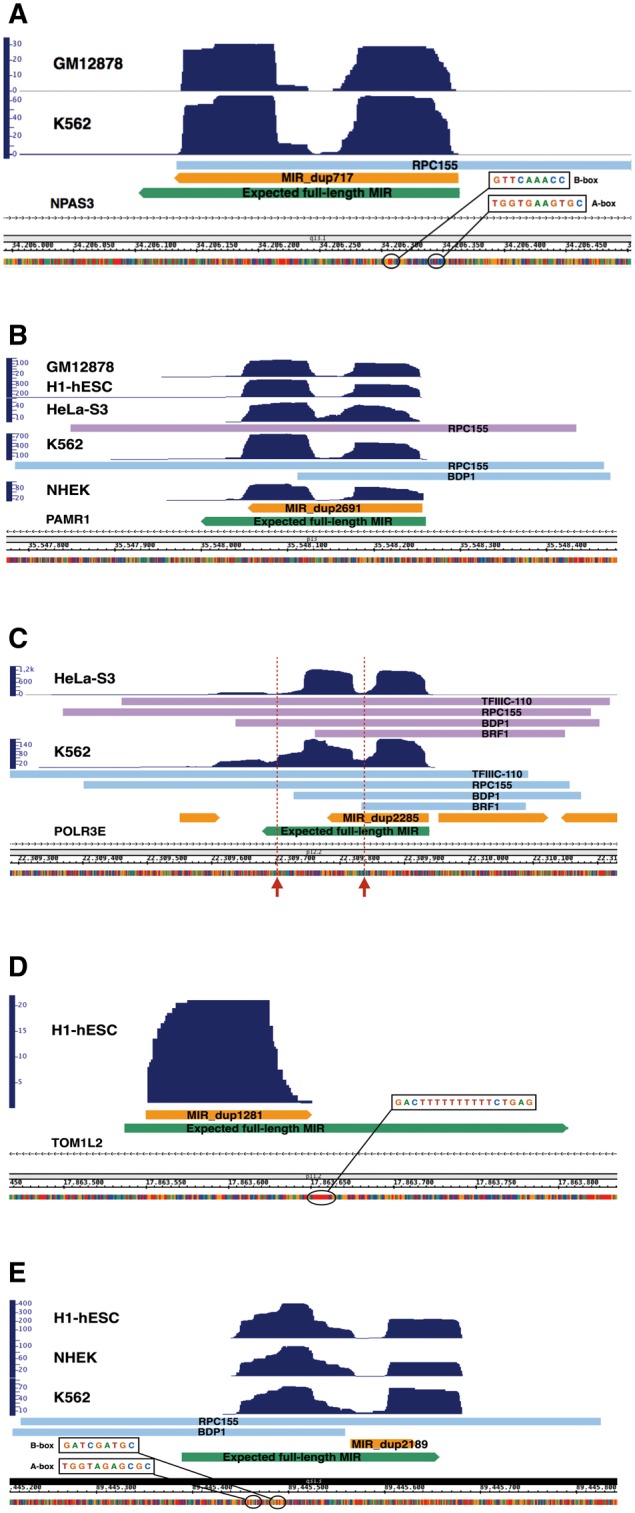
Base-resolution expression profiles for five representative MIRs. See text for descriptions. Bars labeled with the names of Pol III transcription components (RPC155, TFIIIC110, BDP1, BRF1) indicate regions of enrichment of the corresponding proteins according to publicly available ChIP-seq data; upper arrowed bars represent the annotated MIR elements while the lower arrowed bars represent the corresponding expected full-length MIR elements which does not correspond to annotated elements and are reported merely to locate the alignment position of the annotated MIRs within the corresponding consensus sequence. Arrows in panel C point to the positions of strong Pol III terminators. (A) MIR_dup717 chr14:34206132-34206363; (B) MIR_dup2691 chr11:35548054-35548257 which resides within an intron of the PAMR1 gene in sense orientation; (C) MIR_dup2285 chr16:22309780-22309939; (D) MIRb_dup1281 chr17:17863550-17863651; (E) MIRc_dup2189 chr14:89445565-89445634
The MIR reported in Figure 3A (chr14:34206132-34206363 MIR_dup717) was scored as expression-positive in Gm12878 and K562 cells. It also displayed a specific, yet very low coverage, below the threshold for a positive score, in H1-hESC, HeLa-S3, HepG2, and NHEK cells. This representative MIR is full-length and maps to intron 6 or 7 (depending on which transcript isoform is considered) of the NPAS3 gene, in antisense orientation. It has an expression profile that almost entirely covers the annotated element with uniquely mapped sequence reads. The double-humped expression coverage profiles shown in Figure 3 are due to cDNA size selection during library preparation and sequencing specifications, as previously reported.17 A- and B- boxes are conserved at 13 and 50 bp downstream of the TSS, respectively. Unusually, the annotated MIR contains a strong termination signal (T4) in its sequence 104 nt from the beginning of the element, which is clearly skipped by the Pol III machinery, given the coverage signal in the downstream moiety of the element. We investigated the possibility that the genomes of the cell lines analyzed had a sequence variation with respect to the reference GRCh37/hg19 sequence, in correspondence of these Pol III strong terminators. To this end, we reconstructed the corresponding consensus genome sequences from RNA sequence reads (see Supplementary Methods). We did not find any sequence variant, thus indicating the presence of a termination signal skipped by the Pol III machinery. Another terminator of 4Ts is present in the downstream moiety of the MIR, leading to a transcript of ∼220 nt. Strong support for the genuine nature of this Pol III-derived MIR RNA comes from the association with this locus of the Pol III subunit RPC155 in K562 cells (Fig. 3A).
Figure 3B shows a MIR fully contained within an intron of the PAMR1 gene in the sense orientation that has been found to be expressed in 5 cell lines. The Pol III-dependent character of this transcription unit is supported by its association with the Pol III subunit RPC155 and the Pol III transcription factor BDP1 in K562 and HeLa cell lines, also confirming the ability of Pol III machinery complex to access and transcribe Pol II genomic loci.
Reported in Figure 3C is the transcript coverage profile of an incomplete MIR element of 159 bp (chr16:22309780-22309939 MIR_dup2285), which was expressed in all 7 cell lines. This element aligns to the left portion of the MIR consensus sequence, but its transcription continues downstream of the region annotated as MIR (in orange in Fig. 3C) for ∼200 nt before encountering a non-canonical termination signal (T3CT). This previously characterized MIR11 is located in antisense orientation in the first intron of the POLR3E gene, encoding a subunit (RPC5) of human RNA polymerase III. Its transcription by Pol III is supported by the presence of canonical A- and B-boxes as well as by association with components of the Pol III machinery in HeLa-S3 and K562 cell lines. Here, again, the annotated MIR portion contains a strong termination signal (T4) early in its sequence, 121 nt downstream the 5′ end of the element. Another potential terminator is located 82 nt downstream of the 3′ end of the annotated element. In this case, the coverage signal tends to decrease temporarily but slightly increases again downstream, until a non-canonical terminator is encountered. The skipping of the first terminator by Pol III is supported by the expression coverage levels upstream and downstream, which are almost the same, strongly suggesting that they arise from the same transcript. In contrast, lower coverage downstream of the second terminator suggests the occurrence of only limited read through of this termination signal by Pol III. We also considered the possibility of a mutation in the strong terminator sequences in the DNA of these cells (see above), but we did not find any sequence variant, thus confirming the ability of Pol III to skip strong terminator signals within certain sequence contexts.19 This result was partially confirmed by in vitro transcription analysis (see Fig. 5).
Figure 5.

In vitro transcription for selected expression-positive MIRs. In vitro transcription reactions were performed in HeLa nuclear extract using 0.5 μg of the indicated MIR templates (lanes 4–9, 13–18). A previously characterized Alu producing a 355-nt RNA (lanes 2, 11) and a human tRNAVal gene producing a known transcript pattern due to heterogeneous transcription termination (lanes 3, 12)19 were used as positive controls for in vitro transcription and, at the same time, as a source of RNA size markers. Negative control reactions contained empty pGEM®-T Easy vector (lanes 1, 10). For each MIR, both the wild type, B box-mutated (Bmut) and 5′-flanking region (5′ del) version were tested. Indicated by arrows on the gel image are the migration positions (with lengths) of bands corresponding to the expected transcripts in Table 3.
In Figure 3D, we report a MIR element (chr17:17863550-17863651 MIRb_dup1281), found to be expressed at low levels in in H1-hESC cell line, originating from an incomplete MIR element antisense to the TOM1L2 gene. By inspecting the coverage profile in other cell lines, we could find signals of much lower expression in at least one replicate of each of the analyzed cell lines. The source element of this transcript corresponds to a MIRb left fragment carrying in its sequence functional A- and B- boxes. The transcript coverage precisely spans the length of the annotated element up to a strong terminator (T10) right at the end of it, leading to a transcript of ∼100 nt.
Figure 3E shows a MIR, found as expression-positive in H1-hESC, K562 and NHEK cell lines, whose transcription initiates in an upstream MIR-unrelated region (chr14:89445565-89445634 MIRc_dup2189) and ends ∼50 nt downstream of the annotated element corresponding with a strong termination signal (T5). By inspecting the MIR-unrelated upstream region we found canonical A- and B- boxes, indicating that this MIR element, lacking Pol III promoters in its sequence, is transcribed from an upstream MIR-unrelated region providing functional control elements. Also in this case we noted a termination signal (T3AT4) at the beginning of the annotated element which seems to be (partially) skipped in vivo by Pol III (as for MIR_dup2285 in POLR3E gene in Fig. 3C) and is located right at the end of the first coverage hump. Indeed the second hump has a slightly lower coverage signal (at least in H1-hESC and NHEK cells) suggesting a shorter primary transcript as confirmed by in vitro transcription (see below). The binding of Pol III TFs throughout this region in K562 cells, together with the transcript coverage profile, strongly support the Pol III-dependence of this MIR transcript.
As a further note on the MIR in Figure 3C, a Polr3e antisense-oriented MIR with the same location and similar sequence of MIR_dup2285 is also present in the mouse genome, where it has also been shown to be transcriptionally active, and to be associated with an unusual second point accumulation of RNA Pol II, in addition to the expected one in the Polr3e promoter, which abuts precisely on the Pol III occupancy peak reflecting MIR transcription on the opposite strand.14 These observations, along with the fact that this MIR is conserved in human and mouse, led the authors to hypothesize a functional role of this MIR in POLR3E gene regulation. We therefore asked if this second RNA Pol II accumulation point also occurs in the ENCODE cell lines, and found Pol II peaks for all 7 cell types (see Supplementary Table S5) thus strengthening the hypothesis that this transcriptionally active MIR plays a regulatory role in Pol II transcription of POLR3E. Figure 4 shows Pol II in three of the seven cell lines along with the Pol III signals.
Figure 4.
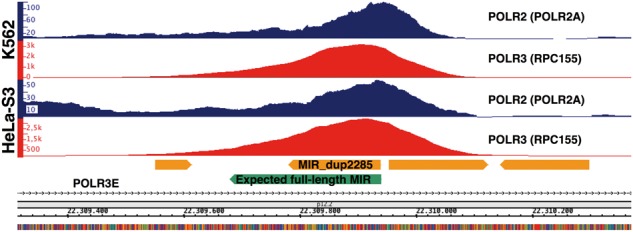
Simultaneous Pol III and Pol II accumulation signals at MIR_dup2285. Shown are the ChIP-seq signals of Pol II (POLR2A) (blue) and Pol III (RPC155) (red) in HeLa-S3 and K562 cell lines across the expression-positive MIR located within the first intron of POLR3E gene in antisense orientation (cf. Fig. 3C).
Finally, we asked if there was a correlation between the expression of the 78 MIRs overlapping with Pol II protein-coding genes in antisense orientation and the expression of the overlapping genes themselves. To this end, we compared normalized read counts of the MIRs in each replicate with those of the overlapping Pol II genes using the Pearson correlation coefficient (See Supplementary Methods) and found only positive correlation (r > 0.7 with P < 10 − 5) for few genes (ARHGEF3, DSCAM, GNA15, JAKMIP2, LRP2, NBPF10, PLXDC2, TMEM177) (data not shown). We also asked whether the protein-coding genes characterized by the presence of intronic MIRs (mapping in either antisense or sense orientation) are enriched in any particular gene ontology functional category. Interestingly, we found a strong enrichment (P < 10 − 139) for genes producing multiple proteins due to alternative splicing, suggesting a possible influence of intronic MIRs on this process. When expression-positive MIR only were considered, such a GO enrichment was less marked, although still remarkable (P value 1.8 × 10 − 24).
3.4. Association of the Pol III machinery with expression-positive MIRs
Before the use of RNA-Seq data to help identify expression-positive SINEs, genome-wide studies based on ChIP-Seq approaches were used in humans and mouse models with the aim of producing inventories of loci inferred to be transcribed by Pol III from their association with one or more component of the Pol III machinery.11,12,14,20,21 Such studies revealed a limited number of Pol III-associated SINE elements, and most of them were Alus. The availability of genome-wide ChIP-Seq data from ENCODE/Stanford/Yale/USC/Harvard (SYDH) and from ENCODE data uniformly processed by the ENCODE Analysis Working Group for key components of Pol III transcription machinery (BDP1, BRF1/2, RPC155, and TFIIIC110) allowed us to readdress this issue, by investigating whether a significant enrichment of these TFs could be found in the expression-positive intergenic/antisense MIRs of HeLa and K562 cells. Supplementary Table S2 lists the expression positive MIRs found to be associated with Pol III components in HeLa and K562 cells. In HeLa cells, of the 32 intergenic/antisense expression-positive MIRs, only 5 were found associated with one or more Pol III TFs. The three transcription components whose association was found to be statistically significant, with a P < 5.5x10 − 5 (calculated using Fisher’s exact test), were BDP1, TFIIIC-110 and RPC155 when considering the fraction of the intergenic/antisense expressed MIRs against the total intergenic/antisense annotated ones. When the Pol III component association with the 156 intron-hosted sense oriented expression positive MIRs was investigated, only one (MIR_dup2691 reported in Fig. 3B) was found associated with the Pol III subunit RPC155. When K562 cells were considered, a higher percentage (36%, 15 MIRs) of intergenic/antisense expression-positive MIRs where found bound by one or more Pol III components, while the percentage drop down to 3% (4 MIRs) when considering the 119 MIRs fully contained within introns of Pol II genes in sense orientation (data not shown). In K562 cells we found significant enrichment for BDP1, TFIIIC110 and RPC155 (P values 5.2 × 10 − 9, 9.7 × 10 − 6 and <2.2 × 10 − 16, respectively) in intergenic/antisense expression-positive MIRs. It is interesting to compare these results with those previously reported for Alus.17 K562 cells were found to have a higher number of Alu loci bound to components of the Pol III machinery complex than HeLa cells, thus suggesting a more permissive environment for Pol III association in K562 cells. The lower percentage of Pol III-associated elements among intronic sense-oriented expression-positive MIRs suggests that their expression is Pol II-dependent in most cases.
Interestingly the MIR antisense to POLR3E, expressed in all 7 cell lines, is the one bound by the highest number of Pol III TFs in both HeLa and K562 cells (BDP1, BRF1, TFIIIC-110, RPC155) thus further confirming its genuine character of Pol III transcription unit.
3.5. Association of expression-positive MIRs with TFs
In order to assess whether the upstream region of MIR elements could influence their transcription through TFs specifically interacting with it, we took advantage of the availability of ChIP-Seq data for several TFs within ENCODE datasets. We asked if intergenic/antisense MIRs identified as expressed through our pipeline tend to be associated with one or more Pol II TFs, in addition to the known components of the Pol III machinery. The results of this analysis are reported in detail in Supplementary Table S2. Highly variable TFs associations were observed among the 7 cell lines, both in terms of the total number of TF-bound MIRs (ranging from 7 to 21) and in terms of the number of TFs associated to each MIR, mostly depending on the number of available TF ChIP-seq data for each cell line (ranging from 3 in NHEK cells to 100 in K562 cells). Among the Pol II-related transcription proteins most significantly associated with expression-positive intergenic/antisense MIRs we found RNA polymerase II itself (POLR2A), TBP, MAX, MAZ, and YY1 (Supplementary Table S3). HeLa, HUVEC and NHEK cell lines are those with the lowest number of TFs being measured by ChIP-seq and therefore have very low numbers of intergenic/antisense expression-positive MIRs bound by TFs and with no significant enrichment.
3.6. Expression-positive MIRs and chromatin states
A recent study, showing that MIR elements are highly enriched in the enhancer state of chromatin in K562 and HeLa cell lines9 prompted us to investigate whether our intergenic/antisense expression-positive MIRs found in the same cell lines were among those found by this study. We intersected the coordinates of our expression-positive MIRs with those of the MIR elements found to be enriched in enhancers (lifted over from NCBI36/hg18 to GRCh37/hg19), but we found only one overlapping MIR among the fully intronic sense-oriented dataset. We also tested for enrichment in the enhancer states of expression-positive intergenic/antisense MIRs by using the ENCODE Chromatin State Segmentation data. A Fisher’s exact test was performed against all the other annotated intergenic/antisense MIRs used in the pipeline. We found significant enrichment only in Gm12878 and HUVEC cell line in strong enhancer state (P values 6.35e − 06 and 1.08e − 05, respectively) (Supplementary Table S5). Extending the test to the fully intronic sense-oriented expression-positive MIRs, we did not find any significant enrichment in any of the seven cell lines.
Considering the results from another study15 which shows that MIR sequences provide insulators in the genome of CD4+ T cells, we verified if any of our expression-positive MIRs found in all the seven cell lines analysed were among the MIR-derived insulators, but we found only three overlaps. Again, we decided to check if our expression-positive MIRs were enriched in the insulator state of chromatin but we found very few not statistically significant (using Fisher’s exact test) overlaps between the coordinates of our expression-positive MIRs, both intergenic/antisense (no overlaps) and fully intronic sense-oriented (one in GM12878, two in H1-hESC, three in HepG2, and two in NHEK cell lines), and those of genomic regions marked as insulators in ENCODE Chromatin State Segmentation by HMM data (data not shown).
3.7. In vitro transcription analysis of expressed MIR elements
Our bioinformatic pipeline permitted us to detect in vivo expression of individual MIR elements that could be transcribed by the Pol III machinery. To confirm Pol III transcription and make a precise promoter characterization of these transcriptional units, we conducted their in vitro transcription in a HeLa nuclear extract. We focused our attention on a small subset of MIR loci, which are expressed in at least three cell types (listed in Table 3). One of them is MIR_dup2285 (chr16:22309780-22309939), expressed in all the seven investigated ENCODE cell lines and associated with at least 3 components of the Pol III machinery in both HeLa-S3 and K562 cells (see Fig. 3C and Supplementary Table S2). Another locus subjected to in vitro transcription analysis, MIR_dup3493 (chr1: 34943459-34943727), was found to be expressed in four cells lines (GM12878, H1-hESC, K562 and NHEK) and has A- and B- boxes perfectly matching the consensus.22 The remaining two in vitro tested loci (MIRb_dup5848 chr2:71762977-71763215 and MIRc_dup2189 chr14:89445565-89445634) were found to be expressed in at least three cell types (GM12878, H1-hESC, HepG2, NHEK and H1-hESC, K562, NHEK respectively). Based on ENCODE ChIP-seq data, only MIRc_dup2189 is associated with two components of the Pol III machinery (RPC155 and BDP1).
The above MIRs were transcribed in vitro using a HeLa cell nuclear extract in the presence of α-amanitin (2 µg/ml) to completely inhibit RNA polymerase II activity (Fig. 5). Transcription reactions were also programmed in parallel with two mutant versions of each MIR element: one in which the B-box internal promoter element was inactivated by site-specific mutagenesis, and the other in which the upstream flanking region was deleted. Control transcription reactions were set with empty pGEM-T-Easy plasmid (Fig. 5, lanes 1 and 10) and the same vector carrying either a previously characterized Alu (AluSq2 chr1:61523296–61523586)17 (lanes 2 and 11) or a tRNAVal (AAC) gene (TRNAV18, chr6) (lanes 3 and 12) whose transcription produces three different primary transcripts (of 87, 112 and 142 nt) because of heterogeneous termination at one of three consecutive termination signals.19 Each of the tested MIR elements produced a distinct pattern of transcription, in which the sizes of the most abundant transcripts agreed with those predicted on the basis of sequence inspection of the Pol III termination signals, either canonical (a run of at least four Ts) or non-canonical, both internal and in the 3′-flanking region (see Table 3). In particular, the MIR_dup2285 transcription unit has a predicted non-canonical terminator (T3CT) ∼200 bp downstream the 3′ end of the annotated element, as well as two strong early terminators, along with another non-canonical terminator, that seems to be skipped by the Pol III machinery (Supplementary Fig. S1). The first strong terminator (T4) is located within the annotated element 121 nt from the start coordinate, while the other one (T4), located 240 nt downstream of the 5′ end, is downstream of the annotated element but still within the corresponding expected full-length MIR. As shown in Figure 5 (lane 4) all four transcripts, corresponding to the four termination signals, are identified during in vitro transcription, supporting the hypothesis of terminator skipping by Pol III. However it is worth noting that the most abundant signal arises from the transcript ending at the first strong Pol III terminator, suggesting a reduced tendency of the Pol III machinery complex to skip terminators of this MIR in vitro than in vivo (see profile in Fig. 3C). A fifth transcript, migrating between 112 nt and 142 nt markers, is also identified as likely corresponding to a transcript ending at the first strong terminator but using an alternative A-box (see Table 3).
The global transcription outputs of the four MIRs were roughly comparable (cf. lanes 4, 7, 13, and 16) indicating that their varying expression levels in cultured cells are not due to differences in cis-acting elements recognized by the basal Pol III transcription machinery. When the MIR B-box was mutationally inactivated (by substituting CG for the invariant TC dinucleotide of the B box consensus sequence GWTCRAnnC), a clear reduction in MIR transcription efficiency was observed in each case, thus confirming the importance of this element for MIR transcription. Remarkably, however, in no case was transcription abolished by B box inactivation. This was most evident for MIR_dup2285, whose B box-independent transcription was only 2.4-fold less than full transcription (cf. lanes 4 and 16). These data demonstrate that MIRs are efficiently transcribed by Pol III and confirms a key role for B box recognition by TFIIIC, even though the appreciable levels of residual transcription observed with B box-mutated MIRs provide evidence of a key role of A box (and possibly of 5′-flanking region) as a core promoter element in the assembly of the basal transcription machinery, even in the absence of a B box, as previously observed for other Pol III-transcribed genes in yeast.23
To understand if upstream regions influence MIR transcription as they do in the case of Alus,17 we compared the in vitro transcriptional activity of the four isolated MIRs with that of the corresponding 5′-deletion mutants. As shown in Figure 5, upstream sequence deletion negatively affected transcription to different extents for the different MIRs. Transcription of upstream deleted MIRs was reduced by 1.8- to 2.5-fold in the case of MIR_dup2285, MIR_dup3493 and MIRb_dup5848 (cf. lanes 4, 7, and 13 with lanes 6, 9 and 15, respectively), while it was only moderately reduced (∼1.3-fold) in the case of MIRc_dup2189 (cf. lanes 16 with 18). Overall these data reveal that MIR transcription is generally influenced by the upstream region, albeit at a lower extent than Alu transcription.
Discussion
This work provides, for the first time, a comprehensive account of transcriptionally active MIR loci in human cells and identifies MIR-derived transcripts representing novel ncRNAs in which a MIR fragment is fused with a MIR-unrelated, unique sequence. It also confirms the existence of ubiquitously transcribed MIR elements that could play a role in the regulation of their Pol II-transcribed overlapping genes.
The analysis of publicly available ENCODE RNA-seq datasets using an improved bioinformatic pipeline derived from our previous work17 allowed us to define the Pol III-driven MIR transcriptome at single locus resolution using a search strategy that worked well especially for intergenic/antisense MIRs, whose coverage expression profiles where less biased than in the case of intronic sense MIRs by signals arising from sequencing of unknown ncRNA or hnRNA introns.
While a few previous studies on MIR elements focused on their genomic role as regulatory sequences, none of them tried to asses MIR expression in vivo, presumably because the only known role of MIR transcripts was that of retrotransposition and they ceased to amplify ∼130 myr ago.2 The only previous experimental evidence of MIR transcription by Pol III in mammals consists of the results of genome-wide localization analyses of the Pol III machinery in human cells11,12 and in mouse.13,14 Our results reveal that MIR elements, although retrotranspositionally inactive, are widely transcribed and thus contribute to the ncRNA transcriptome of human cells.
Pol III transcription of MIRs occurs extensively both in intergenic regions and within introns of Pol II genes both in antisense and sense orientation. Pol III thus generates a MIR transcriptome composed mainly of cell-specific transcripts and, to a lesser extent, of a tiny subset of MIR-derived RNAs expressed in more than one cell line. Many of the observed MIR-derived transcripts arise from complete MIR elements that are transcribed along their entire length. However, we also found frequent cases of expressed MIRs corresponding to the left or right portion of the canonical full-length MIR whose transcription gives rise to chimeric RNAs composed of a MIR portion fused with a MIR-unrelated unique sequence from the genomic moiety downstream or upstream of the annotated MIR fragment. The genuine, Pol III-dependent character of expression-positive intergenic/antisense MIRs identified by our analysis is supported by their statistically significant enrichment of associated Pol III components within ENCODE ChIP-seq datasets for HeLa-S3 and K562 cells.
As previously observed for Alus, we confirm the ability of the Pol III machinery to access SINE elements located in genomic regions transcribed by Pol II, both in the sense and antisense orientation, suggesting the possibility that Pol III-dependent MIR transcription might contribute to the regulation of the overlapping Pol II-transcribed genes. On the other hand, the high percentage of expression-positive MIRs within introns of both protein-coding and noncoding Pol II genes in antisense orientation (see Table 1) might explain their significant association with components of the Pol II machinery (see supplementary Tables S2–S3) and strengthens the notion of a close, functionally relevant association of Pol II and its TFs with Pol III genes.24
Particularly intriguing in this respect is the MIR element located within the first intron of POLR3E gene. This MIR was found to be expression-positive in all the seven cell lines we analyzed, and its intronic location within this gene appears to be conserved in mouse and other mammals (data not shown). Furthermore, its Pol III transcription is conserved at least in mouse.14 This MIR produces a chimeric transcript containing a MIR-unrelated downstream moiety, and its Pol III transcription is confirmed by the high number of associated Pol III components (see Fig. 3C). Since the POLR3E gene codes for a subunit of Pol III (RPC5), it is tempting to speculate that a MIR-mediated negative autoregulation occurs at this locus, whereby Pol III transcribes the MIR element whose transcription in turn downregulates Pol III abundance through reduced Pol II-dependent expression of its RPC5 subunit.14
In an effort to elucidate basic features of MIR transcription mechanism and control, we studied the in vitro transcription properties of four expression-positive MIR loci, and we could establish three distinctive features of Pol III-dependent MIR transcription: (i) the importance of B-box promoter recognition by TFIIIC, but also the possibility of some levels of B box-independent MIR transcription; (ii) the influence of 5′-flanking sequence on MIR transcription; (iii) the strong epigenetic control of MIR expression in vivo, allowing for cell type-specific expression of MIRs containing equally strong basal promoters. Of the four MIRs, whose transcription properties were analyzed in vitro in this study, all showed a marked reduction of transcription ranging from 2.4 to 5.2-fold following mutation of the B-box promoter. Moreover three of them exhibited a 1.8 to 2.5-fold reduction of transcription, while only one was almost unaffected, upon deletion of the 5′-flanking region. The impact of the 5′-flanking region on MIR transcription efficiency is less marked than in the case of Alus17 thus suggesting the possibility that the MIR internal promoter is intrinsically stronger.
The appreciable levels of residual transcription upon B box disruption, together with the modest reduction of transcription upon deletion of the 5′-flanking sequence, suggests that the assembly of the Pol III transcription machinery on MIRs might take place through alternative pathways, involving either the 5′ flank-A box pair or the A box-B box pair of cis-acting elements. We also cannot exclude that other factors, such as Pol II, TBP, and YY1, which were found to be enriched at expression-positive MIR loci, could take part in Pol III transcription of MIR elements in vivo.
The idea of an epigenetic control used by the cell to domesticate transposable elements has been widely accepted and used to explain their overall low expression levels despite their genomic abundance (reviewed in25,26). Recently, SINE silencing has been proposed to more strictly depend on histone methylation rather than DNA methylation.27 This in vivo epigenetic silencing can be deduced from the same in vitro transcription levels of all the four cloned MIRs, whose expression profiles completely differ from each other in cell lines. Epigenetic control could also be responsible for the higher rate of strong terminator signals skipping by Pol III whose ability is only partially maintained in vitro.
Until now, MIRs have been studied from a genomic perspective while no efforts have been carried out to obtain insight into their actual in vivo expression and the role of Pol III machinery in their transcription. The present work reveals that these ancient transposable elements that have lost their retrotranspositional activity million years ago, are still widely transcribed in human cells. Their genome-wide expression profiling at single locus resolution represents another step toward the understanding of the mechanisms through which the cells have domesticated them, as well as of the possible roles played by both the act and the products of MIR transcription. The differential expression in different cell types of a large fraction of expression-positive MIR elements is suggestive of a strong epigenetic control which is specific to each cell type and condition, thus opening the possibility to exploit MIR expression profiling to monitor and discover patterns of epigenomic alterations that may accompany pathological states. The ever-increasing availability of RNA-seq datasets for many cell types in different physiological and pathological states represents a valuable source of information for our bioinformatic pipeline to assess, as anticipated for Alu RNAs,28 MIR RNA profiles and their possible roles as a novel type of highly specific molecular signature of diseases.
Supplementary Material
Acknowledgements
This work used computational and storage services associated with the Hoffman2 Shared Cluster provided by UCLA Institute for Digital Research and Education’s Research Technology Group. We thank Martin Teichmann (IECB, Pessac, France) for HeLa cell nuclear extract. This work was supported by Italian Association for Cancer Research (AIRC, Grant IG16877 to G.D.).
Conflict of interest
None declared.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
References
- 1.Jurka J., Zietkiewicz E., Labuda D. 1995, Ubiquitous mammalian-wide interspersed repeats (MIRs) are molecular fossils from the mesozoic era, Nucleic Acids Res., 23, 170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smit A.F., Riggs A.D. 1995, MIRs are classic, tRNA-derived SINEs that amplified before the mammalian radiation, Nucleic Acids Res., 23, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vassetzky N.S., Kramerov D.A. 2013, SINEBase: a database and tool for SINE analysis, Nucleic Acids Res., 41, D83–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batzer M.A., Deininger P.L. 2002, Alu repeats and human genomic diversity, Nat. Rev. Genet., 3, 370–9. [DOI] [PubMed] [Google Scholar]
- 5.Terai Y., Takahashi K., Okada N. 1998, SINE cousins: the 3'-end tails of the two oldest and distantly related families of SINEs are descended from the 3' ends of LINEs with the same genealogical origin, Mol. Biol. Evol., 15, 1460–71. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert N., Labuda D. 1999, CORE-SINEs: eukaryotic short interspersed retroposing elements with common sequence motifs, Proc. Natl. Acad. Sci. USA, 96, 2869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalei M. B., Korotkov E. V. 2001, Evolution of MIR Elements Located in the Coding Regions of Human Genome, Mol Biol (Mosk), 35, 1023–31. [PubMed] [Google Scholar]
- 8.Sironi M., Menozzi G., Comi G.P., et al. 2006, Gene function and expression level influence the insertion/fixation dynamics of distinct transposon families in mammalian introns, Genome Biol., 7, R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jjingo D., Conley A. B., Wang J., Marino-Ramirez L., Lunyak V.V., Jordan I.K. 2014, Mammalian-wide interspersed repeat (MIR)-derived enhancers and the regulation of human gene expression, Mob. DNA, 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schramm L., Hernandez N. 2002, Recruitment of RNA polymerase III to its target promoters, Genes Dev., 16, 2593–620. [DOI] [PubMed] [Google Scholar]
- 11.Canella D., Praz V., Reina J.H., Cousin P., Hernandez N. 2010, Defining the RNA polymerase III transcriptome: Genome-wide localization of the RNA polymerase III transcription machinery in human cells, Genome Res., 20, 710–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oler A. J., Alla R. K., Roberts D.N., et al. 2010, Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors, Nat. Struct. Mol. Biol., 17, 620–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carriere L., Graziani S., Alibert O., et al. 2012, Genomic binding of Pol III transcription machinery and relationship with TFIIS transcription factor distribution in mouse embryonic stem cells, Nucleic Acids Res., 40, 270–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canella D., Bernasconi D., Gilardi F., et al. 2012, A multiplicity of factors contributes to selective RNA polymerase III occupancy of a subset of RNA polymerase III genes in mouse liver, Genome Res., 22, 666–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J., Vicente-Garcia C., Seruggia D., et al. 2015, MIR retrotransposon sequences provide insulators to the human genome, Proc. Natl. Acad. Sci. USA, 112, E4428–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Djebali S., Davis C.A., Merkel A., et al. 2012, Landscape of transcription in human cells, Nature, 489, 101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conti A., Carnevali D., Bollati V., Fustinoni S., Pellegrini M., Dieci G. 2015, Identification of RNA polymerase III-transcribed Alu loci by computational screening of RNA-Seq data, Nucleic Acids Res., 43, 817–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dignam J.D., Lebovitz R.M., Roeder R.G. 1983, Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei, Nucleic Acids Res., 11, 1475–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orioli A., Pascali C., Quartararo J., et al. 2011, Widespread occurrence of non-canonical transcription termination by human RNA polymerase III, Nucleic Acids Res., 39, 5499–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moqtaderi Z., Wang J., Raha D., et al. 2010, Genomic binding profiles of functionally distinct RNA polymerase III transcription complexes in human cells, Nat. Struct. Mol. Biol., 17, 635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barski A., Chepelev I., Liko D., et al. 2010, Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes, Nat. Struct. Mol. Biol., 17, 629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orioli A., Pascali C., Pagano A., Teichmann M., Dieci G. 2012, RNA polymerase III transcription control elements: themes and variations, Gene, 493, 185–94. [DOI] [PubMed] [Google Scholar]
- 23.Guffanti E., Ferrari R., Preti M., et al. 2006, A minimal promoter for TFIIIC-dependent in vitro transcription of snoRNA and tRNA genes by RNA polymerase III, J. Biol. Chem., 281, 23945–57. [DOI] [PubMed] [Google Scholar]
- 24.Raha D., Wang Z., Moqtaderi Z., et al. 2010, Close association of RNA polymerase II and many transcription factors with Pol III genes, Proc. Natl. Acad. Sci. USA, 107, 3639–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ichiyanagi K. 2013, Epigenetic regulation of transcription and possible functions of mammalian short interspersed elements, SINEs, Genes Genet Syst., 88, 19–9. [DOI] [PubMed] [Google Scholar]
- 26.Roy-Engel A.M. 2012, LINEs, SINEs and other retroelements: do birds of a feather flock together?, Front Biosci. (Landmark Ed), 17, 1345–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varshney D., Vavrova-Anderson J., Oler A.J., Cowling V.H., Cairns B.R., White R.J. 2015, SINE transcription by RNA polymerase III is suppressed by histone methylation but not by DNA methylation, Nat. Commun., 6, 6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carnevali D., Dieci G. 2015, Alu expression profiles as a novel RNA signature in biology and disease, RNA Dis., 2:e735. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.