

Abstract
Trait loss is a widespread phenomenon with pervasive consequences for a species’ evolutionary potential. The genetic changes underlying trait loss have only been clarified in a small number of cases. None of these studies can identify whether the loss of the trait under study was a result of neutral mutation accumulation or negative selection. This distinction is relatively clear-cut in the loss of sexual traits in asexual organisms. Male-specific sexual traits are not expressed and can only decay through neutral mutations, whereas female-specific traits are expressed and subject to negative selection. We present the genome of an asexual parasitoid wasp and compare it to that of a sexual lineage of the same species. We identify a short-list of 16 genes for which the asexual lineage carries deleterious SNP or indel variants, whereas the sexual lineage does not. Using tissue-specific expression data from other insects, we show that fifteen of these are expressed in male-specific reproductive tissues. Only one deleterious variant was found that is expressed in the female-specific spermathecae, a trait that is heavily degraded and thought to be under negative selection in L. clavipes. Although the phenotypic decay of male-specific sexual traits in asexuals is generally slow compared with the decay of female-specific sexual traits, we show that male-specific traits do indeed accumulate deleterious mutations as expected by theory. Our results provide an excellent starting point for detailed study of the genomics of neutral and selected trait decay.
Keywords: Leptopilina clavipes, Wolbachia, parthenogenesis, deleterious variants, sexual trait decay
Introduction
When selective pressures shift, traits may become redundant. Such redundant traits tend to degenerate over time and may eventually be lost entirely. Trait loss is widespread, both phylogenetically and in terms of trait types, and has important evolutionary consequences. For example, when a trait is lost because its function is compensated by an ecological interaction, the species may become dependent on the ecological partner (Ellers et al. 2012). Another common pattern of trait loss is seen when sexually reproducing organisms switch to asexual reproduction. Such lineages quickly lose their ability to attract mates and fertilize eggs, effectively blocking a reversal to sexual reproduction (van der Kooi and Schwander 2014).
The molecular causes of trait loss are diverse. First, trait loss may result from pseudogenization of key genes through deleterious amino acid changes or mutations that disrupt gene function. Examples of trait loss caused by such loss-of-function mutations are the loss of vitamin C synthesis in several groups of mammals (Cui et al. 2011; Drouin et al. 2011; Hiller et al. 2012), loss of taste receptor genes in whales (Feng et al. 2014) and loss of a phospholipid transporter in horses and guinea pigs (Hiller et al. 2012). Second, mutations in regulatory sequences may alter the expression of genes underlying the trait. For example, the loss of pelvic spines in the three-spined stickleback Gasterosteus aculeatus is caused by deletion of a tissue-specific enhancer of the Pitx1 gene (Chan et al. 2010). Comparable deletions of regulatory elements are responsible for the loss of penile spines and forebrain growth arrest in humans (McLean et al. 2011). Last, redundant genes may be lost from a genome completely. Ortholog losses appear to be widespread (Wyder et al. 2007; Suen et al. 2011), although the true absence of a (pseudo)gene is difficult to prove. For example, bird genomes appear to have lost several genes involved in insulin sensitivity, without leaving them as detectable pseudogenes (Dakovic et al. 2014).
Trait integrity may be selectively neutral or under negative selection. This distinction is often difficult to make in real systems, but it is relatively clear-cut in the loss of sexual traits in asexual organisms. Upon the switch from sexual to asexual reproduction, redundant female-specific sexual traits tend to decay rapidly and consistently, suggestive of negative selection (van der Kooi and Schwander 2014). Redundant male-specific traits, on the other hand, are not expressed in asexual females, are consequently not exposed to selection and tend to remain functional for extended lengths of time (van der Kooi and Schwander 2014). Asexual organisms thus provide excellent models to study the dynamics of selected vs. neutral trait decay. An important challenge is to identify the genetic changes underlying the decay of sexual traits in asexuals. Mutations resulting in the decay in female-specific sexual traits may enhance fitness of asexual females and thus have a high chance of getting fixed in the population. In contrast, mutations affecting neutral male-specific traits would only become fixed through genetic drift. As a result, mutations affecting female-specific traits may be more prevalent than mutations affecting male-specific traits in asexual lineages. The parasitoid wasp Leptopilina clavipes provides a promising study species in which to address this issue. L. clavipes features both sexual and asexual reproducing lineages and its asexual lineages have decayed female-specific as well as male-specific traits (Pannebakker et al. 2005; Kraaijeveld et al. 2009).
Here, we present a draft genome assembly of an asexual lineage of the parasitoid wasp Leptopilina clavipes. We aligned whole-genome shotgun sequences of a sexual lineage of the same species to this draft genome. Using this alignment, we compare the genetic load of the sexual and asexual lineages. Tissue-specific expression patterns of homologous genes in Nasonia vitripennis and Drosophila melanogaster were used to identify candidate genes underlying the observed decay of sexual traits in L. clavipes. Given this information, we address the question of whether negative selection on female-specific sexual traits results in fixation of a larger number of deleterious variants in the underlying genes than found in genes encoding selectively neutral male-specific sexual traits. We investigated single-nucleotide polymorphism and insertion–deletion (indel) variants and identified variants likely to decrease the function of a given gene product. For a small set of candidate loci, we additionally examined whether independently evolved asexual lineages of L. clavipes have accumulated identical or comparable trait-loss mutations. This represents the first genome-wide assessment of sexual trait decay in an asexual organism.
Material and Methods
Study System
We sequenced the genome of the haplodiploid wasp Leptopilina clavipes (Hymenoptera: Figitidae), a parasitoid of Drosophila larvae. Asexual reproduction in this species is caused by Wolbachia endosymbionts that induce diploidy through gamete duplication (Pannebakker et al. 2004b). This meiotic alteration results in completely homozygous L. clavipes offspring (Kraaijeveld et al. 2011). L. clavipes occurs in both haplodiploid sexual (arrhenotokous) and asexual (thelytokous) populations, which are geographically separated. Northern European populations of this species have diverged from a Spanish population about 12,000–43,000 generations ago (this species has one or two generations a year in Northern Europe) and have become infected with a parthenogenesis-inducing Wolbachia during this period (Kraaijeveld et al. 2011). Wolbachia has infected multiple female lineages and the northern populations of L. clavipes consequently comprises a series of genetically distinct clones (Kraaijeveld et al. 2011).
Isofemale lineages of L. clavipes were maintained at Leiden University (The Netherlands) as described previously (Kraaijeveld et al. 2009). Three females were used to initiate each subsequent generation for at least 65 generations, thus likely resulting in high inbreeding levels in these isofemale lineages. We chose one asexual lineage (GBW) for whole genome shotgun sequencing and genome assembly. For comparison, we also obtained whole-genome shotgun sequences for one sexual lineage (EPG), which were aligned to the draft reference assembly [see Kraaijeveld et al. (2011) for collection details].
Genome Size Estimation
Flow cytometric genome size estimation was done with an Accuri C6 system following a standard protocol (Hare and Johnston 2011). D. melanogaster (estimated genome size 175 Mb; Animal Genome Size Database; http://www.genomesize.com; last accessed November 15, 2016) was used as reference for co-staining. Heads were removed from frozen animals (−80 °C), transferred into Galbraith buffer and ground using a Dounce tissue ginder. Both L. clavipes and D. melanogaster samples were filtered through a 20 μm nylon mesh and stained with propidium iodide (50 μg/ml) by incubating for 2 h at 4 °C. To compare 2C (and 4C) peak fluorescence signals, samples were run both separately and combined. All flow cytometry estimates are based on minimum counts of at least 1,000 nuclei each (i.e., 2C peaks).
In addition to our flow cytometry estimate, we estimated genome size from the sequence data (see below for details). Scaffolds containing sequences matching the putatively single-copy genes Ef-1a and RNApolII were identified using blast (Altschul et al. 1990). Both scaffolds had a fairly even coverage by HiSeq data of 87×. Genome size can then be estimated as (number of reads * average read length)/87. Furthermore, kmer-based methods provide an alternative method for estimating genome size (Liu et al. 2013). We employed two such methods: SGA (Simpson 2014) and KmerGenie (Chikhi and Medvedev 2014).
Sequencing
DNA was extracted from pools of ten L. clavipes females for Illumina sequencing and 30 females for Pacific Biosciences SMRT sequencing using the DNAeasy Blood and Tissue Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol.
All next-generation sequencing was performed at the Leiden Genome Technology Center (LGTC) at the Leiden University Medical Center (The Netherlands). The GBW and EPG lineages were first sequenced on Illumina GAIIx as described by (Kraaijeveld et al. 2012). To obtain a high-quality reference genome, the GBW lineage was additionally sequenced on Illumina HiSeq 2000 and Pacific Biosciences (see supplementary table S1, Supplementary Material online for details on output).
For Illumina sequencing, genomic DNA was sonicated using the Covaris Instrument (Covaris Inc., USA). Paired-end libraries were prepared following Illumina’s protocol (Illumina DNA sample kit). Briefly, fragments were end-repaired, 3′-adenylated, and ligated with Illumina adapters. Ligation products of 600–700 bp were gel-purified and PCR-amplified using Illumina adapter-specific primers. Libraries were purified and quantified using a Qubit Fluorometer (Thermo Fisher, USA) and evaluated using an Agilent 2100 bioanalyzer (Agilent Technologies, USA). GBW and EPG libraries were sequenced using 75-bp paired-end read chemistry on an Illumina GAIIx (Illumina, USA). The subsequent GBW library was sequenced using 100-bp paired-end read chemistry on Illumina HiSeq 2000 (Illumina, USA).
For Pacific Biosciences SMRT sequencing of the asexual GBW lineage, SMRTbell DNA template libraries were prepared according to the manufacturer’s specification after the fragmentation with G-tubes (Covaris, USA). SMRTbell template libraries of different insert sizes (1.5, 4, 6.4, and 7 kb) were prepared. The fragmented DNA was end-repaired and ligated to hairpin adapters. SMRT sequencing was carried out on the Pacific Biosciences RS according to standard protocols, 16 SMRT cells with the C1 chemistry (diffusion loading, 2 × 45 min, 1 kb fragment size) and four SMRT cells with XL-P4 chemistry (Magbead loading, 1 × 120 min, 1 kb fragment size). All runs were processed using the standard primary data analysis.
Genome Assembly
The Illumina HiSeq (HiSeq) and Pacific Biosciences RS I (PacBio) data were used to assemble the genome of the asexual GBW lineage. First, filtered PacBio subreads >500 bp with a read quality >0.80 were error corrected using the PacBioToCA pipeline available in Celera Assembler 7.0 (Myers et al. 2000) (parameters merSize = 14, utgErroRate = 0.25, utgErrorLimit = 4.5, cnsErrorRate = 0.25, cgwErrorRate = 0.25, ovlErrorRate = 0.25, doOverlapBasedtrimmin = 0). This procedure maps the short, high-quality Illumina HiSeq reads to the long, low-quality PacBio reads and determines the consensus sequence. From the raw PacBio data, read correction removed 24.6% of reads and 35.6% of bases and shortened the average read length by 14.6%. The error-corrected PacBio reads and the HiSeq reads were used for hybrid de novo assembly using the Celera Assembler 7.0 (parameters merSize = 14, unitigger = bogart, toggleNumInstances = 0, cgwDemoteRBP = 0).
As a first validation of the de novo assembly, we re-mapped the HiSeq reads that were used in the de novo assembly to the final assembly using Bowtie2 (Langmead and Salzberg 2012) (parameters –N 1, –mp 4).
To assess the completeness of the assembled gene space, we mapped a set of Core Eukaryotic Genes (CEGs) to the assembly using the Core Eukaryotic Gene-Mapping Approach (CEGMA) pipeline (Parra et al. 2007, 2009). CEGs are highly conserved and thought to be present in every genome of a multicellular eukaryote in low copy numbers (Parra et al. 2009). Therefore, the percentage of CEGs that are present in a given sequenced genome can be taken as an estimator for the completeness of the sequenced gene space. Furthermore, we compared the gene space of the draft assembly to that of the parasitoid wasp N. vitripennis (genome build nvit_2.1) using blastp at an e-value cut-off of 1e-5.
To characterize any co-sequenced symbionts, parasites and contaminants, we employed the Blobology pipeline (Kumar et al. 2013). Briefly, all scaffolds were compared with a local install of NCBI’s nt database using BLASTn (megaBLAST, e-value cut-off = 1e−5). We aligned Illumina GAIIx reads from the sexual lineage and the asexual lineage [described in Kraaijeveld et al. (2012)] to the reference assembly using Bowtie2 (Langmead and Salzberg 2012) with parameters –N 1 –mp 4. Duplicate reads were removed using Picard-tools (http://broadinstittute.github.io/picard; last accessed November 15, 2016) and indels were realigned using GATK (McKenna et al. 2010). The bam files from these two alignments were used to calculate coverage for each scaffold. These were then plotted against the GC content of the scaffolds. Scaffolds and parts of misassembled scaffolds matching prokaryotic endosymbionts were removed from the final assembly.
Annotation
Protein-coding genes in the genome of L. clavipes were automatically annotated using MAKER2 version 2.31.6 (Holt and Yandell 2011). MAKER2 is an annotation pipeline that uses a combination of ab initio and evidence-based approaches to infer gene models with high confidence. We applied a two-pass, iterative workflow that aims to maximize the number of true positives in both gene predictions and annotations. The following information was used as input for the first MAKER2 run: transcriptome data (74,639 transcript sequences) generated as part of the 1KITE project (http://www.1kite.org/; last accessed November 15, 2016); Uniprot reference proteomes for Apis mellifera and Atta cephalotes (17.04.2014, without isoforms); gene predictions generated using the tools CEGMA (version 2.4; Parra et al. 2007), GeneMark-ES (version 2.3c; Lomsadze et al. 2005) and SNAP (release 29.11.2013; Korf 2004), each with default settings; repeat libraries obtained from RepeatMasker (arthropods) and generated de novo using Recon, as implemented in RepeatModeler (version 1.0.7; http://www.repeatmasker.org/RepeatModeler.html; last accessed November 15, 2016); transposable element library provided by MAKER2. The results from the first MAKER2 run were used to train Augustus (version 3.0.1; Stanke and Waack 2003) and SNAP. MAKER2 was then run a second time using the same input files as in the first run, except that we used the improved Augustus and SNAP files.
Functional annotation was carried out using InterProScan 5.7.48 (Jones et al. 2014). We searched the proteins predicted in the L. clavipes genome in the following databases: TIGRFAM 13.0 (Haft et al. 2003), ProDom 2006.1 (Servant 2002), SMART 6.2 (Letunic et al. 2009), HAMAP 201311.27 (Pedruzzi et al. 2013), ProSitePatterns 20.97 (Sigrist et al. 2013), SuperFamily 1.75 (Wilson et al. 2007), PANTHER 9.0 (Mi et al. 2013), Gene3D 3.5.0 (Sillitoe et al. 2015), PIRSF 284 (Wu et al. 2004), Pfam-A 27.0 (Finn et al. 2015), ProSiteProfiles 20.97 (Sigrist et al. 2013), and Coils 2.2 (Lupas et al. 1991). For proteins with matches, we extracted the Gene Ontology (GO) terms. We used OrthoMCL-DB (Chen et al. 2006) to assess orthology of gene models. OrthoMCL conducts blastp (Altschul et al. 1990) searches of all proteins against themselves and against proteins in the OrthoMCL database (e-value cut-off: e−5, 50% match). Proteins with matches above the threshold are assigned to orthologous groups. The remaining proteins are then compared with each other to find putative paralogous pairs, which are then clustered into paralog groups.
Comparison of Coding Variants
To compare the genome of the asexual L. clavipes lineage to that of the sexual lineage, we generated a preliminary list of variants (SNPs and indels) in vcf format using samtools and bcftools from the aligments described above. The vcf file was then filtered for QUAL ≥ 20 (phred-scaled quality score for the variant call) and read depth ≥10. To limit the influence of sequencing or assembly artifacts, we removed all variants that were also present in the alignment of the HiSeq data of the asexual lineage.
Trait loss may result from disruptions at various places in the transcript, leading to loss-of-function variants. Disruptions may appear as premature stop codons, at splice-sites or as insertion/deletions (indels) that break the transcript’s reading frame (Macarthur et al. 2012). We therefore annotated all variants using snpEff (Cingolani et al. 2012) and filtered the resulting list of candidate loss-of-function variants on highly repetitive sequences, variants affecting non-canonical splice sites and transcripts whose underlying gene model did not contain a start codon. We further removed candidates whose protein was predicted to be short (<100 amino acids), that showed no significant similarity to proteins of other hymenopteran insects (assessed via BLASTP search) or where such BLASTP hits were based on repetitive or transposase domains (manual curation). Variants found in the sexually reproducing lineage were considered to be potentially involved in trait loss in the asexual lineage if they removed a stop codon from or caused a frame shift in the reference sequence (of the asexual lineage). We further selected candidates in genes related to sexual functions. For this, we exploited the fact that tissue-specific gene expression is well conserved between insects (Baker et al. 2011), and selected only variants in genes for which the expression of N. vitripennis or D. melanogaster homologs was enriched in one of the tissues related to sexual functions. This expression enrichment was determined by identifying the top blastp hit among N. vitripennis and D. melanogaster genes in the Waspatlas (Davies and Tauber 2015) and Flyatlas (Chintapalli et al. 2007) databases, respectively. Expression data was available for testes in N. vitripennis and testes, accessory glands and spermathecae in D. melanogaster. We attempted to predict whether the variant carried by the sexual lineage would result in a more optimal protein than produced by the variant carried by the asexual lineage by investigating sequence conservation among hymenopteran insects, analogous to the SIFT analysis described below. This assumes that variations on conserved amino acid sequences will usually result in a sub-optimal protein.
In addition to loss-of-function mutations, non-synonymous base substitutions could result in suboptimal protein function. At a given residue, amino acids that optimize protein function should be favored by selection and thus show a higher degree of conservation among related species than amino acids that reduce protein function. To predict whether an amino acid substitution affects protein function, we generated a SIFT (Ng and Henikoff 2001) database for the L. clavipes reference genome. SIFT predicts whether an amino acid substitution is likely to be deleterious to protein function based on sequence homology and the physical properties of amino acids. SIFT uses multiple alignment information to calculate normalized probabilities for all possible substitutions. Positions with normalized probabilities less than 0.05 are predicted to be non-tolerated (deleterious) and those greater than or equal to 0.05 are predicted to be tolerated. We then used SIFT 4G (http://sift4g.org; last accessed November 15, 2016) to annotate all single-nucleotide polymorphisms (SNPs) between the asexual and the sexual L. clavipes genomes. For the variants that were predicted to be non-tolerated in the asexual genome but not in the sexual genome, or vice versa, we searched the protein against the N. vitripennis and D. melanogaster genomes using blastp and determined tissue-specific expression enrichment as above.
For all non-synonymous amino acid differences between the asexual and the sexual genomes, we predicted whether either the asexual or the sexual variant would result in a more stable protein using MUpro (Cheng et al. 2006). MUpro uses machine learning to predict how a single-site amino acid mutation affects protein stability and achieves about 84% accuracy. A confidence score is calculated, taking values between −1 and 1. Negative values indicate a decrease in protein stability and positive values an increase in protein stability. Values closer to −1 or 1 have higher confidence than values closer to 0. Proteins that were predicted to be more stable in the sexual lineage versus the asexual lineage at high confidence were searched against the N. vitripennis and D. melanogaster genomes using BLASTP. Tissue-specific expression enrichment was then determined as above.
Downstream Analysis of Candidate Decayed Genes
To examine whether genetically different asexual lineages all carried the same putative trait-loss variants, we sequenced four variants (two in genes enriched in testes and two in genes enriched in accessory glands) identified from our SIFT analysis in twelve asexual and nine sexual lineages of L. clavipes. These lineages were selected from a larger set of lineages, because microsatellite analysis had previously identified them as between genetically different (Kraaijeveld et al. 2011).
Results
The Leptopilina clavipes Genome
The draft genome assembly of L. clavipes consists of 36,601 scaffold with a size larger 200 bp and spans 255 Mb. The largest scaffold had a size of 419,8 kb and N50 was 13,759. A summary of the assembly statistics is presented in supplementary tables S1 and S2, Supplementary Material online. Overall, 92.7% of Hiseq reads aligned to the genome assembly. 54.6% of read pairs aligned concordantly exactly once and 30.1% more than once. Of the 15.3% read pairs that did not align concordantly, 13.6% aligned discordantly once. Discordantly mapping reads were found on many (28,570) scaffolds and visual inspection showed most of these reads to be spread evenly within scaffolds. The read coverage was unimodal (supplementary fig. S1, Supplementary Material online).
Flow cytometry yielded a genome size estimate of 321 Mb for L. clavipes (supplementary fig. S2, Supplementary Material online). Our read-based method estimated genome size as 318 Mb, whereas the k-mer based methods SGA and KmerGenie yielded estimates of 293.8 Mb and 255.1 Mb, respectively. Based on these various estimates, the draft genome assembly represents 79.5–99.9% of the genome.
We found 230 (93%) of the 248 Core Eukaryotic Genes (CEGs) to be present and seemingly complete in the L. clavipes genome assembly. An additional 15 CEGs (6%) were found incomplete. These CEGs tend to occur as single copies in eukaryote genomes (Parra et al. 2009). The average number of orthologs identified for this set of CEGs in the L. clavipes genome assembly was 1.23 (1.38 when including incomplete CEGs), indicating that the level of redundancy was low. We found 90.1% of the predicted proteins of N. vitripennis to be represented in the L. clavipes genome assembly.
Most scaffolds exhibited local similarity (indicated by BLAST hits) to genomic sequences of eukaryotes (mostly Hymenoptera and other insects; fig. 1). A subset of 90 scaffolds was classified as Rickettsiales, and all but one of these matched various Wolbachia genomes. Most of these scaffolds (n = 53) had very low coverage (<1×) in the sequenced sexual lineage (fig. 1), but above-average coverage (>70×) in the asexual lineage (fig. 1), consistent with the absence of Wolbachia from the sexual lineage. A small number of scaffolds (n = 37) classified as Rickettsiales had coverage within the range of the scaffolds classified as insect in both the sexual and asexual lineage (fig. 1). In twelve of these scaffolds, the Wolbachia hit was flanked by hits to insect genomes, potentially indicative of horizontal transmission of Wolbachia DNA to the nucleus. However, closer inspection revealed that in 15 out of 37 cases, the region corresponding to the Wolbachia hit were not covered by reads from the sexual lineage, suggesting that these regions were not part of the sexual genome. Furthermore, these same regions showed above-average coverage by reads from the asexual lineage, suggesting that they were likely misassembled. The remaining regions were all short (<500 bp) and probably represented spurious hits to Wolbachia. In conclusion, we have no compelling evidence for horizontal transmission events from Wolbachia to the nuclear genome of L. clavipes. We also identified seven scaffolds and two partial (i.e., misassembled) scaffolds matching the WO phage of the wVitB Wolbachia of N. vitripennis. These sequences had > 200× coverage in the asexual lineage, but no coverage in the sexual lineage. A further 18 scaffolds matched other bacteria and 220 scaffolds matched other viruses (mostly an Ichnovirus isolated from the wasp Hyposoter didymator) and had comparable coverage in the asexual and sexual lineage.
Fig. 1.—

“Blobology” plots of read coverage against GC content per scaffold for Wolbachia-infected asexual lineage and uninfected sexual lineage. Dots are colored according the top hit from a BLAST search against the NCBI’s nt database. Only scaffolds for which a significant BLAST hit was obtained are shown.
MAKER2 annotated a total of 49,568 genes, 50,004 transcripts, 186,194 exons and 15,426 untranslated regions (UTRs). We found 16,562 predicted proteins that had at least one match with any of the protein databases (supplementary information, Supplementary Material online). A total of 8,243 orthologous groups were assigned to proteins in the L. clavipes genome. Furthermore, 1,571 groups of paralogous proteins were identified, each containing between 2 and 246 proteins.
Comparison of Coding Variants
Our initial list of possible loss-of-function variants comprised of 597 SNPs and 997 indels. After stringent filtering (see “Methods” section), we obtained a short-list of five genes that contained possible loss-of-function variants in the reference sequence and for which gene expression for putative homologs in N. vitripennis and D. melanogaster was biased to male reproductive tissue (table 1). We were not able to confirm bioinformatically whether variants carried by the sexual lineage would result in a more functional protein, because of a too low level of nucleotide sequence conservation among the investigated Hymenoptera insects.
Table 1.
Shortlist of Candidate Genes Involved in Sexual Traits Decay in Asexual Leptopilina clavipes
| Mutation Type | Identified Using | Drosophila Homolog | Drosophila Tissue Enrichment | Nasonia Homolog | Nasonia Tissue Enrichment | Annotation | Notes |
|---|---|---|---|---|---|---|---|
| Enriched in reproductive tissue | |||||||
| Loss-of-function | snpEff | NP_648446.1 | Testis | XP_003425377.1 | Female body | Pleckstrin homology-like domain family B member 1 | Frame shift |
| Loss-of-function | snpEff | NP_001015401.1 | Testis | XP_003426117.1 | Testis | Tim17b | Stop codon removed |
| Loss-of-function | snpEff | NP_995777.1 | Testis | XP_008217920.1 | Testis | Ribonuclease H1 | Frameshift |
| Loss-of-function | snpEff | XP_008216187.1 | Testis | RNA-binding protein 4.1-like | Frameshift | ||
| Loss-of-function | snpEff | NP_610943.2 | Testis | XP_008206136.1 | Testis | Ubiquitin specific protease 20/33 | Frameshift |
| Non-tolerated | SIFT | NP_788479.1 | acc | XP_008207671.1 | Testis | ergic53 | validated |
| Non-tolerated | SIFT | NP_727442.1 | spt | XP_008217640.1 | Female body | Raspberry | |
| Non-tolerated | SIFT | NP_788565.1 | acc | XP_001602982.1 | Testis | Isoleucyl-tRNA synthetase | validated |
| Non-tolerated | SIFT | NP_611087.1 | Tubule | XP_001606432.1 | Testis | Cysteinyl-tRNA synthetase | |
| Non-tolerated | SIFT | NP_731238.1 | Testis | XP_008205904.1 | Testis | Dipeptidyl aminopeptidase III | Validated |
| Non-tolerated | SIFT | NP_608533.1 | Testis | XP_003427673.2 | Testis | Uncharacterized | Validated |
| Non-tolerated | SIFT | NP_649645.1 | acc | XP_001607849.1 | Testis | Small ribonucleoprotein particle protein SmD2 | |
| Non-tolerated | SIFT | NP_477412.1 | trachea | XP_001601436.1 | Testis | nop5 | |
| Non-tolerated | SIFT | NP_001261050.1 | XP_008205733.1 | Testis | Quaking related 54B | ||
| Unstable protein | MU-pro | NP_611131.2 | Fat body | XP_008208307.1 | Testis | Uncharacterized | |
| Unstable protein | MU-pro | NP_611350.1 | Tubule | XP_001067690.2 | Testis | Autophagy-related 7 | |
| Not enriched in reproductive tissue | |||||||
| Unstable protein | MU-pro | XP_008204426.1 | Female body | Uncharacterized | |||
| Unstable protein | MU-pro | ||||||
| Non-tolerated | SIFT | NP_611179.3 | XP_008203900.1 | Female body | Eps15 homology domain containing protein-binding protein 1 | ||
| Unstable protein | MU-pro | NP_611223.4 | Trachea | anaphase promoting complex subunit 10 | |||
| Non-tolerated | SIFT | NP_725570.1 | Fat body | XP_008208687.1 | Female head | HMG coenzyme A synthase | |
| Non-tolerated | SIFT | NP_572695.2 | Eye | XP_001604944.2 | Female body | antdh | |
We obtained SIFT scores for a total of 11,874 homozygous SNPs in protein-coding sequences (see fig. 2 for an example). Specifically, we found twelve variants for which the asexual genotype was deleterious, whereas the sexual genotype was not (table 1). The reverse was true for 671 variants, indicating that the sexual genome carried a heavier load of deleterious mutations compared with the asexual genome (Fisher exact test P < 2.2 × 10−16). We assessed the putative function of these genes affected by predicted deleterious variants in both the asexual or sexual lineage by identifying their homologs in D. melanogaster and determining the tissue in which the homologue was most expressed. The few deleterious variants identified using SIFT in the genome of the asexual lineage were found in genes expressed in testes, accessory glands, and spermathecae (fig. 3). While this distribution did not differ from random expectation (Fisher exact tests after FDR correction P > 0.25), it is noteworthy that these are all tissues whose functions are likely to be redundant in asexuals. We searched for homologs in the N. vitripennis genome and confirmed that the two genes for which D. melanogaster homologs were enriched in testes, showed the same pattern in N. vitripennis. We also searched for N. vitripennis homologs for two genes for which no flyatlas data was available. One of these genes was enriched in testes in N. vitripennis, adding an additional candidate trait-loss gene to our list (table 1). In contrast, genes containing deleterious variants in the sexual lineage were more often highly expressed in ovaries and less often in salivary glands than expected by chance (Fisher exact tests after FDR correction P = 0.001). This was not the case for genes expressed in testis (fig. 3). Ovarian genes are less likely to be expressed in males and deleterious mutations in these genes are therefore not purged in sexual haplodiploids.
Fig. 2.—

Alignment of reads from the sexual lineage against the reference genome of the asexual lineage, showing variants in a gene primarily expressed in testis. From parasitoids.labs.vu.nl.
Fig. 3.—

Deleterious variants in the Leptopilina clavipes genome are overrepresented in reproductive tissues. Deleterious (non-tolerated) variants were identified using SIFT and the orthologs of the genes in which they were found were searched for in the genome of Drosophila melanogaster. The tissue in which each of these orthologs show highest expression was identified in Flyatlas (Chintapalli et al. 2007) and is shown in blue for asexual and sexual L. clavipes lineages. The distribution of tissues with most abundant expression for all genes in Flyatlas is shown in grey. Significant Fisher exact P values following FDR correction are indicated with an asterisk.
MUpro analysis yielded comparable patterns as Sift analysis in the abundance and function of affected genes in the sexual and asexual lineage. Of the 9,579 non-synonymous differences found between the genomes of the sexual and the asexual lineages, MUpro predicted 379 differences to result in a less stable protein in the asexual lineage (1.3% predicted at > 0.8 confidence). Waspatlas data was available for three of the five genes predicted at high confidence to be less stable in the asexual lineage (table 1). Two of these were enriched in male reproductive tissue. Flyatlas data was also available for three of the five genes, but none was enriched in a tissue related to sexual function (table 1). In contrast, 9,200 (96%) were predicted to have resulted in a less stable protein in the sexual lineage (54.2% predicted at > 0.8 confidence). Again, the affected genes in the sexual lineage were biased towards those expressed in reproductive tissues (mainly ovaries; supplementary fig. S3, Supplementary Material online).
Downstream Analysis of Candidate Decayed Genes
Four of the putative trait-loss genes identified using SIFT (see above) were selected for further testing: in two of these, the N. vitripennis and/or D. melanogaster homologs were enriched in testes and in the other two, a homolog was enriched in accessory glands. We genotyped twelve asexual and nine sexual lineages of L. clavipes at these four loci. The genetically different asexual lineages did not carry the same putative trait-loss variants. Furthermore, the pattern of presence/absence of the variants across the 12 asexual lineages followed their phylogenetic relationships based on neutral microsatellite markers (fig. 4), with more closely related lineages sharing more variants with the genome-sequenced lineage.
Fig. 4.—
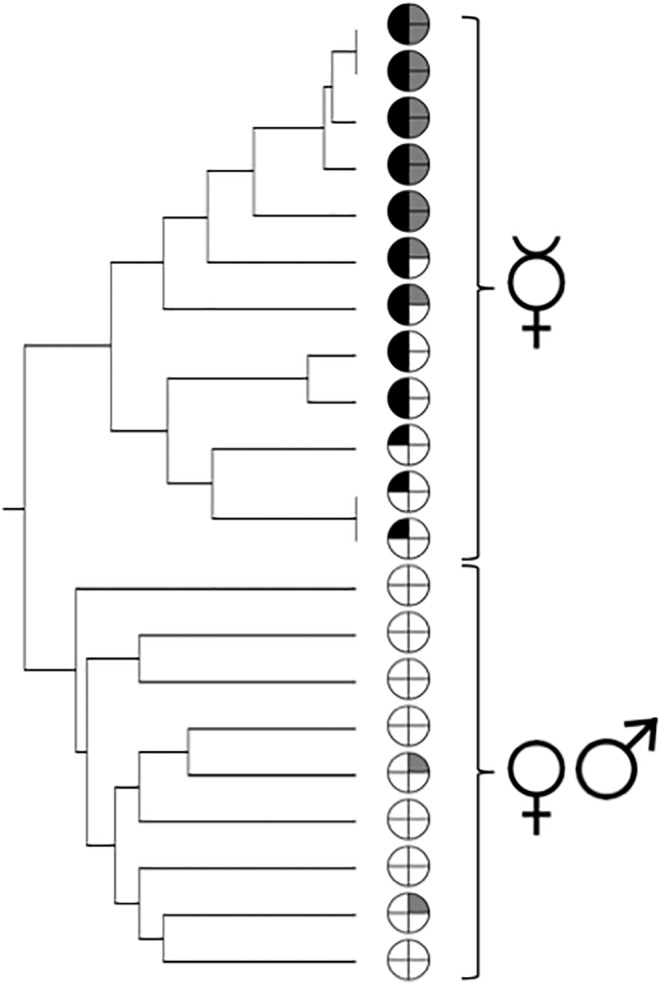
Phylogenetic relationship among twelve asexual and nine sexual lineages of Leptopilina clavipes, based on microsatellite markers; data from (Kraaijeveld et al. 2011). Pie charts indicate the presence (either black or dark grey)/absence (white) of putative trait-loss variants (left: two genes enriched in testes, right: two genes enriched in accessory glands).
The occurrence of putative deleterious variants also differed between asexual and sexual lineages. Both of the putative trait loss variants in a gene enriched in the testes were unique to the asexual lineages (fig. 4). Of the variants in a gene enriched in the accessory glands, one also segregated among the sexual lineages, while the other was only found in the asexual lineages.
Discussion
We sequenced the genome of an asexual lineage of the parasitoid wasp L. clavipes. A small number of variants in coding regions were predicted to be deleterious in this asexual lineage, and these were concentrated in genes expressed in tissues related to redundant sexual functions. We identified a shortlist of deleterious variants in 16 genes that potentially contributed to the observed phenotypic decay of redundant sexual traits in this species. Subsequent analysis of four of these variants showed that not all asexual lineages carry the same deleterious variants.
The patterns of occurrence of deleterious variants in the genome of asexually reproducing L. clavipes are consistent with phenotypic patterns of trait decay observed in L. clavipes. Asexual lineages of this species have degenerated spermathecae (Kraaijeveld et al. 2009) and reduced male fertility (Pannebakker et al. 2005). The spermatheca-specific and testis-specific genes identified as carrying deleterious mutations thus represent candidates underlying these degenerated phenotypes. The genetic basis of reduced male fertility was previously mapped to a single QTL of large effect (Pannebakker et al. 2004a). Subsequent work should focus on the genomic location of the identified candidate genes, and test whether or not they overlap with the QTL region.
Our analysis of gene function is based on tissue-specific expression data of putative homologs in N. vitripennis and D. melanogaster. Tissue-specific expression data for L. clavipes is needed to confirm that our interpretations are correct. However, gene expression patterns tend to be conserved among insects (Baker et al. 2011). Tissue-specific expression data for N. vitripennis covers fewer tissues than that for D. melanogaster, but the patterns of enrichment match for most of our candidate genes (especially when assuming that accessory glands were co-extracted with the testes in N. vitripennis).
It is noteworthy that we identified 15 putatively deleterious variants in genes expressed mostly in male reproductive tissues, but only one in a redundant female-specific tissue (spermathecae). Spermathecae in asexual L. clavipes are heavily degraded and non-functional (Kraaijeveld et al. 2009). Males derived by curing asexual mothers from Wolbachia infection are still fertile—albeit to a reduced degree (Pannebakker et al. 2005). One possible explanation for this apparent discrepancy is that one or more genes crucial for spermathecal development may have been deleted mostly or entirely from the genome and we consequently were unable to detect them in our analysis. Although many genes are known to be upregulated or even specific to mature spermatheca in Drosophila (Prokupek et al. 2008; Schnakenberg et al. 2011), little is known about the genes involved in spermathecal development. The gene Hr39 was shown to be essential for normal spermathecal development in Drosophila (Allen and Spradling 2008) and a homolog of this gene is present in L. clavipes. Female-specific sexual function tends to degrade rapidly upon the switch to asexual reproduction (van der Kooi and Schwander 2014), which might indicate that female-specific trait decay is often caused by few mutations of large effect. Male-specific sexual functions, on the other hand, decay much more slowly (van der Kooi and Schwander 2014). Since we found several candidate variants that could contribute to the decay of male-specific sexual traits, our results suggest that sexual trait decay in L. clavipes males is the result of multiple mutations of small effect.
Our results suggest that the genome of a sexual L. clavipes lineage was more heavily loaded with deleterious variants than that of the asexual lineage. Deleterious variants in the sexual lineage were overrepresented in genes enriched in ovaries, which are probably only expressed in diploid females in which recessive alleles are partially shielded from selection. Our interpretation of the excess of deleterious variants is therefore that prolonged inbreeding exposed recessive deleterious variants that segregated in the ancestral sexual lineage. This interpretation would be consistent with inbreeding effects in other haplodiploid organisms (Brückner 1978; Henter 2003; Tortajada et al. 2009; Tien et al. 2015). Deleterious variants in female-specific tissues were not observed in the asexual lineage, suggesting that these alleles must have been purged by lineage selection during the transition from sexual to asexual reproduction.
We present the first genome-wide assessment of the genetic changes potentially underlying sexual trait decay in an asexual insect. Our results indicate that the genome of asexual L. clavipes was relatively free of deleterious variants and that damaging effects were concentrated in redundant sexual genes. The list of candidate genes we identified will provide an excellent starting point for unraveling the genomics of trait decay in this and similar systems.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We acknowledge Yavuz Ariyurek, Henk Buermans, Emile de Meijer and Kristiaan van der Gaag for help with components of the sequencing work. We thank Pauline Ng and Swarnaseetha Adusumalli for help with the SIFT analysis. Peter Neleman helped with the bioinformatics. JW, ON, TZ, MP acknowledge Dr Alexander Donath for help installing software on the HPC cluster of the Zoological Research Museum Alexander Koenig in Bonn. The research by JW, ON, TZ, MP was supported by the Leibniz Graduate School “Genomic Biodiversity Research”. JE and KK were supported by a Vici grant from the Netherlands Organization for Scientific Research. KK was supported by a Veni grant from the Netherlands Organization for Scientific Research.
Literature Cited
- Allen AK, Spradling AC. 2008. The Sf1-related nuclear hormone receptor Hr39 regulates Drosophila female reproductive tract development and function. Development 135:311–321. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Baker DA, et al. 2011. A comprehensive gene expression atlas of sex- and tissue-specificity in the malaria vector, Anopheles gambiae. BMC Genomics 12:296.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brückner D. 1978. Why are there inbreeding effects in haplo-diploid systems? Evolution (N Y) 32:456–458. [DOI] [PubMed] [Google Scholar]
- Chan YF, et al. 2010. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327:302–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Mackey AJ, Stoeckert CJ, Roos DS. 2006. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34:D363–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Randall A, Baldi P. 2006. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 62:1125–1132. [DOI] [PubMed] [Google Scholar]
- Chikhi R, Medvedev P. 2014. Informed and automated k-mer size selection for genome assembly. Bioinformatics 30:31–37. [DOI] [PubMed] [Google Scholar]
- Chintapalli VR, Wang J, Dow JAT. 2007. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 39:715–720. [DOI] [PubMed] [Google Scholar]
- Cingolani P, et al. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Pan Y-H, Zhang Y, Jones G, Zhang S. 2011. Progressive pseudogenization: vitamin C synthesis and its loss in bats. Mol Biol Evol. 28:1025–1031. [DOI] [PubMed] [Google Scholar]
- Dakovic N, et al. 2014. The loss of adipokine genes in the chicken genome and implications for insulin metabolism. Mol Biol Evol. 10:2637–2646. [DOI] [PubMed] [Google Scholar]
- Davies NJ, Tauber E. 2015. WaspAtlas: a Nasonia vitripennis gene database and analysis platform. Database 2015:bav103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin G, Godin J-R, Pagé B. 2011. The genetics of vitamin C loss in vertebrates. Curr Genomics 12:371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellers J, Kiers ET, Currie CR, McDonald BR, Visser B. 2012. Ecological interactions drive evolutionary loss of traits. Ecol Lett. 15:1071–1082. [DOI] [PubMed] [Google Scholar]
- Feng P, Zheng J, Rossiter SJ, Wang D, Zhao H. 2014. Massive losses of taste receptor genes in toothed and baleen whales. Genome Biol Evol. 6:1254–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, et al. 2015. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, Selengut JD, White O. 2003. The TIGRFAMs database of protein families. Nucleic Acids Res. 31:371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare EE, Johnston JS. 2011. Genome size determination using flow cytometry of propidium iodide-stained nuclei In: Orgogozo V, Rockman MV, editors. Molecular methods for evolutionary genetics.. Totowa (NJ; ): Humana Press. p. 3–12. [DOI] [PubMed] [Google Scholar]
- Henter HJ. 2003. Inbreeding depression and haplodiploidy: experimental measures in a parasitoid and comparisons across diploid and haplodiploid insect taxa. Evolution (N Y) 57:1793–1803. [DOI] [PubMed] [Google Scholar]
- Hiller M, et al. 2012. A ‘forward genomics’ approach links genotype to phenotype using independent phenotypic losses among related species. Cell Rep. 2:817–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C, Yandell M. 2011. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12:491.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, et al. 2014. InterProScan 5: Genome-scale protein function classification. Bioinformatics 30:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraaijeveld K, et al. 2012. Transposon proliferation in an asexual parasitoid. Mol Ecol. 21:3898–3906. [DOI] [PubMed] [Google Scholar]
- Kraaijeveld K, Franco P, De Knijff P, Stouthamer R, Van Alphen JJM. 2011. Clonal genetic variation in a Wolbachia-infected asexual wasp: horizontal transmission or historical sex?. Mol Ecol. 3644–3652. [DOI] [PubMed] [Google Scholar]
- Kraaijeveld K, Franco P, Reumer BM, van Alphen JJM. 2009. Effects of parthenogenesis and geographic isolation on female sexual traits in a parasitoid wasp. Evolution 63:3085–3096. [DOI] [PubMed] [Google Scholar]
- Kumar S, Jones M, Koutsovoulos G, Clarke M, Blaxter M. 2013. Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front Genet. 4:237.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P. 2009. SMART 6: Recent updates and new developments. Nucleic Acids Res. 37:229–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, et al. 2013. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 1308.2012. [Google Scholar]
- Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. 2005. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33:6494–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. 1991. Predicting coiled coils from protein sequences. Science (80-.) 252:1162–1164. [DOI] [PubMed] [Google Scholar]
- Macarthur DG, et al. 2012. A systematic survey of loss-of-function variants in human protein-coding genes. Science (80-.) 205:823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, et al. 2011. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 471:216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Thomas PD. 2013. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 41:D377–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW, et al. 2000. A whole-genome assembly of Drosophila. Science 287:2196–2204. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2001. Predicting deleterious amino acid substitutions. Genome Res. 11:863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannebakker BA, et al. 2005. Sexual functionality of Leptopilina clavipes (Hymenoptera: Figitidae) after reversing Wolbachia-induced parthenogenesis. J Evol Biol. 18:1019–1028. [DOI] [PubMed] [Google Scholar]
- Pannebakker BA, Beukeboom LW, van Alphen JJM, Brakefield PM, Zwaan BJ. 2004a. The genetic basis of male fertility in relation to haplodiploid reproduction in Leptopilina clavipes (Hymenoptera: Figitidae). Genetics 168:341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannebakker BA, Pijnacker LP, Zwaan BJ, Beukeboom LW. 2004b. Cytology of Wolbachia-induced parthenogenesis in Leptopilina clavipes (Hymenoptera: Figitidae). Genome 47:299–303. [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I. 2007. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23:1061–1067. [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Ning Z, Keane T, Korf I. 2009. Assessing the gene space in draft genomes. Nucleic Acids Res. 37:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedruzzi I, et al. 2013. HAMAP in 2013, new developments in the protein family classification and annotation system. Nucleic Acids Res. 41:584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokupek A, et al. 2008. An evolutionary expressed sequence tag analysis of Drosophila spermatheca genes. Evolution 62:2936–2947. [DOI] [PubMed] [Google Scholar]
- Schnakenberg SL, Matias WR, Siegal ML. 2011. Sperm-storage defects and live birth in Drosophila females lacking spermathecal secretory cells. PLoS Biol. 9:e1001192.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant F. 2002. ProDom: automated clustering of homologous domains. Brief Bioinform. 3:246–251. [DOI] [PubMed] [Google Scholar]
- Sigrist CJA, et al. 2013. New and continuing developments at PROSITE. Nucleic Acids Res. 41:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe I, et al. 2015. CATH: Comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 43:D376–D381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT. 2014. Exploring genome characteristics and sequence quality without a reference. Bioinformatics 30:1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Waack S. 2003. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19:ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Suen G, et al. 2011. The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genet. 7:e1002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien NSH, Sabelis MW, Egas M. 2015. Inbreeding depression and purging in a haplodiploid: gender-related effects. Heredity (Edinb) 114:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortajada AM, Carmona MJ, Serra M. 2009. Does haplodiploidy purge inbreeding depression in rotifer populations? PLoS One 4:e8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kooi CJ, Schwander T. 2014. On the fate of sexual traits under asexuality. Biol Rev Camb Philos Soc. 89:805–819. [DOI] [PubMed] [Google Scholar]
- Wilson D, Madera M, Vogel C, Chothia C, Gough J. 2007. The SUPERFAMILY database in 2007: families and functions. Nucleic Acids Res. 35:308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, et al. 2004. PIRSF: family classification system at the Protein Information Resource. Nucleic Acids Res. 32:D112–D114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyder S, Kriventseva EV, Schröder R, Kadowaki T, Zdobnov EM. 2007. Quantification of ortholog losses in insects and vertebrates. Genome Biol. 8:R242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.