

Abstract
The study of enterococcal genomes has grown considerably in recent years. While special attention is paid to comparative genomic analysis among clinical relevant isolates, in this study we performed an exhaustive comparative analysis of enterococcal genomes of food origin and/or with potential to be used as probiotics. Beyond common genetic features, we especially aimed to identify those that are specific to enterococcal strains isolated from a certain food-related source as well as features present in a species-specific manner. Thus, the genome sequences of 25 Enterococcus strains, from 7 different species, were examined and compared. Their phylogenetic relationship was reconstructed based on orthologous proteins and whole genomes. Likewise, markers associated with a successful colonization (bacteriocin genes and genomic islands) and genome plasticity (phages and clustered regularly interspaced short palindromic repeats) were investigated for lifestyle specific genetic features. At the same time, a search for antibiotic resistance genes was carried out, since they are of big concern in the food industry. Finally, it was possible to locate 1617 FIGfam families as a core proteome universally present among the genera and to determine that most of the accessory genes code for hypothetical proteins, providing reasonable hints to support their functional characterization.
Keywords: Enterococcus, food, genomics, niche, probiotics
1. Introduction
The genus Enterococcus (E.) belongs to the lactic acid bacteria (LAB) group and includes nearly 50 species characterized for being facultative anaerobic, non-spore forming and catalase-negative.1,2 They can be associated with humans, animals, plants, water and fermented foods or used as probiotics. Their application in the production of artisanal fermented products such as cheeses, meats and olives, inter alia is founded on their contribution in the typical taste and flavor of the final products as well as on their hygienic quality.1,3 The genus has also the potential to promote health by stimulating the immune system, assisting digestion and maintaining normal intestinal microflora and is thus frequently used in probiotic preparations.4,5 Despite the aforementioned, they are also known for their potential to cause disease being the antimicrobial resistance transferability and virulence phenomena of paramount importance.6
The advent of next-generation technologies has allowed the number of sequenced bacterial genomes to multiply and consequently, has given the possibility of carrying out numerous comparative studies seeking to establish patterns of behavior and evolutionary relationships.7 Tettelin et al.8 introduced the concept of pan-genome. This term refers to the global gene repertoire of a group of organisms consisting of a core genome (genes present in all members of the group) and a dispensable genome (unique and accessory genes present in one or more than one but not all organisms, respectively).8 Numerous studies evidenced that the gene content of a genome and it structure can be affected during organism’s adaptation to a specific niche9 and are the consequence, between others, of processes like horizontal gene transfer (HGT), genome rearrangement and gene loss. For the bacterial family Vibrionaceae, Kahlke et al. showed that unique core genes appear more often in groups of isolates with a common ancestor (monophyletic) and, in the particular case of Vibrio cholerae, play a fundamental role in its adaptation to the ecological niche. Likewise, they indicated that in genophyletic groups of isolates (with no closest common ancestor) core genes are mainly the result of HGT.9 In Pseudomonas putida, meanwhile, the presence of genomic islands carrying genes related to it specific lifestyle pointed out HGT as the driving force of it adaptation to a certain niche.10 In relation to LAB, Douillard and de Vos described that the enormous diversity in the members of this group, as evidenced by comparative genomics studies, is the result of genome-environment interactions. The availability of nutrients in a specific habitat have a direct impact on metabolic properties of LAB. Many species present an enrichment in genes encoding for transporters (ABC or PTS systems) to compensate the loss of genes of biosynthetic pathways.11
The study of enterococcal genomes has grown considerably in recent years with special attention in comparative genomic analysis among clinical relevant isolates. In this sense, several studies have focused on the comparison of the safety aspects of clinical and non-clinical E. faecium and E. faecalis strains12 but, to our knowledge, Enterococcus species of food origin have not been investigated in detail so far, what is surprising given the abundance of these microorganisms in certain fermented food.
Through an exhaustive comparative genomic analysis the goal of our manuscript was to identify common genetic features, features that are specific to enterococcal strains isolated from a certain food related source as well as those present in a species-specific manner. Furthermore, since several studies have shown that foodborne enterococci have a great potential as probiotics,13,14 Enterococcus strains with probiotic properties were also incorporated to the analysis in spite of having or not food as natural niche. In this way, we sought to evaluate, at the genomic level, which were the shared traits between probiotics and food related strains. For this, the genome sequences of 25 Enterococcus strains, from 7 different species, who were the only one with full information about the isolation source by the time of data collection, were selected. Their phylogenetic relationship was determined at nucleotide and amino acid level comparing whole genome alignments and orthologous proteins, respectively. In addition, a search for markers associated with a successful colonization (bacteriocin genes and genomic islands) and genome plasticity (phages and clustered regularly interspaced short palindromic repeats) was performed. Since antibiotic resistance is of big concern in the food industry, the presence of antibiotic resistance genes was also investigated.
2. Materials and methods
2.1. Genome sequences and annotation
According to their isolation source and probiotic potential 25 enterococcal genomic sequences, belonging to seven different species, were retrieved from the NCBI database from December 2014 to July 2015; these included: E. faecalis, E. faecium, E. mundtii, E. hirae, E. malodoratus, E. raffinosus and E. durans. In a next step, the strains were assigned to one of three major groups: dairy isolates, meat isolates and probiotics; and a fourth group was created: probiotics with dairy origin, for those strains with both properties. The dataset thus comprises 3 complete and 22 draft genomes.
In order rely on a directly comparable annotation of genes, we performed a functional annotation combined with a metabolic reconstruction using the RAST (Rapid Annotation Subsystem Technology) webservice.15 For running the analyses the following settings were made: RAST for gene calling, FIGfam Release 70 as the collection of protein families, backfilling of gaps and automatic fixing of errors. Besides rRNAs and tRNAs, RAST identifies protein coding genes and allowed us to achieve a high quality assessment of the gene functions and their genomic context.
2.2. Phylogenetic analysis
2.1.1. Whole genome-based phylogenetic relationships
Whole genomes of all enterococcal species and strains including Lactococcus lactis subsp. cremoris MG1363, Lactococcus garvieae Lg2, Listeria monocytogenes HCC23 and Lactobacillus johnsonii NCC 533 as outgroups were aligned using progressive Mauve.16 All reported alignment blocks present in at least two genomes were concatenated. As using all species and strains at once partially lead to weak bootstrap scores (<10%, probably due to much missing data for incomplete genomes, data not shown), the tree was constructed iteratively in two steps. At species level, we substituted species with multiple strains by a single representative strain that had a fully sequenced genome, namely E. faecalis str. Symbioflor 1 for E. faecalis, E. faecium NRRLB-2354 for E. faecium and E. mundtii ATCC882 for E. mundtii. The resulting 8,602,893 nt long multiple alignment was used in a rapid bootstrap analysis with RAxML v8.1.2017 with 1000 replicates to search for best-scoring maximum likelihood tree with respect to the GTR substitution model and the Gamma model of rate heterogeneity.18 We performed the same analysis at strain level separately for each species resulting in multiple alignments of lengths 3,336,408 nt for E. faecalis, 4,304,502 nt for E. faecium and 5,683,060 nt for E. mundtii. In the latter alignment, E. malodoratus had to be added as phylogenetic trees cannot be reconstructed from three instances (strains) only. It was removed from the resulting tree after reconstruction.
2.1.2. Orthologous proteins-based phylogenetic relationships
We used Proteinortho v5.1.1219 to determine universally conserved 1:1-orthologs in all species and strains (including the four outgroup species as above) with respect to the proteins annotated through RAST (E-value: 1e−10, algebraic connectivity:0). Groups containing any paralogs were removed. Hence, the resulting 455 orthologous groups contained highly similar proteins present once in all species of interest. Amino acid sequences of orthologous proteins were aligned using ClustalOmega v1.2.1.20 The resulting alignment was cropped at both ends till leading and trailing gaps were removed. All alignments were concatenated to single sequences representing one species/strain each resulting in a multiple sequence alignment of 134,771 aa. Based on these, the phylogenetic tree was constructed using RAxML v8.1.2017. A rapid bootstrap analysis with 1000 replicates to search for best-scoring maximum likelihood tree with respect to the LG matrix and the Gamma model of rate heterogeneity18 was performed. To make sure we did not observe effects due to over-representation of some species (E. faecium is present with 13 strains in the dataset comprising 29 genomes) the phylogenetic reconstruction was repeated using only one representative strain for species with multiple strains as described above. Their tree topologies were equivalent (data not shown) indicating no such effect.
3. Niche-specific proteins and core proteome
We directly used the FIGfam annotation classes provided by RAST to group predicted proteins according to their function. We want to note that these classes usually encapsulate multiple instances of different proteins (e.g. paralogs) and thus provide a rough approximation of function rather than reflecting fine-grained homology relations. For each enterococci group (dairy, meat, probiotics) and combination of groups (e.g. dairy and meat and so on), we set a filter such that only those classes remain that are present in at least a given percentage of the respective group but in no species/strain of any other group. Thereby, the two strains that are probiotics but were extracted from a dairy origin, E. faecium L-3 and E. faecalis MB5259, were dynamically assigned to either group (e.g. to dairy when the dairy group was checked). In order to avoid species specific results, we removed all genes solely found in strains of a single species.
Given the amount of incomplete genomes (missing data) and diversity of seven different species it was not surprising that no niche specific genes could be located when applying a presence threshold of 100–60%. At 50% a number of specific gene classes were located but not for dairy and the meat/probiotic combination. We decreased it further to 40% in order to recover specificities of all groups under this study. As statistical evaluation we repeated the analysis 1000 times with random group labels (fixed group sizes) and counted how often a gene family would be reported specific for any group. The results were used as expectation values (E-values), e.g. FIG00632402 was reported 26 times in random 1000 runs and therefore is represented with an E-value of 0.026.
4. Genomic features
4.1. Bacteriocin and antibiotic resistance genes
Bacteriocin-coding genes were searched using BAGEL 321. Mining for modified and non-modified bacteriocins was carried out following two different approaches, one based on the analysis of the gene context, and the other on the identification of the gene itself. Since the gene coding for Enterocin A, entA, could not be located by this program, it was manually assessed by BLAST22 using the sequence firstly described by Aymerich et al.23 (GI: 1296521) as a query and the enterococcal genomes as the database (E-value ≤ 8e−100).
Furthermore, a search for antibiotic resistance genes was performed through the ARG-ANNOT tool.24 For this a BLAST22 in Bioedit25 against the ARG-ANNOT database (April 2015, http://en.mediterranee-infection.com/article.php?laref=23%26titre=arg-annot) was carried out. We used moderately stringent conditions (E-Value ≤ 1e−5), the Blosum 62 substitution matrix26 and allowed for gapped blast hits. To reduce redundancy, only those results with ≥ 70% of query coverage and ≥ 90% sequence identity were taken into account. The antibiotics classes included in the database are: aminoglycosides, beta-lactamases, fosfomycin, fluoroquinolones, glycopeptides, macrolide-lincosamid-estreptogramin, phenicols, rifampicin, sulfonamides, tetracyclines and trimethoprim.
4.2. Prophage sequences and CRISPRs—Cas systems
Intact and incomplete prophage regions were identified through the integrated search and annotation tool PHAST.27 This involved an ORF prediction and translation with GLIMMER 3.0228, a BLAST-based annotation (using a non-redundant bacterial protein library), a tRNA and tmRNA sites localization for attachment sites recognition (tRNAscan-SE29 and ARAGORN30), a phage sequence identification (BLAST against a custom phage/prophage sequence database available at http://phast.wishartlab.com/Download.html) and a density calculation of gene clusters via DBSCAN.27 Only intact regions were analyzed in depth.
Clustered regularly interspaced short palindromic repeats (CRISPRs) were predicted with the CRISPRFinder web service.31 In this way, the highly conserved regions known as direct repeats (DR) (23–55 bp long) and spacers (0.6–2.5 the repeated length) were localized. Putative CRISPR cassettes having at least three motifs (DR + spacer) and a minimum of two identical DRs were considered as confirmed CRISPRs, while the remaining candidates were named as questionable. CRISPRFinder databases are available at http://crispr.u-psud.fr/crispr/(Database status: Last update 2014-08-05).
4.3. Genomic islands
IslandViewer 332 was used for the identification of genes of probable horizontal origin. For localizing genomic islands (GEIs) this tool integrates three different methods: IslandPath-DIMOB,33 SIGI-HMM34 and IslandPick.35 The first two predict GEIs considering the sequence composition, while the latter uses a comparative genomics approach.36,37 For further analysis, the islands predicted for at least one method that were completely included in a contig (in the case of draft genomes), were taken into account. Next, in order to find which GEIs are the same in different organisms we run Proteinortho (all vs all blastp of protein sequences encoded on each GEI) with default options but omitting the final clustering step (algebraic connectivity: 0, E-value ≤ 1e−10). Based on the resulting graph, we extracted the pairwise amount of shared proteins for the respective GEIs and plotted them in a heatmap using the gplots R-package.31 Thereby predicted paralogs could be assigned to a single gene in another GEI (e.g. if one GEI has two paralogs that are orthologous to a gene in another GEI, then both have a partner in this GEI resulting in a similarity of 2/2 = 100% in one direction and 1/1 = 100% vice versa). All islands that did not share at least 50% of their proteins with any other island were removed as were pairs of GEIs with less than four proteins. For sake of simplicity, the names of species were cropped to the end of the strain name followed by the number of the strain-specific genomic island. Thus 19116-i1 means E. faecalis 19116 genomic island 1. All groups of homologous GEIs present in at least four strains were investigated in more detail.
5. Results and discussion
5.1. Overview of the enterococcal genomes
We investigated 25 Enterococcus strains, from 7 different species, that had a food related origin and/or probiotic properties. Among them, a total of 13 E. faecium, 5 E. faecalis, 3 E. mundtii and 1 E. durans, E. hirae, E. malodoratus as well as E. raffinosus strains were included. Based on available data they were next incorporated into four major groups: dairy isolates (11 strains isolated from cheese or milk environment), meat isolates (6 strains of meat origin), probiotics (7 strains known for their ability or potential to be used as probiotics) and dairy isolates and probiotics (2 strains which were probiotics and had a dairy origin as well). In order to avoid erroneous conclusions, the members of the latter group were dynamically assigned to either group dairy isolates or probiotics when comparing.
General genomic features of all the strains analyzed in this study are provided in Table 1. No significant differences (P ≤ 0.05, Kruskal–Wallis statistical test) were observed between the groups in terms of the average number of genes and proteins (RAST annotation as stated above), genome size and GC content. Genome sizes ranged from approximately 2.5–4.6 Mb and GC contents from 36.8 to 39.9%. In agreement with our results, genomic similarities were previously described among E. faecalis strains irrespective of geographical, host temporal or clinical/non-clinical origin of the isolates.38 Singularly, the number of protein encoding genes and ncRNAs detected in E. malodoratus ATCC 43197 (dairy origin) and E. raffinosus CFTRI 2200 (probiotic) draft genomes were the highest, and this was in accordance with their genome sizes that were about 1.6 Mb bigger than the average size of the genomes of the rest of the organisms under study. Interestingly, the strains with the highest GC% in each group were those with a higher number of coding DNA sequence (CDS) in the respective group (E. faecium E1613 for meat isolates).
Table 1.
Relevant genomic features and isolation source of the enterococcal species under study
| Origin/feature | Organism | Genbanka accession Nº | Status | Sizeb (Mbp) | GC% | Genes (NCBI) | Proteins (NCBI) | CDS (RAST) | Source |
|---|---|---|---|---|---|---|---|---|---|
| Dairy isolates | Enterococcus durans IPLA 655 | AOSM00000000.1 | Contig | 3.1 | 37.7 | 2941 | 2789 | 2978 | Cheese |
| E. hirae INF E1 | JMIG00000000.1 | Scaffold | 2.8 | 36.8 | 2501 | 2404 | 2537 | Cultured milk | |
| E. faecium CRL1879 | AOUK00000000.1 | Contig | 2.9 | 37.7 | 2931 | 2616 | 3118 | Artisanal cheese | |
| E. faecium E1604 | AHXD00000000.1 | Scaffold | 2.9 | 38.3 | 2823 | 2749 | 2826 | Cheese | |
| E. faecium NRRL B-2354 | CP004063.1 (chromosome) | Complete | 2.8 | 37.85 | 2771 | 2669 | 2837 | Milk and dairy utensils | |
| CP004064.1 (plasmid) | |||||||||
| E. faecium UC7256 | AWWM00000000.1 | Contig | 2.8 | 38.1 | 2699 | 2507 | 2878 | Pannerone cheese | |
| E. faecium UC7267 | ASAM00000000.1 | Contig | 2.7 | 37.9 | 2674 | 2549 | 2702 | Fontina cheese | |
| E. faecium UC8668 | AWWN00000000.1 | Contig | 2.9 | 38.1 | 2814 | 2667 | 2899 | Cheese | |
| E. malodoratus ATCC 43197 | ASWA00000000.1 | Scaffold | 4.6 | 39.9 | 4581 | 4499 | 4524 | Gouda cheese | |
| E. mundtii ATCC 882 | ASWC00000000.1 | Scaffold | 3.1 | 38.4 | 2986 | 2902 | 2915 | Dairy products | |
| E. mundtii CRL1656 | AFWZ00000000.1 | Contig | 3.1 | 38.4 | 2785 | 2651 | 2765 | Stripping milk of an argentinean cow | |
| E. mundtii CRL35 | JDFT00000000.1 | Contig | 2.9 | 38.3 | 2851 | 2757 | 2759 | Artisanal cheese | |
| Meat isolates | E. faecalis 2924 | AIIV00000000.1 | Scaffold | 3.0 | 37.5 | 2835 | 2788 | 2819 | Turkey meat |
| E. faecalis 19116 | AIIT00000000.1 | Scaffold | 3.0 | 37.3 | 2852 | 2797 | 2816 | Pork meat | |
| E. faecium E1613 | AHXE00000000.1 | Scaffold | 2.9 | 38.2 | 2881 | 2801 | 2849 | Fish burguer | |
| E. faecium UC7251 | ASAL00000000.1 | Contig | 2.7 | 37.9 | 2598 | 2480 | 2680 | Fermented sausage | |
| E. faecium UC8733 | AWWO00000000.1 | Contig | 2.7 | 37.9 | 2628 | 2491 | 2748 | Fermented sausage | |
| E. faecium UC10237 | AWWP00000000.1 | Contig | 2.6 | 37.9 | 2477 | 2390 | 2525 | Llama sausage | |
| Probiotics | E. faecium L-X | JRGY00000000.1 | Contig | 2.7 | 38.3 | 2641 | 2442 | 2728 | Probiotic preparation |
| E. faecalis Symbioflor 1 | HF558530.1 | Complete | 2.8 | 37.7 | 2810 | 2705 | 2724 | Stool specimen of a healthy human adult | |
| E. faecalis PC1.1 | ADKN00000000.1 | Contig | 2.8 | 37.6 | 2661 | 2560 | 2612 | Pooled fecal sample collected from healthy human | |
| E. faecium T-110 | CP006030.1 (chromosome) | Complete | 2.7 | 38.46 | 2639 | 2512 | 2653 | Not specified | |
| CP006031.1 (plasmid) | |||||||||
| E. raffinosus CFTRI 2200 | ATKJ00000000.2 | Contig | 4.2 | 39.4 | 4129 | 4016 | 4180 | Infant fecal material | |
| Dairy isolates and probiotics | E. faecium L-3 | JRGX00000000.1 | Scaffold | 2.6 | 38.0 | 2559 | 2367 | 2646 | Probiotic preparation |
| E. faecalis MB5259 | JMEC00000000.1 | Contig | 3.1 | 37.5 | 3051 | 2734 | 3180 | Dairy origin |
All Accession numbers are from genbank.
Genomes sizes were calculated as the total length of contigs for each genome.
5.2. Phylogeny
The phylogenetic relationships of all strains were determined using two independent approaches. First, a genome based phylogenetic tree was built obtaining two main groups at species level (Fig. 1A). One of them, the most ancient, included the E. malodorous and E. raffinosus species represented by the strains E. malodoratus ATCC 43197 and E. raffinosus CFTRI 2200, respectively; and the other was comprised by the rest of the species under study: E. faecalis, E. mundtii, E. durans, E. hirae and E. faecium. In the latter group, E. faecalis formed a separate branch at the very beginning, and later in evolution, the same was observed first with E. mundtti and then with E. faecium. For their part, E. durans IPLA 655 and E. hirae INF E1 clustered together having with E. faecium the last common ancestor. This topology is in line with that of the 16S rRNA tree presented in Bergey’s manual.39 In the same way, the relationships observed when comparing a 1,102-bp fragment of the gene encoding the alpha subunit of ATP synthase atpA, from 91 Enterococcus strains, were in agreement with the phylogeny inferred from whole genomes in our study.40 According to Naser et al.40 this gene has an optimal discriminatory power that enables enterococcal species differentiation.
Figure 1.

Phylogenetic relationships among food isolated and probiotic enterococci. Strains belonging to different groups are indicated on the bottom left. As outgroups Lactococcus lactis subsp. cremoris MG1363, Lactococcus garvieae Lg2, Listeria monocytogenes HCC23 and Lactobacillus johnsonii NCC 533, were selected. The scale bar indicates the relative distance of sequences. Bootstrap support values are indicated at the nodes. A) Genome-based phylogenetic tree. Genomes were aligned using pMauve and phylogeny reconstructed using a rapid bootstrap analysis with 1000 replicates with RAxML in two steps. First representative strains for each species were used to retrieve the species topology then the procedure was repeated at strain level to maximize resolution here. B) Protein-based phylogenetic tree. The tree is based on all 1:1-orthologs identified with Proteinortho. As in A, a rapid bootstrap analysis with 1000 replicates to search for best-scoring maximum likelihood tree was performed using RAxML. For details, see Materials and Methods.
To provide an in-depth phylogenetic reconstruction we iterated the procedure at strain level. The E. faecium strains were separated into two major subgroups of which one also exhibited a more diverged subgroup comprising the two probiotic and very similar strains L-X and T-110. These results are in agreement with those reported by Palmer et al.41 which describe the E. faecium strains separation into two clades using orthologous groups but without including the two probiotic strains used here. In addition, non-clinical E. faecium strains clustered into two clades according to an analysis carried out by Kim et al.12
In a second approach, we used protein sequences commonly shared in the species and strains under this study (Fig. 1B). Both analyses are in good agreement. However, in contrast to the genome based analysis, E. faecalis is reported as the most ancient branch of enterococci followed by the E. raffinosus and E. maloduratus cluster. As both trees branch with fairly high bootstrap values (0.96 and 0.86) a different evolutional pattern at nucleic and amino acid level can be assumed, leading to this result. Similarly, we find minor differences in the topology of the E. faecium group. We want to note that these findings were highly reproducible in all genome- and protein-based bootstrap runs. As for the genome-based tree, E. mundtti branched separately before E. faecium who shared it last ancestor with E. durans IPLA 655 and E. hirae INF E1.
Both approaches led to the conclusion that non correlations between isolation source/probiotic properties and phylogenetic signal exist neither at species or strain levels. This indicates that niche specific adaptations do not affect the genome and proteome as a whole. In this direction, and analyzing strains of the same species individually, it was only seen that E. faecalis 2924 and 19116 from meat on the one hand, and the probiotic strains E. faecium L-X and E. faecium T-110, but not L-3, on the other hand, clustered together. We thus suppose adaptations to be located in the part of the proteome and genome that is not commonly shared among all strains of a certain species.
5.3. Functional annotation
As a result of the functional annotation conducted with RAST it was possible to determine the features, assigned to subsystems that were (i) present in all organisms, (ii) related to the isolation source or strain property and (iii) species-specific (Fig. 2, Dataset 1). Among the strains of diary origin the average number of annotated protein encoding genes (PEGs) was 2,968; 2,739 for the meat isolates and 2,960 for the probiotics. In addition, it was seen that, for the former group, an average of 46.64% of the identified CDS were assigned to subsystems, 48.50% in case of the strains isolated from meat products and 49.29% for probiotics (Fig. 2A). Of those CDS assigned to subsystems, carbohydrates metabolism was the most enriched metabolic category. This is in accordance with the enterococcís ability to grow in different environments motivated by their capability of metabolizing at least 13 different kinds of sugar.42 Over 30 more can be utilized by at least two members of the genus. These carbohydrates sources are not limited to monomers, they are also usable as polymers.42 The second highest percentage of PEGs was in the protein metabolism category for E. faecium, E. hirae and E. mundtii, strains some of which share the dairy environment as source of isolation; meanwhile for E. malodoratus, E. raffinosus and E. faecalis species it was in the amino acids and derivatives category (Fig. 2B). In this regard, the Lactobacillus adaptation to the dairy niche has been associated with an increase in genes for peptide transport and hydrolysis.43
Figure 2.

Coding sequences dispensed in subsystems and their distribution in categories of metabolic function. A) Percentage of predicted CDS assigned and not assigned to subsystems. B) Distribution of metabolic functions in categories based on the subsystems features counts.
None of the features of a certain functional role were exclusive of the members of one of the three major groups (dairy isolates, meat isolates, probiotics) under consideration (Dataset 1). This observation was valid when taking into account the 100, 60 and 40% of the members of each group for the respective comparison. However, it should be emphasized that 115 of a total of 254 subsystems analyzed were present in all the organisms and can thus be regarded as basic equipment of this genus. The number of CDS for the beta-glucoside metabolism was the most abundant in all groups (Dataset 1). Two beta-glucosides operons were present in all strains, the Bgl (beta-glucoside) and Cel (cellobiose) operons. The genes alsS and alsD of the alpha-acetolactate operon, encoding for the acetolactate synthase and alpha-acetolactate decarboxylase, respectively, were also found in all the Enterococcus strains; both genes are involved in acetoin production from piruvate.32 Seventy-four out of the 115 subsystems were highly conserved as there was a difference of ≤ 1 in the average number of features assigned to the strains of the different groups (Dataset 1). Interestingly, a higher number of genes of the beta-glucoside metabolism, lactose and galactose uptake and utilization, and fructose utilization subsystems were found, in most cases, in the dairy isolates and in probiotics; meanwhile, meat isolates had a greater abundance of genes involved in the oxidative stress response. Dairy bacteria had, at the same time, on average more CDS related to chitin and N-acetylglucosamine utilization than the member of the other groups and probiotics, for their part, were in general more enriched in genes coding for the ABC transporter alkylphosphonate and the energy-coupling factor transporters.
In parallel, an analysis aimed at evaluating the existence of species-specific genomic fingerprints sheds more conclusive results. Specifically, for those species that included more than one strain, it was seen that E. faecalis, for instance, had several features nucleated in subsystems of five different categories, some of them part of the lactate and ethanolamine utilization machinery. Ethanolamine utilization is considered a survival advantage for bacteria that inhabit the gastrointestinal tract and was also related to pathogens virulence.44 E. faecium strains had, on the other hand, genes for the late competence event, fundamental for exogenous DNA acquisition, not localized in any other species; and E. mundtii strains had CDS of the carotenoids subsystem, responsible of their particular color, and one gene encoding a NG, NG-dimethylarginine dimethylaminohydrolase 1.
5.4. Niche-specific proteins and core proteome
We used the RAST annotation to determine genes of certain classes that could be located in a niche specific pattern and that were present in all species, irrespective of their origin (Fig. 3, Supplementary Table S1, Data set 2). The least specific group in this regard was represented by the strains isolated from meat (all belonging to E. faecium or E. faecalis). This group exclusively exhibited the protein IbrB, a co-activator of gene expression which is most likely derived from prophage, and a methyltransferase that modifies adenine bases of the 18S rRNA (in eukaryotes). Both genes must thus be results of horizontal gene transfer related to the environment. On the other hand, most genes specific to dairy and probiotic strains were hypothetical proteins. In the dairy group, these were most pronounced as homologs were located in strains of four different species. Among others, this included an E. faecium strain, a species that is otherwise present in all three groups (dairy, meat, probiotics), indicating these hypothetical proteins to be highly niche specific adaptations, however, of unknown function. Interestingly, one of the niche-specific genes codes for a membrane transport protein that passively transfer molecules or ions through the cell membrane and could thus be related to the peculiarities of the dairy environment. For probiotic strains we have additionally located a putative DNA methyltransferase. Homologs are mainly known from the Haemophilus genus.45
Figure 3.
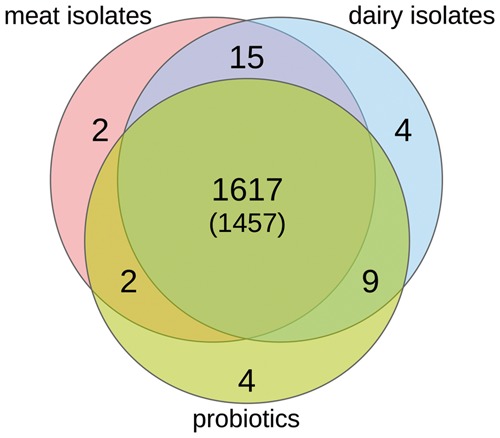
Venn diagram showing i) the number of unique FIGfam protein classes that were present in at least 40% of the strains and in at least two different species of the respective group but in no species/strain of any other group (unique), ii) the number of classes that were similarly present in two or more groups (overlapped), iii) the number of protein classes that were similarly present in all groups (in the center of the diagram), and iv) the subset of classes that were found to be present in all groups and in at least four different Enterococci species rather than two (indicated in parentheses).
Looking at the overlap of shared genes, the meat and probiotic group had the least in common. Above of a phage lysin found in both groups, the fumarate reductase probably represented a true functional finding. Fumaric acid is a food additive used for antimicrobial preservation of meat to reduce the spread of pathogens.46 With respect to this background, it is reasonable to conclude that the respective strains from the group are able to degrade this acid in their habitat (meat isolates). Probiotic strains, on the other hand, might be in contact with fumaric acid in the gastrointestinal habitat, while it does not play a role for strains from the dairy group.
The overlap of the dairy and the probiotic groups can also be seen as set of genes that were not present in the meat group. It contained three phosphonate ABC transporter proteins, all found linked in the same strains, indicating that phosphonates play an important role here but not in meat related environments. In addition two proteins related to galactoside metabolism were located. Both were reported with an E-value slightly above the 0.05 significance threshold (0.076). Again, this energy-source is obviously not important for meat related strains. Alongside with galactoside related proteins, a not further specifiable membrane transporter was identified to be present. Similarly the alpha-glycosyltransferase was found solely in the dairy and probiotic groups.
With 15 shared proteins, the overlap of the meat and the dairy group seems rather large. Most of them, however, were related to phages (six) or were hypothetical proteins (five) often exhibiting weaker E-values closer to or slightly above 0.05. The remaining specific proteins were the ATP-binding protein p271, an ATPase component of an uncharacterized ABC-type transport system and, most interestingly a probable extracellular solute-binding protein that was present in the same strains as an arabinofuranosidase. Both might thus be related, indicating that arabinose might not be as relevant for probiotic strains.
Finally, we determined the core proteome of all enterococci used in this study by asking which genes were present in 40% of all strains in all groups (see Materials and Methods). As a result, 1617 FIGfam families were located to be universally present. Out of them 1457 were found to be present in at least four different enterococci species. A detailed list can be found in Supplementary Table S1.
6. Genomic features among food-related and probiotic enterococcal strains
6.1. Bacteriocins
Genes encoding bacteriocins are widely disseminated among enterococci from different origins.47 For probiotic bacteria, the ability to successfully outcompete undesired species is often due to, or enhanced by, the production of bacteriocins.48 With this background, a search for genes related to bacteriocins was carried out through BAGEL 3 and BLAST (Table 2).
Table 2.
Number of antibiotic resistance and bacteriocin genes in the enterococcal genomes
| ARG for each antibiotic class |
Bacteriocin class |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | Strain | AGly | Gly | MLS | Tet | Tmt | ∑ | I | II | III | ∑ |
| Dairy isolates | IPLA 655 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| INF E1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | |
| CRL 1879 | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 6 | 1 | 7 | |
| E1604 | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 1 | 1 | 2 | |
| NRRL B-2354 | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 6 | 1 | 7 | |
| UC7256 | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 0 | 1 | 1 | |
| UC 7267 | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 0 | 1 | 1 | |
| UC8668 | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 0 | 1 | 1 | |
| ATCC 43197 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| ATCC 882 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 2 | |
| CRL1656 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | |
| CRL35 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | |
| Meat isolates | 2924 | 0 | 7 | 4 | 2 | 1 | 14 | 0 | 0 | 0 | 0 |
| 19116 | 5 | 0 | 4 | 1 | 0 | 10 | 2 | 1 | 1 | 4 | |
| E1613 | 1 | 0 | 1 | 0 | 0 | 2 | 1 | 4 | 1 | 6 | |
| UC7251 | 3 | 0 | 3 | 2 | 0 | 8 | 0 | 0 | 1 | 1 | |
| UC8733 | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 8 | 2 | 10 | |
| UC10237 | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 1 | 0 | 1 | |
| Probiotics | PC1.1 | 0 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 1 | 1 |
| Symbioflor 1 | 0 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | |
| L-X | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 3 | 1 | 4 | |
| T-110 | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 2 | 1 | 3 | |
| CFTRI 2200 | 1 | 0 | 1 | 2 | 0 | 4 | 0 | 1 | 0 | 1 | |
| Dairy isolates and probiotics | L-3 | 1 | 0 | 1 | 0 | 0 | 2 | 0 | 3 | 1 | 4 |
| MB5259 | 0 | 0 | 2 | 0 | 0 | 2 | 2 | 1 | 0 | 3 | |
ARG: antibiotic resistance gene; AGly: aminoglycosides; Gly: glycopeptides; MLS: macrolide-lincosamidestreptogramin; Tet: tetracyclines; Tmt: trimethoprim.
Of all species under examination, E. faecium was the one that harbored the larger number of bacteriocin genes in general, being the meat isolate E. faecium UC8733 the strain with the highest bacteriocinogenic potential with eight genes encoding for class II and two for class III bacteriocins. Reinforcing the idea, in previous studies we could identify six class II bacteriocin gene clusters in E. faecium CRL 187949 coincident with those found in NRRL B-2354, also of dairy origin. Moreover, and in agreement with our previous findings related to non-clinical and clinical E. faecium47, no correlations between the presence of enterocin structural genes and strains origin were observed in the present analysis. Özdemir et al.50 also described the same phenomenon among enterococcal species isolated from different origins (including river, treatment plant, spring and garbage water, soil, animal and vegetables).
Enterocins A and B were always considered as hallmark of this genus. However, as we have shown previously, not all strains carrying enterocin A genes (entA) also harbor a enterocin B gene (entB).49 The first one (entA) was detected in CRL 1879, NRRL B-2354, UC8733, L-3, L-X and UC10237, all belonging to the E. faecium species. Surprisingly, the class III bacteriocin enterolysin A, a bacteriolysin, first characterized in E. faecalis, was not found in all strains belonging to this species. However, this gene was present in all E. faecium belonging to the three different groups (dairy isolates, meat isolates and probiotics) except in E. faecium UC10237 (Supplementary Table S2). Meanwhile, E. faecium E1613 (meat isolate) was the only with a CDS for Subtilosin A. As previously described51,52 E. mundtii CRL35 and E. mundtii CRL1656 (diary isolates) have the CDS for Enterocin CRL35 and Mundticin CRL1656, respectively. According to BAGEL, these were the only bacteriocin encoding genes that they carried within their genome (Supplementary Table S2). Regarding E. faecalis, the strains 19,116 and MB5259 (meat and dairy/probiotics, respectively) were the only harboring genes encoding for Cytolysin structural subunits CylLS and CylLL (Supplementary Table S2). In animal models of enterococcal infections it was observed that Cytolysin contributes to virulence,53 rendering this strain potentially pathogenic. Finally, no CDS related to bacteriocin production were found in the genomes of E. malodoratus ATCC 43197 (incomplete genome) and E. faecalis Symbioflor 1 (complete genome).
6.2. Antibiotic resistance genes
Enterococci have been defined as increasingly resistant to multiple antibiotics in recent years.54 They have an intrinsic resistance to many antibiotics and can also acquire resistance to many others, including those of clinical use. E. faecium and E. faecalis are the most successful strains evolving as multi-resistant pathogens because of their ability to acquire and share adaptive traits, including antimicrobial resistance genes, encoded by mobile genetic elements.55 In this study, genes that conferred resistances to glycopeptides, macrolide-lincosamide-streptogramin, tetracyclines and trimethoprim were detected. The in silico prediction of antibiotic resistance genes revealed that E. faecalis 2924, isolated from turkey meat in 2005, was the only strain carrying genes involved in vancomycin resistance, which is of high clinical importance. At the same time, this was the strain with the highest number of antibiotic resistance genes (14 protein-coding genes, see Table 2 and Supplementary Table S3). This finding is in line with the idea that the increased use of antibiotics in healthcare and animal husbandry is linked with the emergence, spreading and persistence of resistant strains in animal products.56 For example, some enterococci isolated from Portuguese traditional fermented meat products were resistant to erythromycin, nitrofurantoin, rifampicin and tetracycline, but in this case none was to vancomycin.57 With ten resistance genes E. faecalis 19116 was the second in abundance (Table 2). In contrast, strains belonging to E. mundtii, E. durans and E. maloduratus species did not contain antibiotic resistance related CDS at all (Table 3). Remarkably, all of them were isolated from dairy origin indicating that at least in some instance, antibiotic resistances are of minor relevance in dairy environment compared to the other groups where no strain investigated completely lacks antibiotic resistance genes.
Table 3.
Distribution of the intact prophage regions among the enterococcal strains
| Group | Strain | Regiona | Lengthb (Kb) | Nº CDS | GC% | Most common phage (hit genes count)c |
|---|---|---|---|---|---|---|
| Dairy isolates | IPLA 655 | 1 | 40.7 | 55 | 35.4 | Lister 2389 (16) |
| 2 | 44.8 | 64 | 36.8 | Entero phiEf11 (13) | ||
| 3 | 16.5 | 27 | 35.1 | Bacill G (2) | ||
| INF E1 | 1 | 38.8 | 54 | 33.4 | Entero phiFL1A (10) | |
| CRL 1879 | 1 | 52.5 | 61 | 37.5 | Entero EfaCPT1 (11) | |
| E1604 | 1 | 37.6 | 51 | 36.8 | Entero IME EF4 (11) | |
| NRRL B-2354 | 1 | 56.8 | 59 | 36.64 | Lactob phig1e (12) | |
| 2 | 46 | 58 | 34.96 | Lactob phig1e (13) | ||
| UC7256 | 1 | 48.7 | 54 | 36.8 | Lactob phig1e (12) | |
| UC 7267 | 1 | 31 | 36 | 38.5 | Bacill BCJA1c (10) | |
| UC8668 | 1 | 25.8 | 31 | 36.8 | Lactob phig1e (8) | |
| 2 | 47 | 59 | 36.1 | Lister 2389 (15) | ||
| ATCC 43197 | — | — | — | — | — | |
| ATCC 882 | 1 | 46.5 | 66 | 36.7 | Bacill BCJA1c (10) | |
| CRL1656 | — | — | — | — | — | |
| CRL35 | 1 | 38.5 | 50 | 36.3 | Lactob PL 1 (13) | |
| 2 | 44.4 | 68 | 37.3 | Lactoc Tuc2009 (13). Entero phiEf11 (13) | ||
| 3 | 44.1 | 53 | 37.6 | Bacill BCJA1c (9) | ||
| Meat isolates | 2924 | 1 | 36.8 | 46 | 34.5 | Strept phi3396 (8). Lister LP 101 (8) |
| 19116 | 1 | 68.8 | 72 | 36.2 | Entero EFC 1 (38) | |
| E1613 | 1 | 37.1 | 50 | 35.4 | Lister 2389 (14) | |
| UC7251 | 1 | 70.1 | 64 | 35.9 | Lister 2389 (16) | |
| UC8733 | 1 | 31.9 | 35 | 37.9 | Entero IME EF4 (12) | |
| UC10237 | 1 | 21.8 | 23 | 37.5 | Lister B025 (5) | |
| Probiotics | PC1.1 | 1 | 21.6 | 27 | 36.8 | Lister LP 101 (9) |
| Symbioflor 1 | 1 | 41.6 | 54 | 35.83 | Entero phiEf11 (12). Temper phiNIH1 1 (12) | |
| 2 | 45.2 | 66 | 34.63 | Entero phiEf11 (63) | ||
| L-X | 1 | 25.5 | 30 | 34.4 | Bacill BCJA1c (7) | |
| 2 | 41.9 | 55 | 36.1 | Lactoc Tuc2009 (13). Entero phiEf11 (13). Lactoc TP901 1 (13) | ||
| T-110 | 1 | 41.9 | 56 | 36.16 | Lactoc Tuc2009 (13). Entero phiEf11 (13) | |
| CFTRI 2200 | 1 | 50.1 | 71 | 37.1 | Entero phiFL3A (18) | |
| 2 | 18.8 | 32 | 40.5 | Entero EFC 1 (3) | ||
| Dairy isolates and probiotics | L-3 | — | — | — | — | — |
| MB5259 | 1 | 30.8 | 22 | 33 | Lactob phiAT3 (2) | |
| 2 | 43.9 | 68 | 34.4 | Entero phiEf11 (63) | ||
| 3 | 42.5 | 60 | 34.5 | Entero phiFL1A (15) | ||
| 4 | 35.3 | 55 | 35.2 | Entero phiFL3A (13) | ||
| 5 | 41.7 | 31 | 37 | Staphy StauST398 4 (2) |
Intact prophage region.
Intact prophage region length.
Phage with the highest number of CDS in the region and the number og gene counts in brackets.
Genes related to resistance to macrolide-lincosamide-streptogramin (msrC) and aminoglycosides (aac (6')-Ii) were found in all E. faecium strains but not in E. faecalis (Supplementary Table S3). The aac(6′)-Ii gene, is species-specific and encodes for a chromosomal aminoglycoside acetyltransferase specific for E. faecium strains.58 The 6′-N-aminoglycoside acetyltransferases [AAC (6′)s] are of particular interest because they can modify a number of clinically important aminoglycosides and some of them are often present in integrons, transposons, plasmids, genomic islands and other genetic structures. AAC (6′)-I-type enzymes effectively acetylate amikacin but not gentamicin. To date 45 such genes have been characterized.59 On the other hand, msrC encodes a putative efflux pump of the ABC transporter family and it seems that the presence of this gene confers an advantage for the members of this species.58 Similarly, the genes lsaA and mphD were found exclusively in the E. faecalis strains (Supplementary Table S3). Lsa gene, encodes a putative ABC protein that confers resistance to quinupristin-dalfopristin and clindamycin60 and is located in the chromosome. The gen mphD codes for a phosphorylase and belongs to the group of inactivating enzymes61 (Supplementary Table S3). The meat isolate E. faecium UC7251, unlike the other strains of this species, additionally had genes for resistance to antibiotics of the tetracyclines class, apart from the MLS resistance genes ermB and lnuB, and aminoglycosides resistance genes aph3-III and sat4A (Supplementary Table S3).
6.3. Prophage sequences and CRISPRs—Cas systems
Lysogenic phages are an important source of information related to bacterial pathogenesis.62 Comparative genomic analysis evidenced that polylysogeny is a common phenomenon in the Enterococcus genus.62 Particularly, in E. faecalis V583 temperate phages constitute the main source of horizontally acquired DNA representing nearly 10% the size of the genome.62
Within the scope of this study we search for prophage-like sequences in the genomes. In this sense, as a result of the analysis performed with PHAST, we evidenced in all strains but E. mundtii CRL1656, E. malodoratus ATCC 43197 and E. faecium L-3, several intact regions related to different prophages associated with the Lactobacillus, Lactococcus, Listeria, Bacillus, among other genera (Table 3). The temperate bacteriophages EFC-1 and PhiEf11, originally described in E. faecalis, had the highest number of CDS. PhiEf11 contained 63 from a total of 68 phage CDS in the probiotic strain MB5259, and EFC-1 38 CDS from a total of 72 in the meat isolate 19116. At the same time, the φEf11 phage was present in organisms belonging to four different species: E. durans IPLA 655, E. faecalis MB5259, E. mundtii CRL35, E. faecalis Symbioflor 1, E. faecium L-X and E. faecium T-110. Additionally, E. faecalis MB5259, a strain with probiotic properties and a dairy origin had a total of five intact phage related regions with CDS from different phages (Table 3).
It is interesting to note that all strains from meat origin harbored only one intact prophage region and that E. faecium UC7251 contained the largest intact phage-related region (70.1 kb) with the bacteriophage Lister 2389, isolated from Listeria monocytogenes strain Scott A,63 having 16 from a total of 64 CDS located in that region. In E. faecalis 2924 and PC1.1 another Listeria phage, LP-101, was located. Interestingly, this phage was first obtained from silage samples collected on a dairy farm.64 Then Listeria phage B025 related coding sequences were localized on the intact region of E. faecium UC10237 (Table 3). Bacillus phage BCJA1c had the highest protein counts in UC7267, ATCC-882, CRL35 and L-X, but none in those meat related strains and Lactococcus phages related proteins were the most abundant in E. mundtii CRL35 and E. faecium L-X intact phage regions (Table 3).
Related to the acquisition of phages and other mobile elements, enterococci that possess CRISPR-Cas systems, are less likely to acquire phages and other mobile elements.65 We found that E. raffinosus CFTRI 2200, with four individual gene cassettes, was the strain with the highest number of these prokaryotic immune systems among all under evaluation (Table 4). At the same time, one of these four CRISPRs, with a size of 1215 bp, was the largest identified. E. faecium UC7256 and E. hirae INF E1, both dairy isolates, had two confirmed CRISPRS systems each. Regarding meat isolates, E. faecalis 2924, E. faecium E1613 and E. faecium UC7251, had confirmed clusters with five, three and three spacers, respectively. All members of the probiotics group but E. faecium T-110 had confirmed CRISPR systems (Table 4). Among the confirmed CRISPR systems of all organisms DR sizes ranged from 23 to 37 bp and the number of spacers from 3 to 18.
Table 4.
Confirmed CRISPR structures in the enterococcal genomes predicted with CRISPRFinder
| Group | Strain | Confirmed CRISPR | Lengtha | DR lengthb | Nº of spacers |
|---|---|---|---|---|---|
| Dairy isolates | IPLA 655 | — | — | — | — |
| INF E1 | 1 | 300 | 37 | 4 | |
| 2 | 683 | 37 | 9 | ||
| CRL 1879 | — | — | — | — | |
| E1604 | 1 | 199 | 24 | 3 | |
| NRRL B-2354 | — | — | — | — | |
| UC7256 | 1 | 199 | 24 | 3 | |
| 2 | 201 | 28 | 3 | ||
| UC7267 | — | — | — | — | |
| UC8668 | 1 | 201 | 28 | 3 | |
| ATCC 43197 | — | — | — | — | |
| ATCC 882 | — | — | — | — | |
| CRL1656 | — | — | — | — | |
| CRL35 | 1 | 227 | 28 | 3 | |
| Meat isolates | 2924 | 1 | 365 | 36 | 5 |
| 19116 | — | — | — | — | |
| E1613 | 1 | 199 | 24 | 3 | |
| UC7251 | 1 | 199 | 24 | 3 | |
| UC8733 | — | — | — | — | |
| UC10237 | — | — | — | — | |
| Probiotics | PC1.1 | 1 | 365 | 36 | 5 |
| Symbioflor 1 | 1 | 629 | 36 | 9 | |
| L-X | 1 | 199 | 24 | 3 | |
| T-110 | — | — | — | — | |
| CFTRI 2200 | 1 | 484 | 23 | 6 | |
| 2 | 221 | 27 | 3 | ||
| 3 | 1215 | 30 | 18 | ||
| 4 | 537 | 24 | 8 | ||
| Dairy isolates and probiotics | L-3 | 1 | 199 | 24 | 3 |
| MB5259 | 1 | 431 | 36 | 6 |
Total length of the CRISPR cassette.
Length of the repeated sequences.
Certain enterococcal lineages have recently emerged as one of the main causes of hospital infection outbreaks around the world. These are characterized by abundant mobile DNA, including phages, plasmids and transposons that carries multiple antibiotic resistances. Palmer et al. found that antibiotic resistance and possession of complete CRISPR loci are inversely related and that members of recently emerged high-risk enterococcal lineages lack complete CRISPR loci suggesting that antibiotic therapy inadvertently selects for enterococci with compromised genome defense.65 However, we cannot confirm these findings with our results regarding food-related strains. Especially for dairy isolates we frequently found no confirmed CRISPR system but also no antibiotic resistance genes (IPLA 655, ATCC 43197, ATCC 882, CRL1656). Vice versa the probiotic isolate CFTRI 2200 was heavily armed both with four confirmed CRISPR systems and at least four antibiotic resistance genes (see Tables 2 and 4), observations that are opposite to the findings described for the strains of clinical relevance.
Interestingly, the analysis of the gene context of all confirmed CRISPR systems identified by CRISPRFinder evidenced the existence of CRISPR associated genes (cas) upstream the CRISPR cassette only in INF E1 (confirmed CRISPR #2) and CFTRI 2200 (confirmed CRISPR #3). The former has genes exclusive of a Type II CRISPR-Cas system subtype II-A, while the latter related to a Type I CRISPR-Cas system subtype I-B.66,67 These results suggest that the systems detected in the other strains might not be functional or their specific cas related genes are located further than 2000bp upstream or downstream the CRISPR cassette.
On the other hand, No confirmed CRISPR system was detected in CRL1879, NRRL B-2354, UC7267, 19116, UC8733 and UC10237 strains.
6.4. Genomic islands
Horizontal gene transfer (HGT) is one of the phenomena responsible of the diversification and adaptation of microorganisms, and has a direct impact on the genome plasticity and size; it is believed that the majority of the HGT has been facilitated by genomic islands (GEIs).68 In this respect, a detailed analysis of the enterococcal genomes allowed us to identify many GEIs and to correlate them according to the similarity of the encoded CDS (Fig. 4). From the GEI detected, all groups found in at least five different strains were further evaluated. With the exception of the GEI number 5, all four remaining islands were E. faecium-related to the major extend. A correlation with respect to the isolation origin cannot be observed nor can an exclusive occurrence in strains of a certain species (Supplementary Table S4).
Figure 4.

Heat map showing the percentage of shared proteins between the genomic islands identified with IslandViewer3. All islands with at least 50% shared proteins to any other island were considered. Dark colour indicates nearly perfect overlaps while white means no detectable relation at all.
GEIs play a crucial role in dissemination of variable genes, such us antibiotic resistance and virulence genes, and can contribute to the generation of the currently well-known named hospital ‘superbugs’. However, none of the five GEIs here evaluated contains CDS related to known pathogenic markers of clinical relevant enterococcal species. With 33 to 36 CDS GEI 1 (Supplementary Table S4a) was the biggest island containing mainly ribosomal proteins and translation factors along with accessory proteins for transcription and translation and protein secretion (e.g. RNA polymerase, Preprotein translocase secY).
In agreement with one of the ideas that considers GEIs an overarching family of elements, as is the case of mobile DNA elements constituted by integrative and conjugative elements (ICEs), conjugative transposons and some prophages, we found for instance integrases in GEI 2 (XerD, Supplementary Table S4b) and GEI 5 (Supplementary Table S4e) and phage-related proteins in GEI 4 (Supplementary Table S4d). It has been previously described that the coding capacity of GEIs is not limited to pathogenicity functions and can be very diverse including such traits as symbiosis, sucrose and aromatic compound metabolism, mercury resistance and siderophore synthesis.69,70 Thereof especially GEIs 2 and 3 (Supplementary Table S4b and c) encoded some interesting CDS. Among others, some examples were the ferric uptake regulation protein (oxidative stress), an enolase (pyruvate metabolism) and a triosephosphate isomerase (sugar metabolism). Finally, the third group of genes was that of hypothetical proteins with unknown function which was highly abundant in GEI 4 and 5 (Supplementary Table S4d and e) rendering any interpretation futile.
7. Conclusion
In this work we performed a detailed comparative analysis of food-related and probiotic enterococcal genomes. As previously demonstrated by our group, bacteriocin genes were widely distributed among E. faecium strains. Interestingly, antibiotic resistance genes were predominant in those organisms of meat origin coincidentally with the notion of the unrestricted use of antibiotics in husbandry. A search for regions enriched in phage related coding sequences evidenced that they were extensively propagated throughout the genus. In dairy isolates, phages derived from bacteria commonly associated to the milk environment, such us Listeria, Lactobacillus and Lactococcus. Interestingly, only two strains carried a complete CRISPR-Cas system (CFTRI 2200 and INF E1) but potential orphan cassettes were localized in the rest of the species. Importantly, five GEIs were found to be the same in different organisms and none of them contained CDS related with known pathogenic markers of clinical relevant enterococcus species.
Our in-depth phylogenetic reconstruction yielded that strains form clusters in accordance to their species but not to their isolation origin or probiotic properties. While we determined a set of 1617 FIGfam families as core proteome universally present among the genera, we were also able to identify several proteins that seem to occur in a strain-overarching but niche-specific pattern. Besides some phage-related regions/genes that seemingly occur in a habitat-dependent fashion, there was a specific membrane transport protein found exclusively in dairy isolates. Moreover, our data indicated that only dairy isolates could be found without any antibiotic resistance genes. All strains from the other groups code for at least one such gene. On the other hand, all dairy isolates were missing a gene coding for fumarate reductase generally present in meat isolates and probiotics. Enterococci isolated from meat origins lacked three phosphonate ABC transporters and two proteins related to galactoside metabolism that were otherwise found in most dairy isolates and probiotics. Moreover, arabinose-related proteins were missing from probiotics. Finally, we located a number of additional niche-specific but hypothetical proteins, a finding that might nevertheless aid in their future functional characterization. In principal, any of these specific genes could be used as indicator for the original habitat of a species making it possible to trace e.g. the origin of a strain that developed pathogenicity.
Based on the above described we can suggest that dairy isolates have greater potential than meat isolates for use as probiotics, at least at the genomic level, and their formal application in the future will require deep analysis including biochemical tests and in vitro as well as in vivo experiments.
Other than that, we also observed niche-specific features at species level. For instance, the antibiotic resistance genes lsaA and mphD were found exclusively in the E. faecalis strains that belonged to the probiotics and meat isolates groups but not in those isolated from dairy. Also, only Enterococcus faecalis strains from meat origin and probiotics had a set of genes for ethanolamine utilization.
Regarding the data, parallel evolution via horizontal gene transfer seems a likely cause for many specific genes reported here. We assume that most were acquired after speciation due to similar environments. This is certainly true for phage-related genes, likely being a result of the environment rather than a prerequisite. Also a number of specific genes were only present in less than half of all representatives of a niche and most were located mainly in E. faecalis and E. faecium strains specific to a certain niche (see Table 2). With respect to the phylogenetic tree (Fig. 1) one would expect them to occur in some species in between as well if they were already present in a common ancestor. There are, however, notable exceptions. Specific genes present in five or even six of the seven enterococci species under this study that occur in a niche-specific manner were likely present in the common ancestor and probably got lost in strains that did not gain advantage of them anymore due to specialization, e.g. to a different niche or habitat. Here we especially refer to the phosphonate ABC transporters and the genes related to galactoside metabolism.
Altogether, our data demonstrated that while a few niche-specific genomic features of enterococci can be identified, relevant genomic idiosyncrasies mainly depend on the species in the first place rather than a specific niche or habitat.
Supplementary Material
Acknowledgement
This study was carried out with the financial support from CONICET (PIP0406/12) and MinCyT (PICT2011 N°0175) from Argentina.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Conflict of interest
None declared.
References
- 1.Foulquie Moreno M.R., Sarantinopoulos P., Tsakalidou E., De Vuyst L. 2006, The role and application of enterococci in food and health, Int. J. Food Microbiol., 106, 1–24. [DOI] [PubMed] [Google Scholar]
- 2.Van Tyne D., Gilmore M.S. 2014, Friend turned foe: evolution of enterococcal virulence and antibiotic resistance, Annu. Rev. Microbiol., 68, 337–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhardwaj A., Malik R.K., Chauhan P. 2008, Functional and safety aspects of enterococci in dairy foods, Indian J. Microbiol., 48, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karaseva A., Tsapieva A., Pachebat J., Suvorov A. 2016, Draft genome sequence of probiotic Enterococcus faecium strain L-3, Genome Announ., 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ermolenko E., Gromova L., Borschev Y., et al. 2013, Influence of different probiotic lactic Acid bacteria on microbiota and metabolism of rats with dysbiosis, Biosci Microbiota Food Health, 32, 41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kristich C.J., Rice L.B., Arias C.A. 2014, Enterococcal Infection-Treatment and Antibiotic Resistance In: M.S. Gilmore, D.B. Clewell, Y. Ike, N. Shankar. (eds), Enterococci: From Commensals to Leading Causes of Drug Resistant Infection, Boston. [Google Scholar]
- 7.Blom J., Albaum S.P., Doppmeier D., et al. 2009, EDGAR: a software framework for the comparative analysis of prokaryotic genomes, BMC Bioinformatics, 10, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tettelin H., Masignani V., Cieslewicz M.J., et al. 2005, Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”, Proc. Natl. Acad. Sci. USA, 102, 13950–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kahlke T., Goesmann A., Hjerde E., Willassen N.P., Haugen P. 2012, Unique core genomes of the bacterial family Vibrionaceae: insights into niche adaptation and speciation, BMC Genomics, 13, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X., Monchy S., Taghavi S., Zhu W., Ramos J., van der Lelie D. 2011, Comparative genomics and functional analysis of niche-specific adaptation in Pseudomonas putida, FEMS Microbiol. Rev., 35, 299–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Douillard F.P., de Vos W.M. 2014, Functional genomics of lactic acid bacteria: from food to health, Microb. Cell Factories, 13, S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim E. B., Marco M. L. 2014, Nonclinical and clinical Enterococcus faecium strains, but not Enterococcus faecalis strains, have distinct structural and functional genomic features, Appl. Environ. Microbiol., 80, 154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbosa J., Borges S., Teixeira P. 2014, Selection of potential probiotic Enterococcus faecium isolated from Portuguese fermented food, Int. J. Food Microbiol., 191, 144–48. [DOI] [PubMed] [Google Scholar]
- 14.Haghshenas B., Haghshenas M., Nami Y., et al. 2016, Probiotic assessment of Lactobacillus plantarum 15HN and Enterococcus mundtii 50H isolated from traditional dairies microbiota, Adv. Pharm. Bull., 6, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aziz R.K., Bartels D., Best A.A., et al. 2008, The RAST Server: rapid annotations using subsystems technology, BMC Genomics, 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darling A.C., Mau B., Blattner F.R., Perna N.T. 2004, Mauve: multiple alignment of conserved genomic sequence with rearrangements, Genome Res., 14, 1394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stamatakis A. 2014, RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies, Bioinformatics, 30, 1312–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le S. Q., Gascuel O. 2008, An improved general amino acid replacement matrix, Mol. Biol. Evol., 25, 1307–20. [DOI] [PubMed] [Google Scholar]
- 19.Lechner M., Findeiss S., Steiner L., Marz M., Stadler P.F., Prohaska S.J. 2011, Proteinortho: detection of (co-)orthologs in large-scale analysis, BMC Bioinformat., 12, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sievers F., Wilm A., Dineen D., et al. 2011, Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega, Mol. Syst. Biol., 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Heel A. J., de Jong A., Montalban-Lopez M., Kok J., Kuipers O.P. 2013, BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides, Nucleic Acids Res., 41, W448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D. J. 1990, Basic local alignment search tool, J. Mol. Biol., 215, 403–10. [DOI] [PubMed] [Google Scholar]
- 23.Aymerich T., Holo H., Havarstein L.S., Hugas M., Garriga M., Nes I. F. 1996, Biochemical and genetic characterization of enterocin A from Enterococcus faecium, a new antilisterial bacteriocin in the pediocin family of bacteriocins, Appl. Environ. Microbiol., 62, 1676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta S. K., Padmanabhan B. R., Diene S. M., et al. 2014, ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes, Antimicrobial Agents Chemother., 58, 212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall T. 2013, BioEdit: biological sequence alignment editor for Win95/98/NT/2K/XP. pp. p 95–98
- 26.Henikoff S., Henikoff J.G. 1992, Amino acid substitution matrices from protein blocks, Proc. Natl. Acad. Sci. USA, 89, 10915–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Y., Liang Y., Lynch K.H., Dennis J.J., Wishart D.S. 2011, PHAST: a fast phage search tool, Nucleic Acids Res., 39, W347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salzberg S.L., Delcher A.L., Kasif S., White O. 1998, Microbial gene identification using interpolated Markov models, Nucleic Acids Res., 26, 544–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowe T. M., Eddy S. R. 1997, tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence, Nucleic Acids Res., 25, 955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laslett D., Canback B. 2004, ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences, Nucleic Acids Res., 32, 11–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grissa I., Vergnaud G., Pourcel C. 2007, CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats, Nucleic Acids Res., 35, W52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dhillon B.K., Laird M.R., Shay J.A., et al. 2015, IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis, Nucleic Acids Res., 43, W104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsiao W., Wan I., Jones S.J., Brinkman F.S. 2003, IslandPath: aiding detection of genomic islands in prokaryotes, Bioinformatics, 19, 418–20. [DOI] [PubMed] [Google Scholar]
- 34.Waack S., Keller O., Asper R., et al. 2006, Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models, BMC Bioinform., 7, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langille M.G., Hsiao W.W., Brinkman F.S. 2008, Evaluation of genomic island predictors using a comparative genomics approach, BMC Bioinform., 9, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhillon B.K., Chiu T.A., Laird M.R., Langille M.G., Brinkman F.S. 2013, IslandViewer update: Improved genomic island discovery and visualization, Nucleic Acids Res., 41, W129–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langille M.G., Brinkman F.S. 2009, IslandViewer: an integrated interface for computational identification and visualization of genomic islands, Bioinformatics, 25, 664–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zischka M., Kunne C. T., Blom J., et al. 2015, Comprehensive molecular, genomic and phenotypic analysis of a major clone of Enterococcus faecalis MLST ST40, BMC Genomics, 16, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ludwing W., S K.-H., Whitman WB. 2009, Family IV. Enterococcaceae Bergey's Manual of Systematic Bacteriology 2nd Ed., Heidelberg: Springer, 600. [Google Scholar]
- 40.Naser S., Thompson F.L., Hoste B., et al. 2005, Phylogeny and identification of Enterococci by atpA gene sequence analysis. J. Clin. Microbiol., 43, 2224–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palmer K.L., Godfrey P., Griggs A., et al. 2012, Comparative genomics of enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. mBio, 3, e00318–00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramsey M., Hartke A., Huycke M. 2014, The Physiology and Metabolism of Enterococci. [PubMed]
- 43.Cai H., Thompson R., Budinich M.F., Broadbent J.R., Steele J.L. 2009, Genome sequence and comparative genome analysis of Lactobacillus casei: insights into their niche-associated evolution, Genome Biol. Evol., 1, 239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fox K.A., Ramesh A., Stearns J.E., et al. 2009, Multiple posttranscriptional regulatory mechanisms partner to control ethanolamine utilization in Enterococcus faecalis, Proc. Natl. Acad. Sci. USA, 106, 4435–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atack J.M., Srikhanta Y.N., Fox K.L., et al. 2015, A biphasic epigenetic switch controls immunoevasion, virulence and niche adaptation in non-typeable Haemophilus influenzae. Nat. Commun., 6, 7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doores S. 2002, pH control agents and acidulants In A. L. Branen, P. M. Davidson, S. Salminen, J. H. Thorngate., III (eds), Food Additives, Marcel Dekker Inc, NY, 988. [Google Scholar]
- 47.Fontana C., Cocconcelli P.S., Vignolo G., Saavedra L. 2015, Occurrence of antilisterial structural bacteriocins genes in meat borne lactic acid bacteria, Food Control, 47, 53–9. [Google Scholar]
- 48.Gillor O., Etzion A., Riley M.A. 2008, The dual role of bacteriocins as anti- and probiotics. Appl. Microbiol. Biotechnol., 81, 591–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suárez N., Hebert E.M., Saavedra L. 2015, Genome mining and transcriptional analysis of bacteriocin genes in Enterococcus faecium CRL1879, J. Data Mining Genomics Proteomics, 6, 1–8. [Google Scholar]
- 50.Ozdemir G.B., Oryasin E., Biyik H.H., Ozteber M., Bozdogan B. 2011, Phenotypic and genotypic characterization of bacteriocins in enterococcal isolates of different sources, Indian J. Microbiol., 51, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonacina J., Saavedra L., Suarez N.E., Sesma F. 2014, Draft genome sequence of the nonstarter bacteriocin-producing strain Enterococcus mundtii CRL35, Genome Announ., 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Magni C., Espeche C., Repizo G.D., et al. 2012, Draft genome sequence of Enterococcus mundtii CRL1656, J. Bacteriol., 194, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shankar N., Baghdayan A. S., Gilmore M.S. 2002, Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis, Nature, 417, 746–50. [DOI] [PubMed] [Google Scholar]
- 54.Togay S.O., Keskin A.C., Acik L., Temiz A. 2010, Virulence genes, antibiotic resistance and plasmid profiles of Enterococcus faecalis and Enterococcus faecium from naturally fermented Turkish foods, J. Appl. Microbiol., 109, 1084–92. [DOI] [PubMed] [Google Scholar]
- 55.Mikalsen T., Pedersen T., Willems R., et al. 2015, Investigating the mobilome in clinically important lineages of Enterococcus faecium and Enterococcus faecalis, BMC Genomics, 16, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giraffa G. 2002, Enterococci from foods, FEMS Microbiol. Rev., 26, 163–71. [DOI] [PubMed] [Google Scholar]
- 57.Barbosa J., Ferreira V., Teixeira P. 2009, Antibiotic susceptibility of enterococci isolated from traditional fermented meat products, Food Microbiol., 26, 527–32. [DOI] [PubMed] [Google Scholar]
- 58.Portillo A., Ruiz-Larrea F., Zarazaga M., Alonso A., Martinez J. L., Torres C. 2000, Macrolide resistance genes in Enterococcus spp, Antimicrob. Agents Chemother., 44, 967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramirez M.S., Tolmasky M.E. 2010, Aminoglycoside modifying enzymes, Drug Resistance Updates, 13, 151–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh K.V., Murray B.E. 2005, Differences in the Enterococcus faecalis lsa locus that influence susceptibility to quinupristin-dalfopristin and clindamycin, Antimicrob. Agents Chemother., 49, 32–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roberts M.C. 2008, Update on macrolide-lincosamide-streptogramin, ketolide, and oxazolidinone resistance genes, FEMS Microbiol. Lett., 282, 147–59. [DOI] [PubMed] [Google Scholar]
- 62.Duerkop B.A., Palmer K.L., Horsburgh M.J. 2014, Enterococcal bacteriophages and genome defense. [PubMed]
- 63.Zimmer M., Sattelberger E., Inman R.B., Calendar R., Loessner M. J. 2003, Genome and proteome of Listeria monocytogenes phage PSA: an unusual case for programmed + 1 translational frameshifting in structural protein synthesis, Mol. Microbiol., 50, 303–17. [DOI] [PubMed] [Google Scholar]
- 64.Denes T., Vongkamjan K., Ackermann H.W., Moreno Switt A.I., Wiedmann M., den Bakker H.C. 2014, Comparative genomic and morphological analyses of Listeria phages isolated from farm environments, Appl. Environm. Microbiol., 80, 4616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palmer K., van Schaik W., Willems R., Gilmore M. 2014. Enterococcal Genomics. In: M. Gilmore, D. Clewell, Y. Ike and N. Shankar, ed., Enterococci From Commensals to Leading Causes of Drug Resistant Infection, 1st ed. [online] Boston, Massachusetts: The Harvard Medical School, pp. 189-216.
- 66.Makarova K.S., Aravind L., Wolf Y.I., Koonin E. V. 2011, Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems, Biol. Direct, 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Makarova K.S., Wolf Y.I., Alkhnbashi O.S., et al. 2015, An updated evolutionary classification of CRISPR-Cas systems, Nat. Rev. Microbiol., 13, 722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Juhas M., van der Meer J.R., Gaillard M., Harding R.M., Hood D.W., Crook D.W. 2009, Genomic islands: tools of bacterial horizontal gene transfer and evolution, FEMS Microbiol. Rev., 33, 376–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Larbig K.D., Christmann A., Johann A., et al. 2002, Gene islands integrated into tRNA(Gly) genes confer genome diversity on a Pseudomonas aeruginosa clone, J. Bacteriol., 184, 6665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sullivan J.T., Trzebiatowski J.R., Cruickshank R.W., et al. 2002, Comparative sequence analysis of the symbiosis island of Mesorhizobium loti strain R7A, J. Bacteriol., 184, 3086–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.