

Abstract
Arterial blood pressure is oscillatory; whether pulse pressure (PP) regulates cerebral artery myogenic tone (MT) and endothelial function is currently unknown. To test the impact of PP on MT and dilation to flow (FMD) or to acetylcholine (Ach), isolated pressurized mouse posterior cerebral arteries were subjected to either static pressure (SP) or a physiological PP (amplitude: 30 mm Hg; frequency: 550 bpm). Under PP, MT was significantly higher than in SP conditions (p < 0.05) and was not affected by eNOS inhibition. In contrast, under SP, eNOS inhibition increased (p < 0.05) MT to levels observed under PP, suggesting that PP may inhibit eNOS. At a shear stress of 20 dyn/cm2, FMD was lower (p < 0.05) under SP than PP. Under SP, eNOS-dependent production contributed to FMD, while under PP, eNOS-dependent NO was responsible for FMD, indicating that PP favours eNOS coupling. Differences in FMD between pressure conditions were abolished after NOX2 inhibition. In contrast to FMD, Ach-induced dilations were higher (p < 0.05) under SP than PP. Reactive oxygen species scavenging reduced (p < 0.05) Ach-dependent dilations under SP, but increased (p < 0.05) them under PP; hence, under PP, Ach promotes ROS production and limits eNOS-derived NO activity. In conclusion, PP finely regulates eNOS, controlling cerebral artery reactivity.
Keywords: Hydrogen peroxide, myogenic tone, NADPH oxidases, nitric oxide, shear stress, acetylcholine
Introduction
Blood flows in an oscillatory waveform, creating a pulse pressure (PP) that imposes circumferential and tangential forces that make up the primary stress experienced by arterial walls.1 The endothelium is the first component that experiences the PP and given its critical role in regulating vascular tone, it is exceedingly likely that its function is modulated by PP, subsequently influencing organ perfusion. Previous reports have identified that PP is inversely correlated with endothelium-dependent forearm vasodilation in never treated hypertensive patients.2 Similarly, increased PP is associated with essential hypertension3 and the stiffening of large peripheral arteries,4 both of which have been demonstrated to be damaging to the low-resistance brain circulation by promoting hypo-perfusion, microvascular lesions, grey and white matter lesions, and ultimately cognitive deficits.5–8 On the other hand, the absence of pulsatile flow has been associated with metabolic acidosis, increased inflammation, decreased oxygen consumption, loss of vasomotor control, impaired capillary circulation and increased bleeding.9–12 So what are the mechanisms by which PP acutely regulates hemodynamics? Carotid baroreceptors are unloaded in non-pulsatile perfusion, leading to an increase in sympathetic nervous system activity and a rise in blood pressure.13–15 Similarly, non-pulsatile clinical cardiopulmonary by-pass increases sympathetic activity and plasma levels of renin, angiotensin II, norepinephrine, endothelin-1, TNFα and ROS, and inadequate synthesis of NO, inflammation and oxidative stress were also reported in the peripheral circulation ex vivo.12,16–22 While PP may modulate systemic neurohumoral control of hemodynamics, a direct regulatory effect on the endothelium is, however, poorly understood. A seminal study by Vanhoutte's group in 1986 showed that pulsatile flow increased endothelium-dependent relaxation of the canine femoral artery.23 This was confirmed by two studies showing that in rat-isolated coronary arterioles24 and mesenteric arteries,22 cellular deformation induced by pressure, stretch and wall shear stress, potentiated the release of NO resulting in dilation, despite an increase in ROS production. However, Gutterman's group recently reported that in pressurized human peripheral arterioles, a severe increase in intravascular pressure for 30 min induced either ex vivo (in static conditions)25 or in vivo (in pulsatile conditions),26 promoted endothelial dysfunction evidenced by reduced eNOS-dependent NO activity and excessive ROS production. Therefore, both a lack of pulsatile pressure and its excess are damaging to the endothelium as they both impair NO-dependent dilations.
In isolated mouse cerebral arteries and under static pressure (SP), we previously reported that eNOS-derived contributes to FMD,27 while others have demonstrated that FMD is mediated by eNOS-derived NO and/or other endothelium-derived relaxing factors.24,28–31 Such a “switch” in eNOS activity from NO to production is referred to as eNOS uncoupling and is usually associated with cardiovascular diseases.32 Nonetheless, it has been shown that nNOS-derived H2O2 accounted for Ach-induced relaxation of aortic rings isolated from healthy C57Bl6 mice.33 NOS uncoupling can therefore be detected ex vivo, in the absence of pulsatile pressure, in arteries isolated from healthy mice. Our aim was to test whether a physiological PP would optimize endothelium-dependent function and myogenic tone by regulating eNOS coupling and activity, in isolated and pressurized healthy mouse cerebral arteries.
Materials and methods
All protocols and procedures followed the guidelines of the Montreal Heart Institute Animal Ethical Committee and performed in accordance with the Guide for the Care and Use of Laboratory Animals of Canada and the Guide for the Care and Use of Laboratory Animals by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Altogether, 72 C57Bl/6j (Jackson Laboratory) three-month old male mice were used in the study; they were housed one per cage in a specific pathogen free animal facility under controlled light and temperature. Four mice were used to acquire a range of baseline values of heart rate (HR) and blood pressure at rest and during voluntary physical activity in vivo. These data were used to set experimental pressure and pulse frequency values for ensuing ex vivo studies. The remaining 68 mice were euthanized under anaesthesia (ketamine, 44 mg/kg, and xylazine, 2.2 mg/kg) and their cerebral arteries harvested for ex vivo studies. From each mouse, four cerebral artery segments were isolated and tested in the different protocols described below. One segment isolated from one mouse was used per protocol. All sections of this report adhere to the ARRIVE Guidelines for reporting animal research.34
Determination of physiological references of blood pressure and heart rate in conscious mice
For this in vivo study, three-month old mice (n = 4) were instrumented with telemetric blood pressure transducers to monitor blood pressure and HR while conscious at rest and during voluntary running, as previously described.35 Briefly, under anaesthesia, the pressure transducer was implanted in the abdominal subcutaneous space and a sterile saline-filled 0.1 French Teflon catheter was tunnelled under the skin, inserted into the carotid artery and connected to the transducer (Data Sciences International, Arden Hills, MN, USA). Analgesia was provided by repeated injections of buprenorphine (0.05 mg/kg) before and two days post-surgery. Twenty-four hour recordings (5 min every 30 min) started after four days of recovery and lasted for a week; they were analysed (EMKA Technologies, Paris, France), and blood pressure and HR calculated. Running wheel activity was monitored in parallel to determine whether recordings were made at rest or during physical activity.
Cerebral artery isolation and cannulation on the pressure arteriograph
After anaesthesia (laboratory, 8 a.m.) and loss of all reflexes, the mouse was decapitated and the brain removed from the cranium and placed in ice-cold physiological salt solution (PSS; mmol/L: 130 NaCl; 4.7 KCl; 1.18 KH2PO4; 1.17 MgSO4; 14.9 NaHCO3; 1.6 CaCl2; 0.023 EDTA; 10 glucose; pH 7.4) aerated with 12% O2; 5% CO2; and 83% N2 at 37℃. Posterior cerebral arteries (PCA) were isolated, transferred to the arteriograph chamber (Living System Instrumentation, St-Alban, VT, USA), cannulated and pressurized (from 20 to 120 mm Hg during myogenic tone measurements; at 60 mm Hg during endothelial function assessment) as previously described.27,35 Arterial diameter was measured and recorded continuously with a video monitoring system (IonOptix, Milton, MA, USA).
Pulse pressure system
We manufactured a computer-controlled piston pump system to propagate pulse waves in an isolated artery mounted on the pressure arteriograph. The system generates PP of controllable frequency and amplitude in the tubing upstream of the arterial segment (Figure S1, Supplementary material). The actuator generates force that transmits to a lever, which functions to compress a fluid filled pipette, thereby displacing a volume of fluid and creating a pulse wave that propagates through the system and the pressurized arterial segment. In this study, we used a frequency of 9.2 Hz (mimicking the normal HR of 550 bpm in conscious mice) at an amplitude of 31.5 ± 0.1 mm Hg (±15 mm Hg, i.e. an imposed intraluminal pressure of 45 to 75 mm Hg), corresponding to the normal carotid PP we determined in conscious mice (see Results section). In one series of experiments, PP was increased to 50 mm Hg without changing the pseudo-diastolic pressure (the mean intraluminal pressure applied was 70 mm Hg instead of the usual 60 mm Hg), i.e. 45–95 mm Hg at 9.2 Hz.
Endothelial function in cerebral arteries
Pressurized 2–3 mm segments of the right and left PCA were studied after 30 min of equilibration at an intraluminal pressure of 60 mm Hg. Myogenic tone was evaluated either under static pressure (SP) or during PP after a 30-min pre-exposure to PP. Endothelial function was assessed (i) under SP; (ii) under SP following 30 min of PP to measure FMD or acetylcholine (Ach)-dependent dilations, or (iii) under PP following 30 min of PP to measure Ach-dependent dilations. For technical reasons, PP was stopped to allow for intraluminal flow for FMD; in contrast, PP was maintained, or not, during Ach stimulation. To validate that eNOS activity was directly responsible for FMD, cerebral arteries were pre-incubated during the equilibration period with Nω-nitro-L-arginine (L-NNA, 100 μmol/L), or denuded of their endothelium by passing an air bubble through the lumen of the artery.27 To target the enzymes generating ROS, arteries were pre-incubated during the equilibration period with PEG-catalase (100 U/mL), a permeable catalase mimetic inactivating H2O2, or with the NOX-2-specific inhibitor gp91ds-tat (10 μmol/L). Before stimulating with Ach or flow, phenylephrine (10−6 mol/L or 3 × 10−6 mol/L) was added to the bath in order to induce a pre-contraction representing a 40–50% reduction from the maximal diameter (Dmax). In separate arterial segments, a single cumulative dose–response curve to Ach (from 10−12 to 3 × 10−5 mol/L) was performed. Similarly, in other arterial segments, a single cumulative curve to flow (from 0 to 20 dyn/cm2, with steps of 2 dyn/cm2 between 0 and 10 dyn/cm2, followed by two steps of 5 dyn/cm2) was performed, at a constant mean pressure of 60 mm Hg. Shear stress (τ, dyn/cm2) was calculated according to [τ = 4ηQ/πr3], where η represents viscosity (0.009 P), Q is the applied flow rate through the lumen (ml/s), and r is the lumen radius (cm). Data are presented as a percentage of dilation for every concentration of Ach and shear stress value. Posterior cerebral arteries were also used to assess the myogenic tone. The internal diameter (D) was measured after each variation of the intraluminal pressure from 20 to 120 mm Hg, by steps of 20 mm Hg maintained for 2 min. Myogenic responses were assessed without flow to eliminate any responses of endothelial cells to shear stress. Myogenic tone for each intraluminal pressure value was calculated according to the following formula: MT (%) = [(Dmax−D)/(Dmax)]×100, where Dmax is the maximum internal diameter measured in passive condition at the end of each experiment, in a calcium-free PSS containing sodium nitroprusside (3 × 10−5 mol/L).
Fluorescence studies
To test the hypothesis that PP modifies eNOS activity from H2O2 to NO production, cerebral arteries were incubated with 10 μmol/L 4,5-diaminoflorescein diacetate (DAF-2, a NO-sensitive fluorescent dye) or with 5 μmol/L 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA, a H2O2-sensitive fluorescent dye), as previously described.27,36,37 Arterial segments were incubated with or without L-NNA (100 μmol/L) or PEG-catalase (100 U/mL) for 30 min, washed three times with PSS, pre-constricted with phenylephrine and then exposed to a shear stress of 20 dyn/cm2; during the dilation, NO- or H2O2-fluorescence was quantified.
Data analysis
“n” refers to the number of animals. Results are presented as mean±SEM. In the absence of treatment in vivo, no blinding was performed. Arterial segments were assigned to an ex vivo protocol pre-determined by the researchers with no deviation and exclusion of data/animals. All individual data were integrated and analysed by two experimenters (AR, VB) and validated by the supervisor of the study (ET). For MT, a two-way ANOVA with repeated measures was followed, if the group×pressure interaction was significant, by a Bonferroni test to analyse the pressure-dependence of the myogenic response; a two-way ANOVA followed by a Bonferroni test was used to compare groups (static and pulsatile pressure) at each given pressure. A two-way ANOVA followed, if group×shear stress or group×Ach was significant, by a Bonferroni test was used for the flow- and Ach-mediated responses. A value of p < 0.05 was considered statistically significant. An independent statistician performed the statistical analyses using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA).
Results
In vivo pressures and heart rate
We first assessed PP amplitude and frequency in vivo in four conscious mice, at rest and during voluntary running. As expected, variability of these parameters was very low, justifying the low number of animals tested. Diastolic (DAP) and systolic arterial pressure (SAP) increased from 85 ± 1 and 113 ± 2 mm Hg to 100 ± 1 and 130 ± 1 mm Hg at rest and during running, respectively. The PP (SAP-DAP) did not differ from rest (28 ± 2 mm Hg) to active (30 ± 1 mm Hg) state. A PP of 30 mm Hg was therefore chosen for ex vivo testing, although in vivo PP in the PCA might be smaller,38 ranging from ∼20 to 25 mm Hg.39 Resting HR averaged 562 ± 11 beats per minute (bpm) [range: 538–589], and we, therefore, selected a pulse modulation frequency of 9.2 Hz, corresponding to 550 bpm, for ex vivo testing. During voluntary running, peak HR averaged 745 ± 6 bpm [range: 729–754]. In summary, DAP, SAP and HR increased by 19%, 15% and 33% during exercise, respectively.
Impact of pulsatile pressure on myogenic tone
In SP conditions, MT was low and poorly responsive to increases in intraluminal pressure (Figure 1(a)); however, MT was significantly (p < 0.05) increased by both L-NNA and PEG-catalase, and developed with rises in pressure (Figure 1(a)), suggesting that NO and/or H2O2 limit MT. In contrast, under PP alone, MT was significantly greater (p < 0.05), but was of similar magnitude to MT measured in SP conditions with either L-NNA or PEG-catalase (Figure 1(b)), indicating that PP inhibits eNOS. The MT that developed under PP conditions was maximal between 60 and 100 mm Hg and was insensitive to L-NNA or PEG-catalase treatment.
Figure 1.
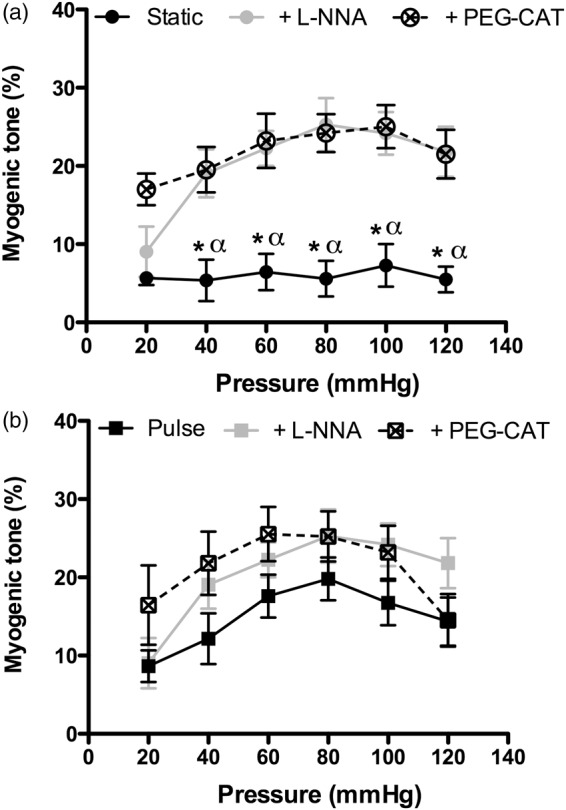
Impact of pulse pressure on myogenic tone regulation in isolated pressurized mouse cerebral arteries, with or without inhibition of eNOS by L-NNA (100 µM) or inactivation of H2O2 by PEG-catalase (PEG-CAT; 100 U/mL) in static (a) or pulse (b) pressure conditions. *p < 0.05 vs. L-NNA and vs. PEG-catalase; α: p < 0.05 vs. myogenic tone measured during pulse pressure. Results are mean±SEM of n = 11 (Static) to 12 (Pulse) mice.
Impact of pulsatile pressure on flow-mediated dilation and eNOS activity
Flow-mediated dilations (FMD) are endothelium- and shear stress-dependent in both SP and PP conditions, as demonstrated by the inhibition of FMD in denuded arteries (Figure 2(a)). In SP conditions, FMD peaked at a shear stress value of 10 dyn/cm2 (Figure 2(a)), while under PP, FMD further increased at 15 and 20 dyn/cm2 (p < 0.05), suggesting that pulsatile pressure increases endothelial shear stress sensitivity. In both SP and PP conditions, inhibition of eNOS with L-NNA fully prevented (p < 0.001) shear stress-induced FMD (Figure 2(b)), demonstrating that eNOS activity is central to FMD. PEG-catalase reduced FMD in SP (p < 0.05), as expected from our previous study,27 signifying that eNOS-derived mediates the dilation. In PP conditions, however, while eNOS inactivation blunted FMD, PEG-catalase treatment did not (Figure 2(b)), suggesting that are not involved under PP and that eNOS produces NO. Therefore, PP may favour eNOS coupling.
Figure 2.

Effects of static pressure (n = 11 mice) and pulse pressure (n = 12 mice) on endothelium-dependent flow-mediated dilation in pressurized (60 mm Hg) mouse cerebral arteries, (a) before or after endothelial denudation (-Endo) and (b) before or after treatment with PEG-catalase (PEG-CAT; static, n = 10 mice; pulse, n = 8 mice) or L-NNA (n = 4 mice). (a) *p < 0.05 vs. Static; α: p < 0.001 vs. with an intact endothelium; (b) *p < 0.05 vs. Static; †p < 0.05 vs. Static in control conditions; α: p < 0.001 vs. untreated conditions. Results are mean±SEM of n mice.
To confirm these results, we measured the fluorescence associated with NO (DAF-2) and H2O2 (DCFDA) release in isolated arteries36,37 during FMD induced by a shear stress of 20 dyn/cm2, in both SP and PP conditions. In all conditions, fluorescence was extinguished by L-NNA confirming the unique involvement of eNOS in FMD (Figure 3). As expected in SP conditions,27,37 FMD was associated with a rise in H2O2-DCFDA fluorescence that was inactivated by PEG-catalase (Figure 3(a) and (c)); FMD was not associated with detectable NO-DAF-2 fluorescence in SP conditions (Figure 3(b) and (d)). In contrast, under PP conditions, FMD was associated with a rise in NO-DAF-2 fluorescence insensitive to PEG-catalase (Figure 3(b) and (f)), and low H2O2–DCFDA fluorescence (Figure 3(a) and (e)). These results support the concept that PP favours eNOS coupling, driving its activity towards NO production rather than .
Figure 3.

Effects of static pressure and pulse pressure on DCFDA-associated H2O2 fluorescence (a, c, e) and DAF2-associated NO fluorescence (b, d, f) during flow-mediated dilation in mouse cerebral arteries before or after treatment with PEG-catalase or L-NNA. Typical recordings of fluorescence (arbitrary units, a.u.) are presented (a, b) during dilation induced by flow (20 dyn/cm2), in pressurized (60 mm Hg) pre-constricted cerebral arteries with phenylephrine (PE). Results are mean±SEM of data collected in arterial segments isolated from 14 mice (n numbers indicated for each of the 12 experimental conditions on the graph). *p < 0.05 as indicated.
Mechanism by which eNOS activity is regulated under PP
Bradykinin-induced dilations of isolated human coronary arteries have been associated with generation and are sensitive to the NOX2 inhibitor gp91ds-tat.40 In our conditions, gp91ds-tat limited FMD under both SP and PP (Figure 4). Therefore, NOX2 recruitment likely contributes to the transduction of the mechanical flow signal to the activation of eNOS, in both static and PP conditions. In addition, combining PEG-catalase and gp91ds-tat did not further antagonize FMD in SP (Figure 4(a)), suggesting that NOX2 contributes to the uncoupling of eNOS. In PP conditions, FMD remained insensitive to PEG-catalase, indicating that eNOS was coupled despite NOX2 inhibition (Figure 4(b)).
Figure 4.
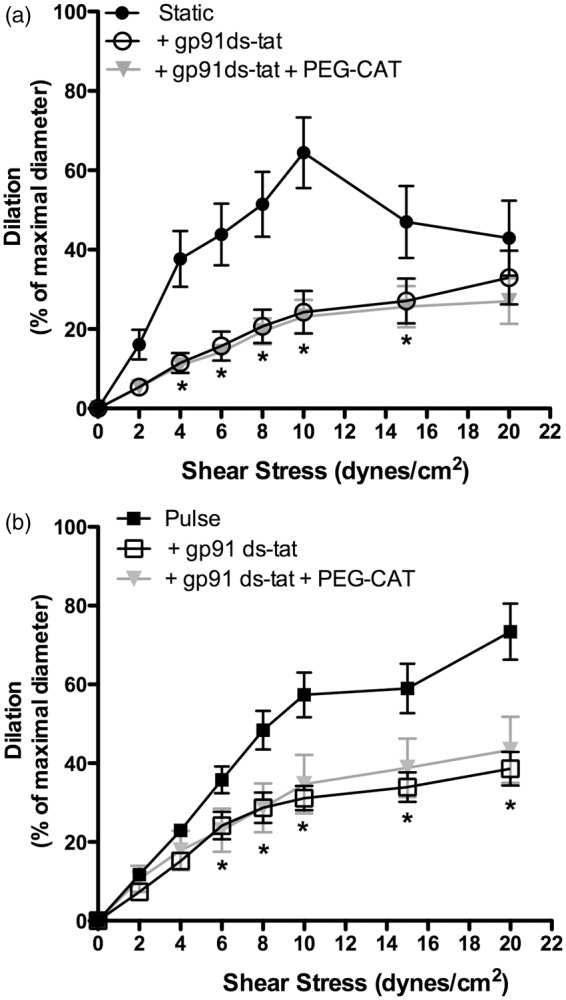
Consequences of NOX2 inhibition with gp91ds-tat (10 µM) combined with or without PEG-catalase (PEG-CAT; 100 U/mL) on flow-mediated dilation in mouse cerebral arteries pressurized in static pressure (a) or pulse pressure (b) conditions. Results are mean±SEM of n mice. *p < 0.05 vs. control conditions (Static or Pulse; n = 6 per group).
Impact of pulsatile pressure on Ach-mediated dilation
Like flow, which elicits a unique endothelial response by transducing a mechanical signal to eNOS activation,41 activation of muscarinic receptors with Ach is a commonly used stimulus to investigate the mechanisms of endothelium-dependent relaxation. In SP conditions, similar to FMD, Ach-mediated dilation was associated with H2O2 production (Figure 5(a)), as previously reported.36 Ach-mediated dilation was indeed significantly reduced by PEG-catalase. However, the muscarinic response (Figure 5) differed from FMD (Figures 2 and 4) with both continuous PP and a pre-conditioning period of PP, like that used to study FMD (Figure S2, Supplementary material). In these conditions (1) maximal Ach-induced dilations were smaller than in SP conditions, contrasting the results of FMD; (2) Ach-induced dilations were increased by PEG-catalase, whereas FMD was insensitive; (3) Ach-induced dilations were increased by the NOX2 inhibitor gp91ds-tat (Figure 5(b)), and the vascular sensitivity to Ach was increased (in opposition to FMD). The data obtained with PEG-catalase and gp91ds-tat suggest that, under PP, Ach-mediated dilation is associated with ROS generation that limits the dilatory response. Because PEG-catalase increased Ach-dilation under PP, likely do not mediate the dilation, and thus muscarinic receptor activation under PP is associated with eNOS-dependent NO production. Therefore, PP promotes eNOS coupling; unlike flow, however, Ach induces the concomitant production of ROS that likely inactivates NO, reducing the potency and efficacy of Ach.
Figure 5.

Influence of pulse pressure on the dilation of isolated and pressurized mouse cerebral arteries induced by acetylcholine (Ach). Experiments were performed in the absence or presence of either (a) PEG-catalase (100 U/mL) or (b) gp91ds-tat (10 µM). Results are mean±SEM of n mice. *p < 0.05 compared to respective control condition; ‡p < 0.05 compared to static pressure conditions. ((a): Static n = 9 mice, Static + PEG-catalase n = 10 mice, PP n = 9 mice, PP + PEG-catalase n = 11 mice; (b) Static n = 9 mice, Static + gp91ds-tat n = 7 mice, PP n = 9 mice, PP + gp91ds-tat n = 6 mice).
Consequences of increased PP amplitude on FMD
Because an abnormal increase in PP is proposed to be damaging to the cerebral microcirculation,42 we increased the amplitude of the PP from 30 to 50 mm Hg. As a consequence, FMD was reduced by ∼40% at physiological shear stresses between 8 and 20 dyn/cm2 (Figure 6). Nonetheless, this impaired dilation was not sensitive to PEG-catalase, suggesting that eNOS activity was reduced, but not uncoupled as it was observed under SP (Figure 2(b)).
Figure 6.
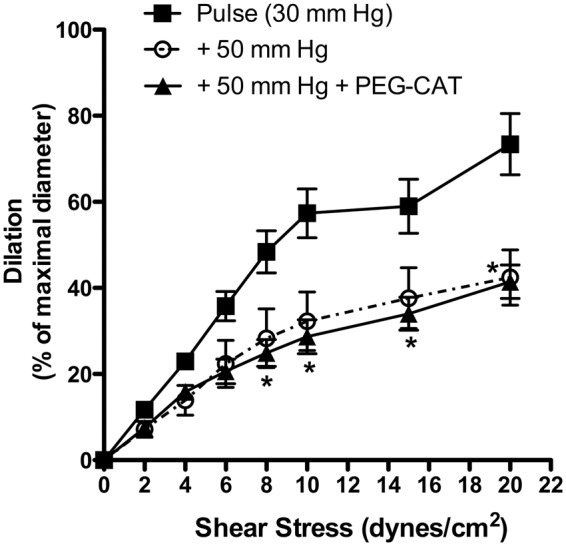
Effects of an increase in pulse pressure from 30 to 50 mm Hg on endothelium-dependent flow-mediated dilation in pressurized mouse cerebral arteries with or without addition of PEG-catalase. Results are mean±SEM of n = 5 mice. *p < 0.05 + 50 mm Hg vs. Pulse (30 mm Hg).
Discussion
Pulsatility of the blood pressure has rarely been considered in ex vivo experiments involving isolated arteries, despite the fact that this oscillatory nature is a fundamental characteristic of arterial blood pressure. We therefore postulated that myogenic tone and flow-mediated responses measured ex vivo in mouse cerebral arteries would be more representative if exposed to pulsatile pressures of physiological amplitude and frequency. Compared to static pressure conditions, we indeed observed that in the presence of pulse pressure, myogenic contractions were enhanced, while endothelial shear stress sensitivity was increased and associated with eNOS coupling and the production of NO. We did not investigate the endothelial mechanosensor to PP, but we did observe that NOX2 inactivation reduced FMD in both pressure conditions. Thus, NOX2 recruitment likely contributes to the transduction of the mechanical flow signal to the activation of eNOS, in both static and PP conditions. In SP conditions, NOX2 may contribute to eNOS activity and its uncoupling, while under PP, NOX2 favours eNOS coupling. We propose that in the absence of PP, NOX2-related eNOS uncoupling maintains a dilatory tone through H2O2 production. In addition, PP revealed a differential endothelial responsiveness to shear stress versus muscarinic receptor activation: acetylcholine stimulates eNOS-derived NO with a concomitant NOX2-derived ROS production that limits eNOS activity, an in vivo observation reported previously.43 The interpretation of our results is summarized in Figure 7; we purport that the cerebrovascular endothelium functions optimally and couples to eNOS under physiological pulse pressure.
Figure 7.

Schematic representation of the hypothetical signalling pathway leading to endothelium-dependent dilation in static versus pulsatile pressure conditions. (a) Under static pressure, flow promotes endothelial-dependent dilation, sensitive to both eNOS inhibition with L-NNA and to PEG-catalase, which degrades H2O2; this suggests that eNOS is uncoupled and produces that, in turn, is converted into the dilatory H2O2 by superoxide dismutase (SOD). Flow-mediated dilation is therefore eNOS-dependent, but mostly NO-independent, as confirmed by the measurement of insignificant NO and large H2O2 production during FMD under static pressure. Flow-mediated dilation is also reduced by the specific NOX2 inhibitor gp91ds-tat, suggesting that NOX2 participates in the dilatory pathway: flow activates an unidentified shear stress sensor, which may stimulate the NOX2/eNOS complex, leading to the production of H2O2 and to dilation. How NOX2 and eNOS are associated remains to be identified. Acetylcholine also promotes a dilation that is reduced by L-NNA, PEG-catalase and gp91 ds-tat, suggesting that FMD and ACh activate similar pathways, i.e. NOX2/eNOS-derived H2O2. Acetylcholine activates the muscarinic receptor, which is associated with the Gq heterotrimeric G-protein; the α-subunit of the Gq protein is coupled to phospholipase C (PLC) that promotes intracellular Ca2+ release, which activates eNOS and, to a lesser extent, endothelium-derived hyperpolarizing factor (EDHF) release. The link between the muscarinic receptor and NOX2/eNOS complex could be through the β-subunit of the Gq protein. (b) Under pulse pressure, flow promotes endothelium-dependent dilation sensitive to L-NNA and gp91ds-tat, but insensitive to PEG-catalase. This suggests that eNOS is coupled and that the NOX2/eNOS complex leads to NO release, as confirmed by the significant NO production during FMD under pulsatile pressure. The nature of the pulse sensor and how it is associated to NOX2/eNOS complex are unknown. Under pulsatile pressure, acetylcholine induces a dilation that is lower than that observed under static pressure; the dilation is reduced by L-NNA, but augmented by gp91ds-tat and by PEG-catalase. The fact that both NOX2 inhibition and favoured H2O2 degradation augment the dilation suggests that the pulsatile pressure promotes production (via NOX2 and possibly other sources) that inactivates eNOS-derived NO. Ach: acetylcholine; EDHF: endothelial-derived hyperpolarizing factor; FMD: flow-mediated dilation; M: muscarinic receptor; NOX: NADPH oxidase; PLC: phospholipase C; PP: pulse pressure; ROS: reactive oxygen species; SOD: superoxide dismutase; SS: shear stress.
The demonstration that a significant myogenic tone developed after exposure to a physiological PP argues in favour of our postulate that cerebral arteries are adapted to a pulsatile environment. In SP conditions, which is the classical methodological approach to investigate MT and FMD ex vivo, C57Bl6 mouse cerebral arteries with an intact endothelium develop little tone44,45 when compared to other species including human cerebral arteries.46–51 The vascular endothelium, however, is known to potently inhibit MT: indeed, after eNOS inhibition, MT is magnified under static pressure (Figure 1), as previously reported in mouse and rat cerebral arteries.51,52 Our data contrast those of a recent study demonstrating that MT was not magnified by PP in middle cerebral arteries isolated from three-month old C57Bl6 mice.53 This latter study investigated the impact of age on PP-dependent regulation of MT, and although not specified, the endothelium may have been removed in order to specifically address this aim. Furthermore, the amplitude of the pulse was greater (50 mm Hg) and the frequency lower (450 bpm), which may also contribute to this difference. In our hands, a PP of 50 mm Hg decreased shear stress sensitivity (Figure 6) and a PP of 70 mm Hg led to a complete loss of reactivity (data not shown). We also observed that when compared to SP, the greater myogenic tone under a physiological PP was insensitive to both L-NNA and PEG-catalase and reached levels of tone measured in static conditions after eNOS inhibition; this suggests that in no-flow conditions, PP inhibits eNOS activity. Altogether, our data demonstrate that pulse pressure regulates myogenic responses in large cerebral arteries with an intact endothelium.
We previously reported that in mouse cerebral, but not gracilis arteries, and under static pressure, eNOS enzymatic cycling generated at rest and during FMD,27 consistent with the present results (Figures 2 and 3). We also previously demonstrated that eNOS is predominantly uncoupled in endothelial cells isolated from mouse cerebral arteries.36 This uncoupling of eNOS in the cerebrovasculature of young and healthy mice, however, is inconsistent and remains conflicts with most of the literature. The difference between our results and the normal eNOS coupling observed by others is not clear. It could be simply due to the fact that most experimenters conclude that if dilatory responses are blocked by NOS inhibitors, endothelium-dependent dilations are mediated by NO.29 It could also be due to specific experimental conditions, notably our choice of gas to aerate the physiological salt solution (12% O2; 5% CO2; 83 % N2 generating a physiological pO2 of 150 ± 10 mm Hg instead of the classical 95% O2; 5% CO2 generating a pO2 of 410 ± 11 mm Hg). We believe that eNOS uncoupling observed under SP permits maintenance of a dilatory tone through H2O2 production. Accordingly, uncoupling of eNOS has been proposed to be a highly conserved defence mechanism.54 In conditions of blood stasis following an arterial occlusion where pulsatile pressure and flow are blunted, eNOS uncoupling may maintain a basal dilatory tone and anti-platelet aggregation activity through generation that would protect against thrombus formation, at least temporarily.
In the present study, it is only under pulse pressure that flow (and Ach) activates eNOS to generate NO (Figures 2 and 3). The second novel finding of our study is, therefore, that a physiological PP favours eNOS coupling (Figure 7). While uncoupling of eNOS may represent a defence mechanism, it is not the only source of dilatory H2O2 in vessels: is the main precursor of H2O2, and NAD(P)H oxidases (NOX) and mitochondria are by far the main producers of .55 Recently, it was shown that a rise in ROS production accounted for the generation of H2O2 and acted as an alternative defence pathway to compensate for the loss of NO production during extra-physiologically high blood pressure stress.25,26 In an attempt to identify the coupling mechanisms linking PP and eNOS activity, we targeted NOX2. NOX2 inhibition with gp91ds-tat limited FMD (and thus eNOS activity) in both SP and PP conditions (Figure 4). Therefore, NOX2 recruitment appears necessary for shear stress sensitivity in both SP and PP conditions. NOX2 may contribute to the flow response by acting as a scaffold, permitting the coupling of the putative flow sensor (or shear stress sensor) and eNOS (illustrated in Figure 7). In the absence of PP, NOX2 appears essential to facilitate eNOS uncoupling; however, NOX2 also contributes to eNOS coupling in the presence of PP. This would presuppose that the shear stress sensor, the stretch/mechano-sensor to PP, eNOS and NOX2 are part of a larger protein complex at the plasma membrane, as speculated in Figure 7. This hypothesis remains to be validated.
Flow41 and Ach56 induce endothelium-dependent dilations through different coupling partners and intracellular pathways. Activation of muscarinic subtype-3 receptors57 coupled to Gq proteins58 increases intracellular Ca2+,56 likely accounting for Ach-mediated dilation in mouse arteries. Flow, however, increases Akt-dependent phosphorylation of eNOS and enhances its activity, while the involvement of intracellular Ca2+ is unclear.27,41 We observed that the impact of PP on Ach-induced dilations was opposite to its effect on FMD. In the presence of PP, (i) maximal Ach-induced dilations were smaller than in static conditions (while maximal FMD were higher); (ii) Ach-induced dilations were increased by PEG-catalase (while FMD were unaffected); and (iii) Ach-induced dilations were increased by gp91ds-tat (while FMD were decreased) (Figure 5). This suggests that under PP, Ach-dilation is associated with a deleterious rise in (originating from NOX2 and/or other sources), limiting the dilatory response to Ach. The fact that Ach-mediated dilation was increased by gp91ds-tat and PEG-catalase suggests that NOX2 is a component of the molecular pathway leading to eNOS-NO release. NOX2-dependent ROS production could be activated by Ach in PP conditions and limit Ach-induced dilation by inactivating NO (Figure 7). Accordingly, Ach-induced dilation of cerebral arteries has been reported to be reduced by NOX2 activity.43 This represents the third novelty of this study: in the presence of PP, Ach-induced NO-dependent dilation is attenuated by a concomitant production of ROS; consequently, Ach has a much lower efficacy than flow, in similar PP conditions.
Our approaches, however, have limitations. First, the sensitivities of the fluorescent dyes used to quantify NO and H2O2 production during FMD are likely suboptimal. NO was not detected in SP conditions despite PEG-catalase only partially preventing FMD, though it is clear that eNOS is responsible since both L-NNA and endothelium removal abolished FMD. Second, our system does not allow the measurement of FMD during PP: arteries were exposed to PP for 30 min, then PP was stopped and flow was applied. On the other hand, Ach-mediated endothelium-dependent dilations can be measured either after (like FMD) or during PP. However, we observed that Ach-mediated dilations were similarly increased by PEG-catalase and gp91ds-tat when PP was applied either before (Figure S2, Supplementary material) or during Ach, suggesting that pre-exposure to PP is sufficient to modify eNOS activity, at least temporarily. Third, because physiological endothelium-dependent vasodilatory mechanisms are redundant,59 it will be difficult to isolate the molecular transduction mechanism involved in PP-dependent eNOS regulation. Pulsatile pressure may induce a dynamic redox-sensitive post-translational modification of eNOS55,60 and this would be difficult to identify with the current technologies.
In conclusion, our data indicate that pulse pressure regulates myogenic tone and flow-mediated responses in cerebral arteries, a process implicating NOX2 and eNOS activity and coupling. Pulse pressure-dependent adjustments in vessel diameter may therefore be implicated in regulating cerebrovascular tone. This is of importance since PP increases with age and prematurely in atherosclerotic and hypertensive patients; by extrapolation, a PP-dependent inappropriate control of cerebrovascular tone, and thus of cerebral blood flow, could contribute in the long term to hypo-perfusion, small vessel rupture, blood–brain barrier disruption and cognitive decline.4,61
Supplementary Material
Acknowledgements
The authors are very thankful to Pierre-François Beauchemin, Anne-Marie Blanchard, Nikita Cobetto, Frédérique Desrochers-Perrault, Quoc-Viet Tran and Jessica Vu who designed the pulse pressure apparatus during their third-year engineering project, under the supervision of Dr F. Lesage at the École Polytechnique de Montréal. We are thankful to Dr. Nathalie Thorin-Trescases for performing the statistical analysis and for her critical review of this work. We are thankful to Eric Duong for his editorial assistance.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Canadian Institutes for Health Research (MOP 89733), the Heart & Stroke Foundation of Canada, the Heart & Stroke Foundation of Quebec and the Foundation of the Montreal Heart Institute.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
AR and VB performed all experiments and contributed to the analysis of the data, their interpretation and the draft of the paper. FL supervised the conception of the pulse pressure system and its validation. ET conceived the research project, supervised AR (MSc student) and VB (PhD student), and is responsible for the data and the finalization of the paper.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Thorin E, Thorin-Trescases N. Vascular endothelial ageing, heartbeat after heartbeat. Cardiovasc Res 2009; 84: 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ceravolo R, Maio R, Pujia A, et al. Pulse pressure and endothelial dysfunction in never-treated hypertensive patients. J Am College Cardiol 2003; 41: 1753–1758. [DOI] [PubMed] [Google Scholar]
- 3.Iadecola C, Davisson RL. Hypertension and cerebrovascular dysfunction. Cell Metab 2008; 7: 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stone J, Johnstone DM, Mitrofanis J, et al. The mechanical cause of age-related dementia (alzheimer's disease): The brain is destroyed by the pulse. J Alzheimer's Dis: JAD 2015; 44: 355–373. [DOI] [PubMed] [Google Scholar]
- 5.Sierra C, de la Sierra A, Chamorro A, et al. Cerebral hemodynamics and silent cerebral white matter lesions in middle-aged essential hypertensive patients. Blood Press 2004; 13: 304–309. [DOI] [PubMed] [Google Scholar]
- 6.Tarumi T, Ayaz Khan M, Liu J, et al. Cerebral hemodynamics in normal aging: Central artery stiffness, wave reflection, and pressure pulsatility. J Cereb Blood Flow Metab: Official journal of the International Society of Cerebral Blood Flow and Metabolism 2014; 34: 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsao CW, Seshadri S, Beiser AS, et al. Relations of arterial stiffness and endothelial function to brain aging in the community. Neurology 2013; 81: 984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitchell GF, van Buchem MA, Sigurdsson S, et al. Arterial stiffness, pressure and flow pulsatility and brain structure and function: the age, gene/environment susceptibility–reykjavik study. Brain: A journal of neurology 2011; 134: 3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halley MM, Reemtsma K, Creech O., Jr Cerebral blood flow, metabolism, and brain volume in extracorporeal circulation. J Thoracic Surg 1958; 36: 506–518. [PubMed] [Google Scholar]
- 10.Wilkens H, Regelson W, Hoffmeister FS. The physiolgic importance of pulsatile blood flow. N Engl J Med 1962; 267: 443–446. [DOI] [PubMed] [Google Scholar]
- 11.Loor G, Gonzalez-Stawinski G. Pulsatile vs. Continuous flow in ventricular assist device therapy. Best Pract Res Clin Anaesthesiol 2012; 26: 105–115. [DOI] [PubMed] [Google Scholar]
- 12.Baufreton C, Intrator L, Jansen PG, et al. Inflammatory response to cardiopulmonary bypass using roller or centrifugal pumps. Ann Thoracic Surg 1999; 67: 972–977. [DOI] [PubMed] [Google Scholar]
- 13.Ead HW, Green JH, Neil E. A comparison of the effects of pulsatile and non-pulsatile blood flow through the carotid sinus on the reflexogenic activity of the sinus baroceptors in the cat. J Physiol 1952; 118: 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cornwell WK, Tarumi T, Aengevaeren V, et al. Nonpulsatile flow among patients with cf-lvads leads to greater levels of sympathetic neural activity through unloading of arterial baroreceptors. Circulation 2014; 130: A15992. [Google Scholar]
- 15.Markham DW, Fu Q, Palmer MD, et al. Sympathetic neural and hemodynamic responses to upright tilt in patients with pulsatile and nonpulsatile left ventricular assist devices. Circulation Heart Fail 2013; 6: 293–299. [DOI] [PubMed] [Google Scholar]
- 16.Milano AD, Dodonov M, Van Oeveren W, et al. Pulsatile cardiopulmonary bypass and renal function in elderly patients undergoing aortic valve surgerydagger. Eur J Cardio-thoracic Surgery: Official journal of the European Association for Cardio-thoracic Surgery 2015; 47: 291–298. [DOI] [PubMed] [Google Scholar]
- 17.Nakano T, Tominaga R, Nagano I, et al. Pulsatile flow enhances endothelium-derived nitric oxide release in the peripheral vasculature. Am J Physiol Heart Circulat Physiol 2000; 278: H1098–H1104. [DOI] [PubMed] [Google Scholar]
- 18.Sezai A, Shiono M, Nakata K, et al. Effects of pulsatile cpb on interleukin-8 and endothelin-1 levels. Artificial Organ 2005; 29: 708–713. [DOI] [PubMed] [Google Scholar]
- 19.Taylor KM, Bain WH, Russell M, et al. Peripheral vascular resistance and angiotensin ii levels during pulsatile and no-pulsatile cardiopulmonary bypass. Thorax 1979; 34: 594–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toda K, Tatsumi E, Taenaka Y, et al. Impact of systemic depulsation on tissue perfusion and sympathetic nerve activity. Ann Thorac Surg 1996; 62: 1737–1742. [DOI] [PubMed] [Google Scholar]
- 21.Kurotobi S, Sano T, Kogaki S, et al. Bidirectional cavopulmonary shunt with right ventricular outflow patency: The impact of pulsatility on pulmonary endothelial function. J Thorac Cardiovasc Surg 2001; 121: 1161–1168. [DOI] [PubMed] [Google Scholar]
- 22.Pinaud F, Loufrani L, Toutain B, et al. In vitro protection of vascular function from oxidative stress and inflammation by pulsatility in resistance arteries. J Thoracic Cardiovasc Surg 2011; 142: 1254–1262. [DOI] [PubMed] [Google Scholar]
- 23.Rubanyi GM, Romero JC, Vanhoutte PM. Flow-induced release of endothelium-derived relaxing factor. Am J Physiol 1986; 250: H1145–H1149. [DOI] [PubMed] [Google Scholar]
- 24.Koller A, Bagi Z. Nitric oxide and h2o2 contribute to reactive dilation of isolated coronary arterioles. Am J Physiol Heart Circulat Physiol 2004; 287: H2461–H2467. [DOI] [PubMed] [Google Scholar]
- 25.Beyer AM, Durand MJ, Hockenberry J, et al. An acute rise in intraluminal pressure shifts the mediator of flow-mediated dilation from nitric oxide to hydrogen peroxide in human arterioles. Am J Physiol Heart Circulat Physiol 2014; 307: H1587–H1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durand MJ, Dharmashankar K, Bian JT, et al. Acute exertion elicits a h2o2-dependent vasodilator mechanism in the microvasculature of exercise-trained but not sedentary adults. Hypertension 2015; 65: 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drouin A, Thorin E. Flow-induced dilation is mediated by akt-dependent activation of endothelial nitric oxide synthase-derived hydrogen peroxide in mouse cerebral arteries. Stroke; a journal of cerebral circulation 2009; 40: 1827–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shipley RD, Kim SJ, Muller-Delp JM. Time course of flow-induced vasodilation in skeletal muscle: Contributions of dilator and constrictor mechanisms. Am J Physiol Heart Circulat Physiol 2005; 288: H1499–H1507. [DOI] [PubMed] [Google Scholar]
- 29.Green DJ, Dawson EA, Groenewoud HM, et al. Is flow-mediated dilation nitric oxide mediated?: A meta-analysis. Hypertension 2014; 63: 376–382. [DOI] [PubMed] [Google Scholar]
- 30.Fujii K, Heistad DD, Faraci FM. Flow-mediated dilatation of the basilar artery in vivo. Circulat Res 1991; 69: 697–705. [DOI] [PubMed] [Google Scholar]
- 31.Joannides R, Haefeli WE, Linder L, et al. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation 1995; 91: 1314–1319. [DOI] [PubMed] [Google Scholar]
- 32.Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006; 113: 1708–1714. [DOI] [PubMed] [Google Scholar]
- 33.Capettini LS, Cortes SF, Gomes MA, et al. Neuronal nitric oxide synthase-derived hydrogen peroxide is a major endothelium-dependent relaxing factor. Am J Physiol Heart Circulat Physiol 2008; 295: H2503–H2511. [DOI] [PubMed] [Google Scholar]
- 34.Kilkenny C, Browne WJ, Cuthill IC, et al. Improving bioscience research reporting: The arrive guidelines for reporting animal research. PLoS Biol 2010; 8: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolduc V, Drouin A, Gillis MA, et al. Heart rate-associated mechanical stress impairs carotid but not cerebral artery compliance in dyslipidemic atherosclerotic mice. Am J Physiol Heart Circulat Physiol 2011; 301: H2081–H2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drouin A, Thorin-Trescases N, Hamel E, et al. Endothelial nitric oxide synthase activation leads to dilatory h2o2 production in mouse cerebral arteries. Cardiovasc Res 2007; 73: 73–81. [DOI] [PubMed] [Google Scholar]
- 37.Yu C, Luo X, Duquette N, et al. Knockdown of angiopoietin like-2 protects against angiotensin ii-induced cerebral endothelial dysfunction in mice. Am J Physiol Heart Circulat Physiol 2015; 308: H386–H397. [DOI] [PubMed] [Google Scholar]
- 38.Faraci FM, Heistad DD. Regulation of large cerebral arteries and cerebral microvascular pressure. Circulat Res 1990; 66: 8–17. [DOI] [PubMed] [Google Scholar]
- 39.Baumbach GL, Faraci FM, Heistad DD. Effects of local reduction in pressure on endothelium-dependent responses of cerebral arterioles. Stroke; a journal of cerebral circulation 1994; 25: 1456–1461. discussion 1461-1452. [DOI] [PubMed] [Google Scholar]
- 40.Larsen BT, Bubolz AH, Mendoza SA, et al. Bradykinin-induced dilation of human coronary arterioles requires nadph oxidase-derived reactive oxygen species. Arterioscl Thromb Vasc Biol 2009; 29: 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou J, Li YS, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscl Thromb Vasc Biol 2014; 34: 2191–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Rourke MF, Hashimoto J. Mechanical factors in arterial aging: A clinical perspective. J Am College Cardiol 2007; 50: 1–13. [DOI] [PubMed] [Google Scholar]
- 43.Girouard H, Park L, Anrather J, et al. Angiotensin ii attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2-derived radicals. Arterioscl Thromb Vasc Biol 2006; 26: 826–832. [DOI] [PubMed] [Google Scholar]
- 44.De Silva TM, Ketsawatsomkron P, Pelham C, et al. Genetic interference with peroxisome proliferator-activated receptor gamma in smooth muscle enhances myogenic tone in the cerebrovasculature via a rho kinase-dependent mechanism. Hypertension 2015; 65: 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomsen AB, Kim S, Aalbaek F, et al. Intracellular acidification alters myogenic responsiveness and vasomotion of mouse middle cerebral arteries. J Cereb Blood Flow Metab: Official journal of the International Society of Cerebral Blood Flow and Metabolism 2014; 34: 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Roldan JL, Bevan JA. Flow-induced constriction and dilation of cerebral resistance arteries. Circulat Res 1990; 66: 1445–1448. [DOI] [PubMed] [Google Scholar]
- 47.Thorin-Trescases N, Bevan JA. High levels of myogenic tone antagonize the dilator response to flow of small rabbit cerebral arteries. Stroke; a journal of cerebral circulation 1998; 29: 1194–1200. discussion 1200–1191. [DOI] [PubMed] [Google Scholar]
- 48.Hill MA, Meininger GA, Davis MJ, et al. Therapeutic potential of pharmacologically targeting arteriolar myogenic tone. Trends Pharmacol Sci 2009; 30: 363–374. [DOI] [PubMed] [Google Scholar]
- 49.Kauffenstein G, Laher I, Matrougui K, et al. Emerging role of g protein-coupled receptors in microvascular myogenic tone. Cardiovasc Res 2012; 95: 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koller A, Toth P. Contribution of flow-dependent vasomotor mechanisms to the autoregulation of cerebral blood flow. J Vasc Res 2012; 49: 375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ngai AC, Winn HR. Modulation of cerebral arteriolar diameter by intraluminal flow and pressure. Circulat Res 1995; 77: 832–840. [DOI] [PubMed] [Google Scholar]
- 52.Drouin A, Farhat N, Bolduc V, et al. Up-regulation of thromboxane a(2) impairs cerebrovascular enos function in aging atherosclerotic mice. Pflugers Archiv: European journal of physiology 2011; 462: 371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Springo Z, Toth P, Tarantini S, et al. Aging impairs myogenic adaptation to pulsatile pressure in mouse cerebral arteries. J Cereb Blood Flow Metab: Official journal of the International Society of Cerebral Blood Flow and Metabolism 2015; 35: 527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rabelink TJ, Luscher TF. Endothelial nitric oxide synthase: Host defense enzyme of the endothelium? Arterioscl Thromb Vasc Biol 2006; 26: 267–271. [DOI] [PubMed] [Google Scholar]
- 55.Sies H. Role of metabolic h2o2 generation: Redox signaling and oxidative stress. J Biol Chem 2014; 289: 8735–8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen H, Kold-Petersen H, Laher I, et al. Impaired endothelial calcium signaling is responsible for the defective dilation of mesenteric resistance arteries from db/db mice to acetylcholine. Eur J Pharmacol 2015; 767: 17–23. . [DOI] [PubMed] [Google Scholar]
- 57.Gericke A, Sniatecki JJ, Mayer VG, et al. Role of m1, m3, and m5 muscarinic acetylcholine receptors in cholinergic dilation of small arteries studied with gene-targeted mice. Am J Physiol Heart and circulatory physiology 2011; 300: H1602–H1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vequaud P, Thorin E. Endothelial g protein beta-subunits trigger nitric oxide-but not endothelium-derived hyperpolarizing factor-dependent dilation in rabbit resistance arteries. Circulat Res 2001; 89: 716–722. [DOI] [PubMed] [Google Scholar]
- 59.Durand MJ, Gutterman DD. Diversity in mechanisms of endothelium-dependent vasodilation in health and disease. Microcirculation 2013; 20: 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heiss EH, Dirsch VM. Regulation of enos enzyme activity by posttranslational modification. Curr Pharmaceut Des 2014; 20: 3503–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Faraco G, Iadecola C. Hypertension: A harbinger of stroke and dementia. Hypertension 2013; 62: 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.