

Abstract
As a tumor suppressor, RASSF5 (NORE1A) activates MST1/2 thereby modulating the Hippo pathway. Structurally, activation involves RASSF5 and MST1/2 swapping their SARAH domains to form a SARAH heterodimer. This exposes the MST1/2 kinase domain which homodimerizes, leading to trans-autophosphorylation. The SARAH-SARAH interaction shifts RASSF5 away from its autoinhibited state and relieves MST1/2 autoinhibition. Separate crystal structures are available for the RA (Ras association) domain and SARAH dimer, where SARAH is a long straight α-helix. Using all-atom molecular dynamics simulations, we modeled the RASSF5 RA with a covalently connected SARAH to elucidate the dynamic mechanism of how SARAH mediates between autoinhibition and Ras triggered-activation. Our results show that in inactive RASSF5 the RA domain retains SARAH, yielding a self-associated conformation in which SARAH is in a kinked α-helical motif that increases the binding interface. When RASSF5 binds K-Ras4B-GTP, the equilibrium shifts toward SARAH’s interacting with MST. Since the RA/SARAH affinity is relatively low, whereas that of the SARAH heterodimer is in the nM range, we suggest that RASSF5 exerts its tumor suppressor action through competition with other Ras effectors for Ras effector binding site, as well as coincidentally its recruitment to the membrane to help MST activation. Thus, SARAH plays a key role in RASSF5’s tumor suppression action by linking the two major pathways in tumor cell proliferation: Ras and the MAPK (tumor cell proliferation-promoting) pathway, and the Hippo (tumor cell proliferation-suppressing) pathway.
Introduction
The classical Ras association domain family (RASSF) proteins such as RASSF1A and RASSF5 (also known as NORE1A) are tumor suppressors, promoting cell apoptosis.1,2 RASSF5 activates mammalian sterile 20-like kinase 1/2 (MST1/2) in the Hippo pathway.3–9 Hippo’s signaling stimulates phosphorylation and thereby activation of a core kinase cascade including MST1/2 and large tumor suppressor 1/2 (LATS1/2), leading to phosphorylation of Yes associated protein 1 (YAP1).10,11 YAP1’s phosphorylation encodes its degradation, thus abolishing its transcriptional activity.12 Overexpression amplifies oncogenic signaling through YAP1’s association with the TEA domain (TEAD) family of transcription factors.13–16 RASSF5 links Ras and the Hippo pathway.17 Ras activates Raf kinase, thus the mitogen-activated protein kinases (MAPK) pathway (Ras/Raf/MEK/ERK). Hippo and MAPK are independent core pathways with similar actions; drug resistance mutations in the MAPK pathway and inactivating mutations in the Hippo pathway lead to similar consequences in tumor cell proliferation.15,16 In vitro, in the absence of cell membrane, RASSF5 promotes cancer; in vivo, it acts as a tumor suppressor. RASSF5 can be considered as an adaptor protein (Fig. 1), connecting Ras to the MST1/2 kinase through a conformational change.17
Fig. 1.
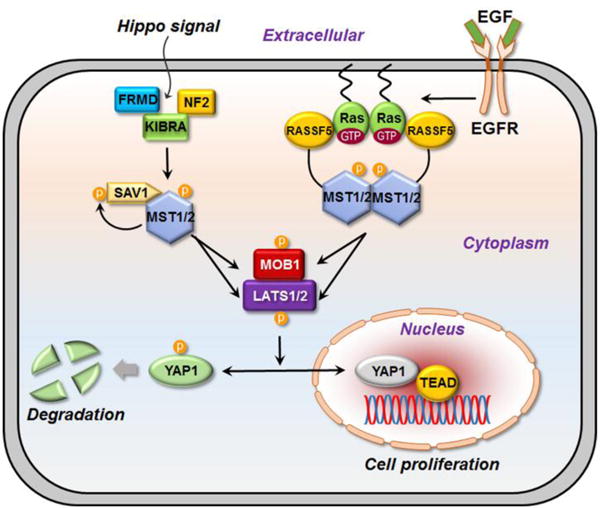
The Hippo pathway controls the cell proliferation and apoptosis. The central kinase is MST. In mammals, MST can be activated by upstream proteins, KIBRA-Ex-NF2 complex, SAV1, and RASSF5. These regulators facilitate the phosphorylation of MST kinase domain. Here we propose a mechanism of how RASSF5, as a tumor suppressor, activates the MST by associating with Ras. Once the epidermal growth factor receptor (EGFR) receives an EGF signal, Ras is activated and, with the GTP bound, anchors into the membrane and forms a cluster. RASSF5 associates with Ras and undergoes structural changes for binding to MST, facilitating its kinase domain dimerization. As MST is activated, the signal cascades down the Hippo pathway and phosphorylates MOB1 and LATS1. MOB1 enhances the activity of LATS1. LATS1 phosphorylates YAP1 resulting its degradation. YAP1 degradation leads to tumor suppression. On the other hand, if YAP1 enters the nucleus together with the transcriptional factor, TEAD, it causes cell proliferation.
RASSF5 interacts with MST1/2 through heterodimerization of the C-terminal SARAH (Sav-RASSF-Hippo) domains.18,19 The RASSF5-MST1/2 SARAH heterodimer promotes homodimerization of the N-terminal kinase domain of MST1/2, followed by trans-autophosphorylation. The C-terminal region of RASSF5 contains the Ras association (RA) and SARAH domains (Fig. 2). The structure of the N-terminal region, including the putative membrane binding C1 domain, is currently unavailable. MST1/2 also contains the SARAH domain, which is structurally similar to that of RASSF5. RASSF5 connects Ras to MST1/2; its RA domain binds the Ras catalytic domain and the SARAH domain forms an antiparallel coiled coil with MST1/2 SARAH. The coiled coil conformation illustrates that SARAH is a long stretched α-helix. The coiled coil can be also observed in the crystal structures of the RASSF5-RASSF5 and MST-MST SARAH homodimers. The affinity of the RASSF5-MST SARAH heterodimer is higher than those of either homodimers, RASSF5-RASSF5 or MST-MST.20
Fig. 2.
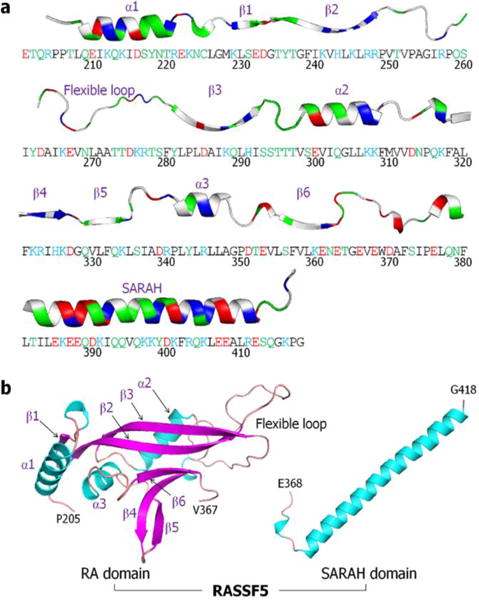
RASSF5 sequence and structure. Human RASSF5 contains 418 residues involving the RA domain (residues 274–364) and SARAH (residues 366–413). (a) The sequence of N-terminal truncated RASSF5 protein with each domain structure embedded in a cartoon representation. In the sequence, hydrophobic/glycine, polar, positively charged, and negatively charged residues are colored black, green, blue, and red, respectively. Similarly, in the ribbon representation for the secondary structures, the same colors were used, except for the hydrophobic/glycine residues which are colored white. (b) Crystal structures of murine RA domain (PDB code: 3DDC) and human SARAH domain (PDB code: 4LGD) as a long straight α-helix defined in a coiled coil with MST2 SARAH domain. In the RA domain, the missing flexible loop (residues 254–278) was constructed by using the Modeller server.30
RASSF5 is of particular importance since it links the MAPK and the Hippo pathways, two core pathways in tumor cell proliferation.17 Drug resistant mutations in Ras or MAPK proteins are often accompanied by mutations in the Hippo pathway or YAP1. Ras is still undruggable, currently with no drug in the clinic. There is also a lack of structural information about RASSF5 functional states and thus the mechanism relating to how exactly Ras activates RASSF5 and how RASSF5 activation stimulates activation of the MST1/2 kinase whose action stimulates the Hippo pathway and thus YAP1 degradation. Currently, crystal structures of the functional regions of RASSF5 are only available for the unlinked SARAH and RA domains due to high fluctuations of the domains connected by a flexible linker. The crystal structures of SARAH RASSF5-MST2 heterodimer and the RASSF5-RASSF5 and MST2-MST2 homodimers exhibit the coiled coil motif. As to the RA domain, the available crystal structure is of murine RASSF5 in complex with a GTP analog-bound H-Ras. Thus, we conclude that in the active state, RASSF5 promotes the SARAH and RA domains interaction with their binding partners, Ras and MST – rather than with each other. Data are unavailable for the inactive state; however, we reasoned that in the absence of Ras and MST2, RASSF5 retracts both domains (RA and SARAH), yielding a self-associated conformation. That inactive ‘closed’ RASSF5 conformation can be regarded as the autoinhibited state. The lack of crystal structure data for such closed autoinhibited conformation suggests that the RA-SARAH inter-domain interaction is transient, not sufficiently stable to permit its crystallization; however, we can expect it to be present in the dynamic conformational ensembles, in which case it could be captured by sampling the broadly dispersed structures populating the free energy landscape of the inactive states. Ras can activate RASSF5 and by crossing over the free energy barrier from the closed toward the open, active state, shift the equilibrium to liberate the SARAH domain.
To corroborate RASSF5 structure in the inactive state, we modelled the self-associated RASSF5 conformation with SARAH covalently connected to the RA domain. Since no crystal structure of the combined form is available, we exploited docking programs to predict possible modes of the interaction between SARAH and the RA domain. In the initial prediction, both long straight α-helical SARAH extracted from the crystal coiled coil, and kinked SARAH sampled from replica-exchange molecular dynamics (REMD) simulations were used. The prediction programs generated multiple decoys of self-associated RASSF5, and a screening process based on the energy score was performed in order to sample the best initial configurations for standard atomistic molecular dynamics (MD) simulations in solution. Our results verify that in the inactive state RASSF5 persists in the self-associated conformation, in which SARAH contacts the RA domain through various interfaces. However, when those same RASSF5 structures interact with GTP-bound K-Ras4B, SARAH tends to disengage from the RA domain, demolishing the weakly self-associated conformation. RASSF5 activation by GTP-bound Ras points to a shift of the RASSF5 ensemble from the inactive to the active state toward the strongly associated SARAH heterodimer. Variants of the RASSF5 conformation suggest that the protein acts as an adaptor between Ras and MST1/2 (Fig. 1). Scaffolding proteins typically control regulation dynamically.21 Our simulated RASSF5 structures in atomic detail reveal how the protein accomplishes the conformational adjustments which are required for MST activation and Hippo pathway signaling, and offer the rational and target for Ras-driven cancer.
Methods
For the SARAH dimer simulations, the crystal structures of SARAH dimers, RASSF5-MST2 SARAH heterodimer (PDB code: 4LGD), MST2-MST2 SARAH homodimer (PDB code: 4OH9), and RASSF5-RASSF5 SARAH homodimer (PDB code: 2YMY) were obtained from the Protein Data Bank (PDB). The CHARMM program22 was used to construct the set of initial conformations and to relax the systems for the atomistic MD simulations. Using the same MD protocols as in our previous works,23–29 the dimer systems were solvated by the modified TIP3P water model and subsequently minimized using the steepest descent (SD) and adopted-basis Newton-Raphson (ABNR) methods, followed by consecutive cycles of dynamics in the pre-equilibrium stage. During the pre-equilibrium simulations, the initial crystal structures of SARAH dimers were gradually relaxed after solvation.
To sample ensembles of monomeric SARAH, the coordinates of RASSF5 SARAH were extracted from the RASSF5-MST2 SARAH heterodimer (PDB code: 4LGD). The isolated RASSF5 SARAH monomer was subjected to REMD simulations for 50 ns. Twelve replicas of the SARAH monomer were simulated at temperatures in the range from 300 K to 360 K, incremented gradually for each replica. During the simulation, temperatures were exchanged between trajectories according to a Metropolis criterion. After the simulations, ensemble configurations of the SARAH monomer with the kinked α-helical motif were determined by analyzing replica trajectories, discarding the high-temperature replicas. Based on the population distribution of the interaction energy as a function of residue pair-distance, five initial kinked SARAH configurations (KS1 to KS5) were obtained.
To construct the N-terminal truncated RASSF5 (residues 205 – 418) structure, the SARAH domain was covalently connected to the RA domain. The crystal structure of the RA domain from murine RASSF5 (PDB code: 3DDC) was obtained from the PDB server and used to model the human RA domain. Residue modifications were made to obtain the human sequence, and the flexible loop with missing coordinates (residues 254 – 278) was constructed by using the Modeller server.30 Both straight α-helical SARAH extracted from the crystal structure and kinked SARAH defined by the REMD simulations were used to predict the self-associated RASSF5 conformation by using the PRISM31,32 and Patchdock33,34 servers. All-atom MD simulations were prepared for the best fourteen configurations of the RASSF5 structure predicted by the docking programs. During the simulations, ensembles of the self-associated RASSF5 conformations in an aqueous environment were obtained. For the simulations of RASSF5 in complex with Ras, seven different RASSF5 conformations among the fourteen configurations were selected to model the dimeric RASSF5/Ras complex. The crystal structure of 3DDC contains the murine RASSF5 RA domain in complex with the GNP-bound H-RasD30E/E31K. By replacements of H-Ras with the GTP-bound K-Ras4B (PDB code: 3GFT) and the murine RASSF5 RA domain with the selected RASSF5 configurations, seven different dimeric configurations for the RASSF5/K-Ras4B-GTP complex, DC1(1), DC2(4), DC3(6), DC4(9), DC5(10), DC6(11), and DC7(14), where DC represents dimeric configuration and the numbers in the parenthesis denote the monomeric RASSF5 configuration, were obtained. The structure of 3GFT contains a point mutation, Q61H, which was replaced with the wild-type residue, and the GNP in 3GFT was replaced by GTP.
The initial fourteen configurations of monomeric RASSF5 and seven dimeric configurations of RASSF5 in complex with K-Ras4B-GTP were solvated by the TIP3P water model and subsequently minimized with the protein backbone rigid for 10,000 steps, followed by a dynamic cycle of 50,000 time steps. All systems were subsequently neutralized, with the number of counterions needed to neutralize the systems. In addition, a number of ions, Na+ and Cl−, were added to the systems to satisfy a total cation concentration near 100 mM, followed by the same minimization and dynamic protocols of previous stage. In the pre-equilibrium stages, harmonic restraints were applied to the heavy atoms (k = 5 kcal/mol/Å2/atom) and gradually relaxed to k = 0 with a full particle mesh Ewald (PME) calculation for long-range electrostatic interactions and a constant temperature (Nosé−Hoover) thermostat/barostat at 310 K. Following pre-equilibrium, a 200 ns production run was performed with the NAMD 2.10 code35 and the CHARMM program22 with version 3636 on the Biowulf cluster at the National Institutes of Health (Bethesda, MD). Averages were taken after 30 ns, discarding initial transients.
Results
Strong interaction of the RASSF5-MST2 SARAH heterodimer compared to the SARAH homodimers
The SARAH domain plays a key role in RASSF5 and MST2 association. The formation of RASSF5-MST2 SARAH heterodimer facilitates the MST2 kinase domain dimerization and trans-autophosphorylation, leading to cell apoptosis. The SARAH-SARAH interaction involves an antiparallel coiled coil formation. Both the RASSF5 and MST2 SARAHs can form heterodimeric and homodimeric coiled coils, but the heterodimeric coiled coil has a stronger dimeric interface.20 The RASSF5 SARAH domain is very similar to the MST2 SARAH; both are long straight α-helices of ~50 residues in a coiled coil organization (Fig. 3a,b). To decipher the relative interface interaction strength for different SARAH dimers, we simulated three SARAH dimers observed in crystals: RASSF5-MST2 SARAH heterodimer (PDB code: 4LGD), MST2-MST2 SARAH homodimer (PDB code: 4OH9), and RASSF5-RASSF5 SARAH homodimer (PDB code: 2YMY). As expected, we observed that RASSF5 SARAH strongly interacts with MST2 SARAH, while its interaction with the same RASSF5 SARAH is weaker (Fig. 3c). Similarly, the MST2 SARAH interaction with the same SARAH is weaker than the heterodimeric SARAH interaction, but it is relatively stronger than the homodimeric RASSF5 SARAH interaction. The strong heterodimeric SARAH interaction is due to strong electrostatic contribution, since RASSF5 SARAH is acidic with the isoelectric point, pI < 7, while MST2 SARAH is basic with pI > 7. These bipolar characteristics favor heterodimer formation with strong electrostatic attraction. To corroborate the SARAH dimer interface, we examined key residues involved in dimer formation. For the RASSF5-MST2 SARAH heterodimer, salt bridge interactions between residues E385-R474, E388-R467, E388-R474, K398-E462, and R403-D456 (former and latter residues denote RASSF5 and MST2, respectively) strongly retain the dimer interface. Similarly, the salt bridge interactions between the residues, D456-R474, E462-R469, R469-E462, and R474-D456 for the MST2-MST2 SARAH homodimer and E385-R403, E388-R403, R403-E388, and R403-E385 for the RASSF5-RASSF5 SARAH homodimer hold the dimer interface. In the salt bridge interactions, we found that residues E385, E388, and R403 in the RASSF5 SARAH domain, and R474 and D456 in the MST2 SARAH domain are frequently involved in the dimer interface. In contrast, for both MST2-MST2 and RASSF5-RASSF5 SARAH homodimers, relatively strong hydrophobic and hydrophilic interactions can be observed (see Table S1, ESI†), although the contributions from these interactions to the total interaction energy are relatively weak. The E388A and E388K mutations in RASSF5 weaken both the heterodimeric and homodimeric SRARAH interactions (Fig. 3b,c). The E388A mutation reduces the salt bridge interactions, and the E388K mutation introduces unfavorable electrostatic repulsion with its binding partners (Table S1, ESI†). Of particular note, based on the data from the catalogue of somatic mutations in cancer (COSMIC), the mutation E388K in RASSF5 can cause cancer.
Fig. 3.
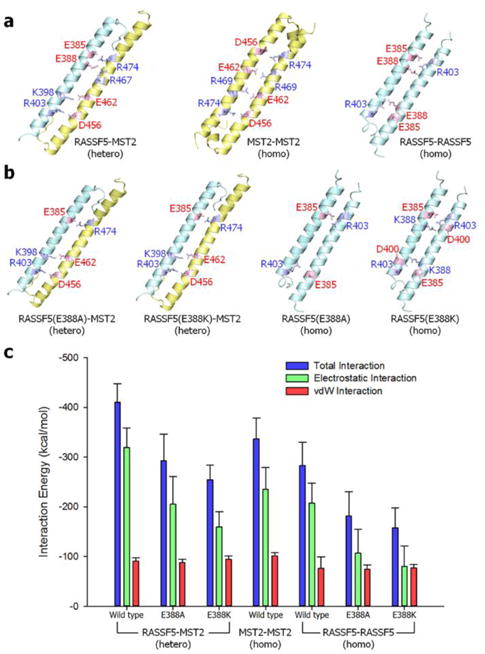
The SARAH-SARAH interaction in the coiled coil dimer. (a) Averaged structures of RASSF5-MST2 SARAH heterodimer (left), MST2-MST2 (center) and RASSF5-RASSF5 (right) homodimers. (b) Averaged structures of RASSF5-MST2 SARAH heterodimers with the E388A (far left) and E388K (second left) mutations in RASSF5, and RASSF5-RASSF5 SARAH homodimers with the E388A (second right) and E388K (far right) mutations in RASSF5. In the ribbon representation, RASSF5 and MST2 are colored cyan and yellow, respectively. The salt bridge pairs are shown in the average structures of each SARAH-SARAH dimer. (c) Interaction energy gauging the SARAH-SARAH association in the coiled coil dimer. Averaged total interaction energy (blue bars), and the contributions from the electrostatic (green bars) and vdW (red bars) interactions for three different SARAH dimers are shown.
A kinked helical motif predominantly observed in the RASSF5 SARAH monomer
In the antiparallel coiled coil motif, SARAH exists as a long straight α-helix. When dissociated from the coiled coil, monomeric SARAH does not persist in a straight α-helical motif. We observed incidents of a kinked helical motif of monomeric RASSF5 SARAH emerging during the simulation (Fig. 4a). To verify the kinked SARAH structure as a representative of the monomeric conformational ensemble and its relative population, we employed HingeProt,37 an algorithm for protein hinge prediction using elastic network models, to predict the position of the kink. The results showed that a kink takes place at Q389. To obtain ensembles of monomeric SARAH conformations, we performed REMD simulations of the RASSF5 SARAH monomer. During the simulations, we observed that about 90% of the SARAH domain structures contain zero and one kink (see Table S2, ESI†). The location of the kink can be identified by analyzing the secondary structure, since residues at the kink have a low α-helical probability. Using STRIDE,38 we found that the kink occurs at the region involving residues 387–389 (Fig. 4b), which is consistent with the predicted location at Q389 by HingeProt.37 We sampled SARAH conformations over trajectories generated from the REMD simulations and sorted SARAH into several groups with similar conformations based on population maps for different SARAH topologies (Fig. 4c). To classify the conformations, we monitored the occurrence frequency of the interaction energy of two α-helical segments, separated at the hinge point, as a function of the distance between two residues located at both ends of SARAH. Thus highly kinked SARAH has a shorter residue pair-distance and stronger interaction. The two-dimensional contour surface of the population map provides highly populated states enclosed by many contour lines. The highly populated state located at the upper right on the map represents the monotonic conformation of straight SARAH, while widely dispersed kinked SARAH states indicate various SARAH conformations. From the contour surface, we extracted five different SARAH conformations with a single kink, from a highly kinked SARAH 1 (KS1) to less kinked SARAH 5 (KS5) (Fig. 4c). The sampled kinked SARAH conformations, as well as the straight SARAHs, were used in the construction of the self-associated RASSF5 model.
Fig. 4.
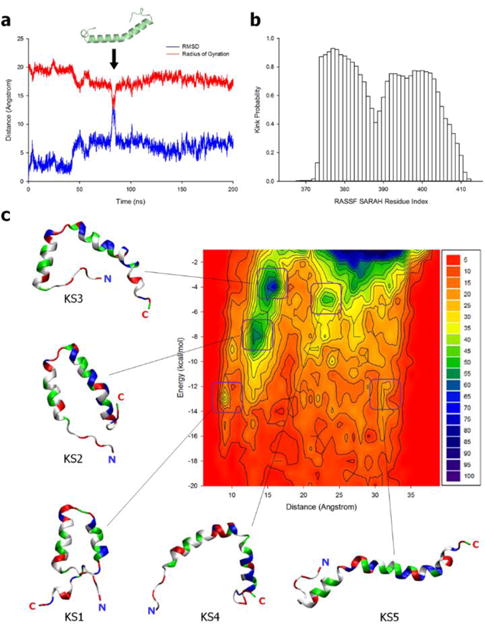
The analysis of RASSF5 SARAH monomer. (a) Time series of the Cα atoms root-mean-squared deviation (RMSD) from the starting point and the radius of gyration of SARAH monomer in the standard MD simulation. The SARAH structure in the graph reflects the kinked motif occurred at the event indicated by arrow. (b) Probability of the secondary structure analyzed by the STRIDE38 along the SARAH residues for the ensembles of SARAH monomer generated from the REMD simulation. (c) Two-dimensional contour map representing the occurrence frequency of the residue pair-distance dependence of the interaction energy between two α-helical segments in the kinked SARAH conformations sampled from the REMD simulation. Five selected kinked SARAH conformations from the population map. The SARAH configurations from KS1 with highly kinked to KS5 with less kinked, where KS denotes kinked SARAH, are marked on the map. In the ribbon representation of SARAH, hydrophobic/glycine, polar, positively charged, and negatively charged residues are colored white, green, blue, and red, respectively.
The self-associated RASSF5 conformation in the inactive sate
We constructed the N-terminal truncated RASSF5 structure (residues 205–418) with SARAH covalently connected to the RA domain. Since the structure of the human RASSF5 RA domain is currently unavailable, we adopted the murine RASSF5 RA domain (PDB code: 3DDC) converting its sequence to human. The murine and human RA have 96% sequence identity. The disordered region with missing coordinates for the residues 254–278 was constructed as a flexible loop by using the Modeller server.30 In the covalently connected SARAH with the RA domain, we considered two possible SARAH conformations; straight and kinked α-helix. Since the linker between the SARAH and RA domains can serve as a hinge point, there are a limited number of modes of SARAH binding to the RA domain. To obtain the self-associated inactive RASSF5 conformation, we exploited a powerful template-based protein-protein complex structure prediction algorithm (PRISM).31,32 By using a straight α-helical SARAH, defined from the crystal structure, we obtained four possible modes of the SARAH interaction with the RA domain (Fig. S1a, ESI†). With the kinked SARAH models (KS1-5 from the REMD simulations in Fig. 4c), we were able to obtain many possible decoys representing the RA-SARAH association by using the Patchdock server.33,34 For each kinked SARAH, the docking program generated 12 decoys of the RASSF5 structure (Fig. S1b–f, ESI†). Based on the energy scoring function, we selected the first two decoys for each kinked SARAH model, gathering 10 additional configurations. Thus, a total of 14 configurations representing the possible ensembles of self-associated RASSF5 were subjected to an all-atom MD simulation in aqueous environment. During the simulations, no immediate dissociation of SARAH from the RA domain was observed. Occasionally, we observed a structural convergence between the configurations. For example, configurations 6 and 7 present similar conformational ensembles, and the structure in configuration 2 resembles configuration 14 (Fig. 5a). To quantify the RA-SARAH interaction, we calculated the interaction energy of SARAH with the RA domain (Fig. 5b). Here, we found that the kinked SARAH interaction with the RA domain is relatively stronger than the straight SARAH interaction. The straight SARAH weakly interacts with the RA domain with fewer residues involved in the interface, while the kinked SARAH with conformational change folds onto the interface increasing the interaction surface. The weaker interaction of straight SARAH indicates that the RA domain does not have a comparable long helix. The long straight α-helical SARAH favors a coiled coil.
Fig. 5.
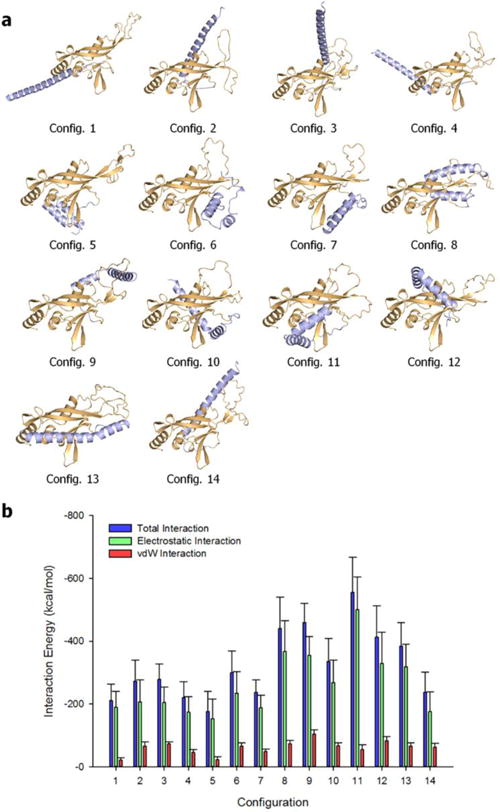
Self-associated RASSF5 conformation. (a) Averaged structures of RASSF5 after 200 ns molecular dynamics simulations. RSSF5 configurations 1 – 4 contain straight SARAH, and configurations 5 – 14 were modeled with kinked SARAH. In the cartoon, the RA and SARAH domains are colored orange and light blue, respectively. (b) Interaction energy of SARAH with the RA domain for the RASSF5 configurations. Averaged total interaction energy (blue bars), and the contributions from the electrostatic (green bars) and vdW (red bars) interactions for the RASSF5 configurations are shown.
To grade the self-associated RASSF5 conformations, we investigated the interacting residue types at the RA-SARAH interface. Unlike the SARAH residues involved in coiled coil formation, we found different residues participating in the RA-SARAH association (see Table S3, ESI†). In particular, for all 14 configurations we observed that the SARAH residues, E366, D370, E376, and E387, and the RA residues, R323, K334, and E353, are commonly involved in strong salt bridge interactions. A number of residue pairs are involved in hydrophobic interactions, additionally supporting the self-associated conformation, but no common residue pairs were observed, nor were hydrophilic interacting residue pairs. However, in the RA domain, hydrophobic residues, P283 and I337, and the hydrophilic residues, Y281, H325, Q329, and Q333, significantly contribute to the interaction with SARAH. Hydrogen bonding (H-bond) residue pairs were rarely observed during the simulations. To delineate the RA domain residues interacting with SARAH, we calculated the backbone amide NMR chemical shift perturbations (CSPs) for the RA domain using the ShiftX2 server39 (Fig. 6). These CSPs40,41 represent the difference in predicted chemical shifts for the trajectory of RA domain alone and as part of the various RASSF5 configurations shown in Figure 4b. For all configurations, strong CSPs are common in the region containing residues 254–278, which represent the flexible loop of the RA domain. In addition, we observed large CSPs for RA residues which are in contact with SARAH, consistent with the interaction pair analysis. We noticed that each configuration has distinct residue pair interactions as reflected in the inhomogeneous distributions of the strong CSPs among the configurations, suggesting that the SARAH interaction with the RA domain might be transient, and multiple modes of interaction are possible, in agreement with the absence of a crystal structure of the self-associated state.
Fig. 6.
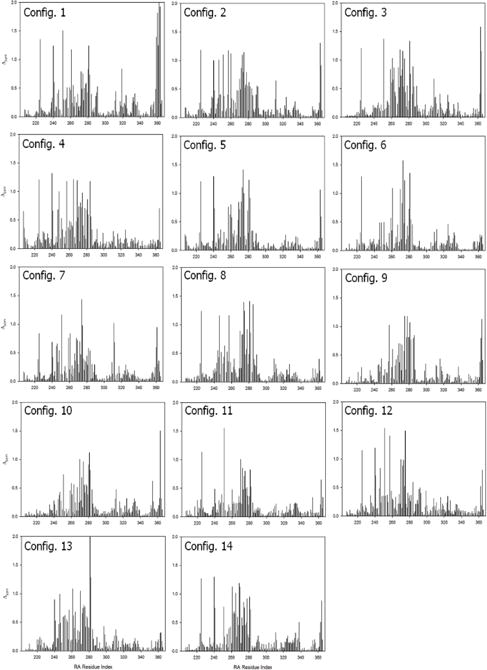
Calculated amide chemical shift perturbations (CSPs) for RA residues in the RASSF5 configurations shown in Fig. 5. The ShiftX2 program39 calculated 1H, 13C, and 15N chemical shifts. The combined amide CSPs for the RSSF5 configurations with respect to the RA domain alone trajectory were calculated during the simulations using the equation , where and denote the 1H and 15N chemical shift differences, respectively, between the RSSF5 and RA domain trajectories. The predicted chemical shifts were averaged over 1500 trajectory snapshots. A scaling factor αN = 0.17 was applied to the 15N chemical shift difference.40,41
Despite the fact that the interaction of SARAH with the RA domain is transient, we attempted to capture the most reliable models of the self-associated inactive RASSF5. Based on the combined residue pair interaction and CSP analyses, we selected configurations 1, 4, 6, 9, 10, 11, and 14 as representative RASSF5 conformations (Fig. 5a). These configurations ensured that the SARAH domain does not block the Ras binding site at the β3 region. With the Ras binding site exposed, we were able to construct initial models of RASSF5/K-Ras4B complex at a later stage. For the straight SARAH model, only configurations 1 and 4 were selected, since in configurations 2 and 3 the SARAH domain interferes with Ras binding. For the kinked SARAH model, configurations 8, 12, and 13 were not considered for the same reason. Configurations 5 and 7 were also omitted, since they have the weakest SARAH interaction among the kinked models (Fig. 5b). We noticed that SARAH dominantly interacts with the flexible loop (residues 254–278) in the RA domain, which may contribute to the RA-SARAH association. Among the representative RASSF5 models, configurations 6, 9, and 10 show strong interaction between the loop and SARAH (Fig. 7). In configuration 6, the loop marginally holds the SARAH domain through the salt bridge interaction of R277 with D370 and the cation-π interaction of Y262 with K416. However, RA residue Y281, near the loop, sequesters SARAH by forming H-bonds with E408 and L410 (Table S3, ESI†). In the RA-SARAH interaction, configuration 6 contains most of the common salt bridge residues, involving R323, K334, and E353 in the RA domain and E366, E368, E370, K386, and E387 in SARAH. Configuration 9 shows the most abundant interactions between the loop and SARAH, including the strong salt bridge interactions of E267-K391, E267-K398, K276-D400, and K276-E407, and the H-bond formation between T274 and R403 (Table S3, ESI†). The hydrophobic and hydrophilic interactions also stabilize the loop interaction with SARAH. Similarly, configuration 10 contains abundant hydrophobic and hydrophilic interactions, as well as strong salt bridge interactions, K266-E407 and E267-R403, but no H-bond formation. In configurations 11 and 14, the loop interacting with SARAH only involves weak hydrophobic interactions. The residue pair interactions illustrate that the flexible loop commonly interacts with the C-terminal portion of SARAH, except configuration 11. We speculate that the loop may act as a hook that transiently holds SARAH onto the RA domain. Figure 6 helps deduce the self-associated inactive RASSF5 conformation. As we noted above, the autoinhibitory RA-SARAH interaction is transient, since SARAH has to be released when RASSF5 is activated by Ras, forming a coiled coil with another SARAH. Thus, the ‘hooked’ SARAH configurations can be candidates RASSF5 conformations in the inactive state.
Fig. 7.
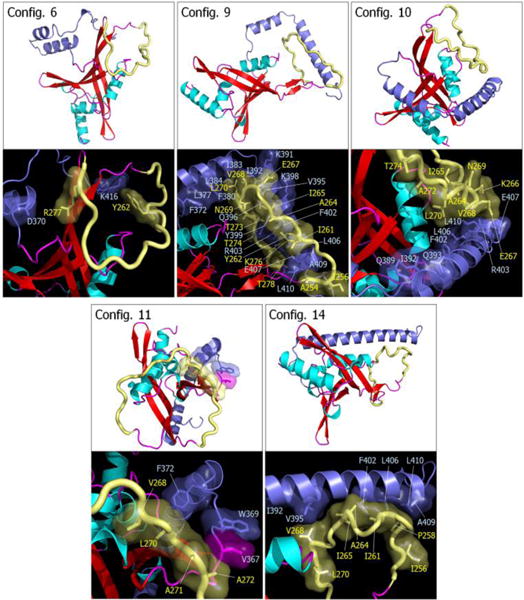
Snapshots representing average conformations of RASSF5. Selected RASSF5 configurations 6, 9, 10, 11, and 14 are shown on the top of each panel. In the cartoon representing the secondary structure, the α-helix and β-sheet structures are colored cyan and red, respectively. The SARAH domain is highlighted by blue, and the flexible loop is shown as yellow tube. Highlighted interfaces between SARAH and flexible loop are shown in the bottom of each panel. Residues involving the interactions are marked.
RASSF5 in complex with GTP-bound K-Ras4B
Active Ras recruits RASSF5, resulting in a dimeric complex at the plasma membrane (PM). This conduces to the MST1/2 kinase being able to translocate to the PM.17 RASSF5 connects between Ras and MST1/2. The RA domain of RASSF5 binds the catalytic domain of Ras, and at the same time the SARAH domain forms an antiparallel coiled coil with the MST1/2 SARAH. Ras dimerization would further facilitate the dimerization of MST1/2 kinase domains and the trans-autophosphorylation. Each Ras molecule requires an active RASSF5 to connect to MST1/2. In inactive RASSF5, SARAH clings to the RA domain, biding its time until the RA domain is enlisted to reside on the Ras catalytic domain. SARAH is released once RASSF5 binds and gets activated by Ras. To understand the mechanism of the conformational aptness of RASSF5, we simulated it in complex with the GTP-bound K-Ras4B. We adopted the crystal structure of murine RASSF5 RA domain in complex with H-Ras (PDB code: 3DDC) as a template for the MD simulations. In the initial construction, those self-associated inactive RASSF5 conformations (configurations 1, 4, 6, 9, 10, 11, and 14 in Fig. 5a) were used to generate dimeric complexes with K-Ras4B-GTP. This resulted in seven different dimeric configurations, DC1(1), DC2(4), DC3(6), DC4(9), DC5(10), DC6(11), and DC7(14), where DC represents dimeric configuration and the numbers in parenthesis denote the self-associated RASSF5 configuration. Remarkably, we observed that the SARAH domain tends to dissociate from the RA domain upon binding to K-Ras4B (Fig. 8a). The release generates straight SARAH conformations, which suggests a clan of coiled coils. The interaction strength of SARAH with the RA domain is significantly reduced as compared to that of the Ras-free RASSF5 configurations (Fig. 8b). In DC6(11), SARAH still holds the RA domain strongly even when bound with K-Ras4B, suggesting that the configuration may not have reached relaxation in the simulation. Nevertheless, SARAH loses a number of interacting residue pairs including DC6(11), as RA residues participate in the interaction with K-Ras4B (see Table S4, ESI†). The mechanism of SARAH release suggests that K-Ras4B promotes RASSF5 activation, allosterically shifting the protein from the fluctuating loosely associated inactive to the strongly bound active state.
Fig. 8.
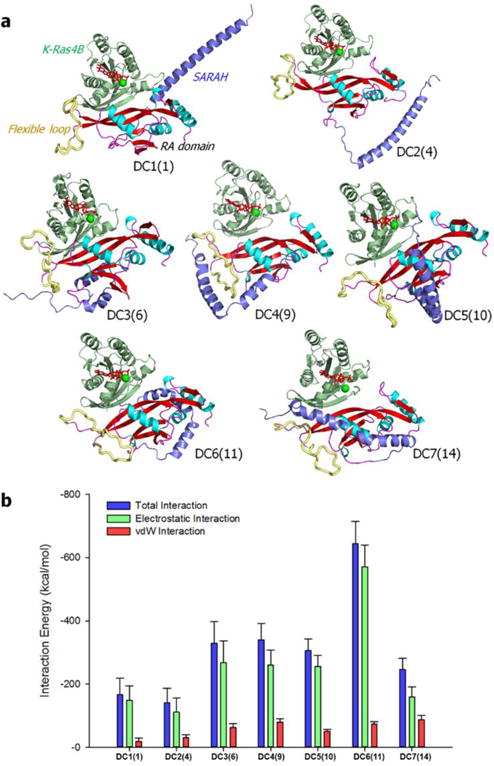
RASSF5 in complex with the GTP-bound K-Ras4B. (a) Averaged structures of RASSF5 interacting with K-Ras4B-GTP after 200 ns molecular dynamics simulations. Seven dimeric complexes, DC1(1), DC2(4), DC3(6), DC4(9), DC5(10), DC6(11), and DC7(14), where DC denotes dimeric configuration and the numbers in the parenthesis correspond to the self-associated RASSF5 configuration, were selected for the simulations. DC1(1) and DC2(4) contain straight SARAH, and other DCs have the kinked SARAH model. In the cartoon representing the secondary structure, the α-helix and β-sheet structures are colored cyan and red, respectively. The SARAH domain is highlighted by blue, and the flexible loop is shown as yellow tube. (b) Interaction energy of SARAH with the RA domain for the dimeric configurations of RASSF5/K-Ras4B-GTP complex. Averaged total interaction energy (blue bars), and the contributions from the electrostatic (green bars) and vdW (red bars) interactions for the dimeric configurations are shown.
To elucidate how K-Ras4B allosterically affects the RASSF5 conformation, we conducted a dynamical network analysis using the NetworkView plugin in VMD. The dynamical network analysis can identify the shortest signal propagation pathway through nodes, represented as residues in a protein, and edges connecting between nodes. In this analysis, weighted implementation of suboptimal paths (WISPs)42 for the allosteric signal transmitted from K-Ras4B to RASSF5 were calculated based on the sum of the shortest edge distances defined by the pairwise dynamic cross-correlation. We calculated over 100 optimal and suboptimal pathways between two selected residues on K-Ras4B and RASSF5 SARAH, and provided two best pathways for the dimeric configurations with the kinked SARAH model (Fig. 9). It can be seen that most of the signaling pathways are connected through the effector binding region of Ras (residues 32–40 of the Ras catalytic domain, see Table S5, ESI†). Interestingly, some pathways pass through GTP, implying that only the GTP-bound state induces conformational changes in RASSF5, facilitating its activation. We noted that the GTP-bound, not the GDP-bound, Ras preferentially binds its effectors, such as Raf and PI3K. The dynamical cross-correlation map (DCCM) reveals apparent residue motions across the K-Ras4B and RASSF5 domains in the highly-kinked SARAH dimeric complex model, with positive (C(i,j) → 1) and negative (C(i,j) → −1) correlations (Fig. S2, ESI†). In contrast, the dimeric complex with the straight SARAH model yields uncorrelated residue motions with C(i,j) → 0, suggesting that no allosteric signal is required for the unconstrained straight SARAH. The dimeric configurations can be ranked from more to less kinked SARAH in the order of DC3(6) > DC6(11) > DC4(9) > DC5(10) > DC7(14) > DC1(1) > DC2(4), which is consistent with the highly correlated residue motions.
Fig. 9.
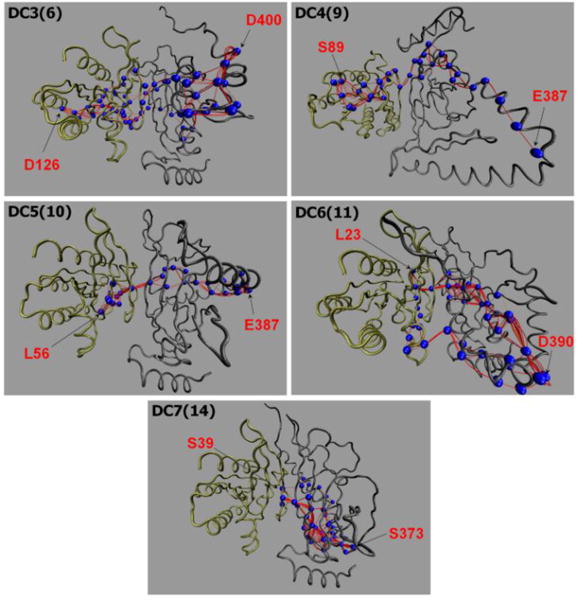
The allosteric pathways induced by the GTP-bound K-Ras4B. The allosteric pathways were calculated by the weighted implementation of suboptimal path (WISP)42 methods for the dimeric configurations, DC3(6), DC4(9), DC5(10), DC6(11), and DC7(14) with kinked SARAH. Blue beads on the paths highlight the residues with high occurrence rate, >50% (Table S5). The source residue on K-Ras4B-GTP (left) and sink residue on RASSF5 (right) are marked.
Discussion
RASSF5 is a mediator that links Ras and MST1/26. Its SARAH domain binds its partners, including the RA and other SARAH domains with varied affinities, thereby playing a critical role in RASSF5’s functions.7,20 RASSF5’s SARAH can interact with the RA domain resulting in autoinhibition and also associate with a cognate SARAH.17 The SARAH-SARAH interaction forms an antiparallel coiled coil, since SARAH adopts a long α-helical motif.43 The RASSF5-MST2 coiled coil SARAH heterodimer has the highest binding affinity, compared to both MST-MST and RASSF5-RASSF5 SARAH homodimers.20 Thus, RASSF5 SARAH can easily compete with the MST-MST SARAH homodimer. Our MD simulations also verified the strong interaction of RASSF5 SARAH with MST2 SARAH, compared to the homo-dimeric SARAH-SARAH interactions. We suspect that the high binding affinity of the SARAH heterodimer is due to the strong electrostatic attraction originating from the bipolar characteristics between RASSF5 and MST2 SARAHs. The isoelectric point calculations of the SARAH domains illustrated that RASSF5 SARAH is acidic, while MST2 SARAH is basic. In the SARAH-SARAH interactions, we observed that RASSF5 SARAH residues, E385, E388, and R403, are commonly involved in both the heterodimer and homodimer. Of particular note is E388, the residue at the kink in the α-helix when dissociated from the coiled coil. COSMIC data showed that the E388K mutation in RASSF5 can cause cancer. We speculate that E388K can disrupt the RASSF5-MST2 SARAH heterodimer, since the opposite basic residue can introduce unfavorable electrostatic repulsion in the SARAH-SARAH interaction. Without activation of MST1/2, Hippo signaling can be suppressed, and as a result, the oncogenic YAP1 signal is amplified.
It is clear that RASSF5 assists in MST1/2 kinase activation.9,17,44 In the inactive state, MST1/2 SARAH blocks its kinase domain, preventing dimerization of the kinase domain with other MST1/2. For MST1/2, kinase domain dimerization is required to conduct trans-autophosphorylation, leading to phosphorylation of YAP1. RASSF5 enables kinase domain dimerization, since RASSF5 SARAH can sequester MST1/2 SARAH, thereby shifting the equilibrium to an exposed kinase domain. The dynamic mechanism of the MST1/2 SARAH can be also applied to RASSF5 SARAH.17 In the inactive state, similar to MST1/2, the RASSF5 SARAH engages the RA domain, resulting in autoinhibition of RASSF5. The engagement of RASSF5 SARAH is dislodged when Ras interacts with the RA domain. To elucidate the mechanism, we modeled the inactive self-associated RASSF5 conformation in atomic detail. We obtained several possible RASSF5 structures with multiple modes of interaction of SARAH with the RA domain, suggesting that the inactive RASSF5 conformation is transient. In the RASSF5 structures, the SARAH domain was dominantly defined as a kinked α-helix. It is reasonable that the long α-helical motif of SARAH can be stabilized in a coiled coil with another SARAH, and that the RA domain does not contain a compatible α-helix to form the coiled coil. Our modeling suggests that the RASSF5 RA domain marginally holds the SARAH domain through the salt bridge interactions. Especially, the interactions of the flexible loop in the RA domain with the C-terminal portion of the SARAH domain constrain the fluctuations of SARAH’s C-terminus. The loop acts a hook that sequestrates SARAH to the RA domain.
The autoinhibited RASSF5 becomes active when Ras binds to the RA domain. Ras induces a conformational change in RASSF5, liberating SARAH from the RA domain. To capture the event, we simulated RASSF5 interacting with the GTP-bound K-Ras4B and monitored allosteric pathways propagating from K-Ras4B-GTP to the RASSF5 SARAH domain. With K-Ras4B-GTP, we observed that RASSF5 generally released the SARAH, removing the autoinhibition. The interaction strength between SARAH and the RA domain is dramatically reduced, compared to that of RASSF5 without K-Ras4B-GTP. We reasoned that K-Ras4B-GTP transmits the allosteric signal releasing SARAH. This shifts the energy landscape of RASSF5 from the inactive to the active state. For K-Ras4B-GTP/RASSF5 complex with kinked SARAH, the DCCM calculations showed that the residue motions between K-Ras4B and RASSF5 are highly correlated. The allosteric signals are promoted over long range, stretching from the K-Ras4B allosteric lobe to the SARAH domain. In contrast, for the complex with straight SARAH, the DCCM shows less correlated residue motions, with weak allosteric signals transmitted over the short distance from the K-Ras4B effector lobe. This suggests that in the case of straight SARAH domain RASSF5 is more likely to be found in or near the active state.
Conclusions
To conclude, here we aim to figure out the mechanism on the conformational level of how Ras binding results in MST1/2 activation, making RASSF5 a tumor suppressor. Ras activates RASSF5, and the activated RASSF5 activates MST1/2. The mechanism of RASSF5 activation deploys the conformational dynamics of the SARAH domain, switching it between states. SARAH’s dynamics characterizes its conformational plasticity: in the active state of RASSF5, SARAH is an elongated α-helix stretching to form a coiled coil with MST1/2 SARAH; in the inactive state, SARAH becomes flexible converting to a kinked α-helix and receding toward the RA domain. The closed, self-associated RASSF5 conformation with the retracted SARAH implies an autoinhibition of the protein; Ras can however easily compete with SARAH, allosterically altering the autoinhibited conformation. Our structural modeling of RASSF5 in atomic detail provides insight into the mechanism of how the structural dynamics of RASSF5 with SARAH can be related to its functional role - connecting oncogenic Ras proteins to cancer, here by suppressing the Hippo pathway. Noteworthy, like other Ras binding proteins, such as Raf, PI3K, and RalGDS, RASSF5 contains a largely undefined N-terminal portion, including the putative membrane binding C1 domain.45,46 A more complete understanding should involve these segments, as well as a mechanistic structural grasp of how MST activates the Hippo pathway and exactly how the Hippo pathway and YAP1 can rescue Ras and MAPK inhibition. Mutations in YAP1 are frequently observed in Ras-driven cancers,10,11,14,47,48 and more.15,16 RASSF5 tumor suppressor is currently among the key drug targets in Ras-related cancers due to its role in linking Ras/MAPK and YAP1.
Taken together, we suggest that when RASSF5 binds K-Ras4B-GTP, the equilibrium shifts. SARAH’s fluctuations increase predominantly toward the open state and heterodimer formation with MST’s SARAH. Since the RA/SARAH affinity is relatively low, whereas that of the SARAH heterodimer is in the nM range, the emerging mechanism of RASSF5 action as a tumor suppressor is through competitive binding to the Ras-GTP effector interaction site, and recruitment to the membrane, where it supports MST kinase domain dimerization and trans-autoactivation. Such a conformational dynamic view suggests how RASSF5 can suppress cell proliferation. Since RASSF5 binds at the effector binding site, it is not Ras isoform-specific.49–52 Its two domains link Ras and the MAPK (tumor cell proliferation-promoting), and Hippo (tumor cell proliferation-suppressing) pathways, thereby enabling RASSF5’s tumor suppressing action. At the same time, its N-terminal region, including its putative C1 domain, anchor it to the membrane, further stimulating MST1/2 activation. Thus ensuring RASSF5 action emerges as an important cancer drug discovery strategy.
Supplementary Material
Acknowledgments
This project has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here].
References
- 1.Volodko N, Gordon M, Salla M, Ghazaleh HA, Baksh S. FEBS Lett. 2014;588:2671–2684. doi: 10.1016/j.febslet.2014.02.041. [DOI] [PubMed] [Google Scholar]
- 2.Avruch J, Xavier R, Bardeesy N, Zhang XF, Praskova M, Zhou D, Xia F. J Biol Chem. 2009;284:11001–11005. doi: 10.1074/jbc.R800073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Praskova M, Khoklatchev A, Ortiz-Vega S, Avruch J. Biochem J. 2004;381:453–462. doi: 10.1042/BJ20040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper WN, Hesson LB, Matallanas D, Dallol A, von Kriegsheim A, Ward R, Kolch W, Latif F. Oncogene. 2009;28:2988–2998. doi: 10.1038/onc.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song H, Oh S, Oh HJ, Lim DS. Biochem Biophys Res Commun. 2010;391:969–973. doi: 10.1016/j.bbrc.2009.11.175. [DOI] [PubMed] [Google Scholar]
- 6.Donninger H, Schmidt ML, Mezzanotte J, Barnoud T, Clark GJ. Semin Cell Dev Biol. 2016;58:86–95. doi: 10.1016/j.semcdb.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez-Sanz G, Tywoniuk B, Matallanas D, Romano D, Nguyen LK, Kholodenko BN, Rosta E, Kolch W, Buchete NV. PLoS Comput Biol. 2016;12:e1005051. doi: 10.1371/journal.pcbi.1005051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez-Sanz G, Matallanas D, Nguyen LK, Kholodenko BN, Rosta E, Kolch W, Buchete NV. Brief Bioinform. 2016;17:593–602. doi: 10.1093/bib/bbv070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang E, Cheong HK, Ul Mushtaq A, Kim HY, Yeo KJ, Kim E, Lee WC, Hwang KY, Cheong C, Jeon YH. Acta Crystallogr D Biol Crystallogr. 2014;70:1944–1953. doi: 10.1107/S139900471400947X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun S, Irvine KD. Trends Cell Biol. 2016;26:694–704. doi: 10.1016/j.tcb.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zanconato F, Cordenonsi M, Piccolo S. Cancer Cell. 2016;29:783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plouffe SW, Hong AW, Guan KL. Trends Mol Med. 2015;21:212–222. doi: 10.1016/j.molmed.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. Genes Dev. 2001;15:1229–1241. doi: 10.1101/gad.888601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, Zwang Y, Roberts TM, Root DE, Jacks T, Hahn WC. Cell. 2014;158:171–184. doi: 10.1016/j.cell.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nussinov R, Tsai CJ, Jang H, Korcsmaros T, Csermely P. Semin Cell Dev Biol. 2016;58:79–85. doi: 10.1016/j.semcdb.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Nussinov R, Tsai CJ, Jang H. Expert Rev Proteomics. 2016;13:711–716. doi: 10.1080/14789450.2016.1209417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao TJ, Tsai CJ, Jang H, Fushman D, Nussinov R. Curr Opin Struct Biol. 2016;41:217–224. doi: 10.1016/j.sbi.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Ni L, Li S, Yu J, Min J, Brautigam CA, Tomchick DR, Pan D, Luo X. Structure. 2013;21:1757–1768. doi: 10.1016/j.str.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creasy CL, Ambrose DM, Chernoff J. J Biol Chem. 1996;271:21049–21053. doi: 10.1074/jbc.271.35.21049. [DOI] [PubMed] [Google Scholar]
- 20.Makbul C, Constantinescu Aruxandei D, Hofmann E, Schwarz D, Wolf E, Herrmann C. Biochemistry. 2013;52:1045–1054. doi: 10.1021/bi3014642. [DOI] [PubMed] [Google Scholar]
- 21.Nussinov R, Ma B, Tsai CJ. Biochim Biophys Acta. 2013;1834:820–829. doi: 10.1016/j.bbapap.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 22.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R. Biochem J. 2016;473:1719–1732. doi: 10.1042/BCJ20160031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jang H, Banerjee A, Chavan TS, Lu S, Zhang J, Gaponenko V, Nussinov R. FASEB J. 2016;30:1643–1655. doi: 10.1096/fj.15-279091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu S, Jang H, Nussinov R, Zhang J. Sci Rep. 2016;6:21949. doi: 10.1038/srep21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakrabarti M, Jang H, Nussinov R. J Phys Chem B. 2016;120:667–679. doi: 10.1021/acs.jpcb.5b11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chavan TS, Jang H, Khavrutskii L, Abraham SJ, Banerjee A, Freed BC, Johannessen L, Tarasov SG, Gaponenko V, Nussinov R, Tarasova NI. Biophys J. 2015;109:2602–2613. doi: 10.1016/j.bpj.2015.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu S, Banerjee A, Jang H, Zhang J, Gaponenko V, Nussinov R. J Biol Chem. 2015;290:28887–28900. doi: 10.1074/jbc.M115.664755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jang H, Abraham SJ, Chavan TS, Hitchinson B, Khavrutskii L, Tarasova NI, Nussinov R, Gaponenko V. J Biol Chem. 2015;290:9465–9477. doi: 10.1074/jbc.M114.620724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiser A, Do RK, Sali A. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogmen U, Keskin O, Aytuna AS, Nussinov R, Gursoy A. Nucleic Acids Res. 2005;33:W331–336. doi: 10.1093/nar/gki585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tuncbag N, Gursoy A, Nussinov R, Keskin O. Nat Protoc. 2011;6:1341–1354. doi: 10.1038/nprot.2011.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duhovny D, Nussinov R, Wolfson HJ. Algorithms in Bioinformatics, Proceedings. 2002;2452:185–200. [Google Scholar]
- 34.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. Nucleic Acids Res. 2005;33:W363–367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emekli U, Schneidman-Duhovny D, Wolfson HJ, Nussinov R, Haliloglu T. Proteins. 2008;70:1219–1227. doi: 10.1002/prot.21613. [DOI] [PubMed] [Google Scholar]
- 38.Frishman D, Argos P. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 39.Neal S, Nip AM, Zhang H, Wishart DS. J Biomol NMR. 2003;26:215–240. doi: 10.1023/a:1023812930288. [DOI] [PubMed] [Google Scholar]
- 40.Tochio H, Hung F, Li M, Bredt DS, Zhang M. J Mol Biol. 2000;295:225–237. doi: 10.1006/jmbi.1999.3350. [DOI] [PubMed] [Google Scholar]
- 41.Williamson MP. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 42.Van Wart AT, Durrant J, Votapka L, Amaro RE. J Chem Theory Comput. 2014;10:511–517. doi: 10.1021/ct4008603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu G, Shi Z, Jiao S, Zhang Z, Wang W, Chen C, Hao Q, Zhang M, Feng M, Xu L, Zhang Z, Zhou Z, Zhang M. J Struct Biol. 2014;185:366–374. doi: 10.1016/j.jsb.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 44.Hwang E, Ryu KS, Paakkonen K, Guntert P, Cheong HK, Lim DS, Lee JO, Jeon YH, Cheong C. Proc Natl Acad Sci U S A. 2007;104:9236–9241. doi: 10.1073/pnas.0610716104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Das J, Rahman GM. Chem Rev. 2014;114:12108–12131. doi: 10.1021/cr300481j. [DOI] [PubMed] [Google Scholar]
- 46.Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, Zhang J. Chem Rev. 2016;116:6607–6665. doi: 10.1021/acs.chemrev.5b00542. [DOI] [PubMed] [Google Scholar]
- 47.Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, Chang Q, Chu GC, Al-Khalil R, Jiang S, Xia H, Fletcher-Sananikone E, Lim C, Horwitz GI, Viale A, Pettazzoni P, Sanchez N, Wang H, Protopopov A, Zhang J, Heffernan T, Johnson RL, Chin L, Wang YA, Draetta G, DePinho RA. Cell. 2014;158:185–197. doi: 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin L, Bivona TG. Mol Cell Oncol. 2016;3:e1021441. doi: 10.1080/23723556.2015.1021441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nussinov R, Muratcioglu S, Tsai CJ, Jang H, Gursoy A, Keskin O. Expert Opin Ther Targets. 2016;20:831–842. doi: 10.1517/14728222.2016.1135131. [DOI] [PubMed] [Google Scholar]
- 50.Nussinov R, Tsai CJ, Chakrabarti M, Jang H. Cancer Res. 2016;76:18–23. doi: 10.1158/0008-5472.CAN-15-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nussinov R, Muratcioglu S, Tsai CJ, Jang H, Gursoy A, Keskin O. Mol Cancer Res. 2015;13:1265–1273. doi: 10.1158/1541-7786.MCR-15-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nussinov R, Tsai CJ, Muratcioglu S, Jang H, Gursoy A, Keskin O. Expert Rev Proteomics. 2015;12:669–682. doi: 10.1586/14789450.2015.1100079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.