

Abstract
Stringent searches for microRNAs (miRNAs) have so far only identified these molecules in animals, land plants, chlorophyte green algae, slime molds and brown algae. The identification of miRNAs in brown algae was based on the analysis of a single species, the filamentous brown alga Ectocarpus sp. Here, we have used deep sequencing of small RNAs and a recently published genome sequence to identify miRNAs in a second brown alga, the kelp Saccharina japonica. S. japonica possesses a large number of miRNAs (117) and these miRNAs are highly diverse, falling into 98 different families. Surprisingly, none of the S. japonica miRNAs share significant sequence similarity with the Ectocarpus sp. miRNAs. However, the miRNA repertoires of the two species share a number of structural and genomic features indicating that they were generated by similar evolutionary processes and therefore probably evolved within the context of a common, ancestral miRNA system. This lack of sequence similarity suggests that miRNAs evolve rapidly in the brown algae (the two species are separated by ∼95 Myr of evolution). The sets of predicted targets of miRNAs in the two species were also very different suggesting that the divergence of the miRNAs may have had significant consequences for miRNA function.
Keywords: brown algae, Ectocarpus, heterokonts, microRNA, Saccharina japonica, stramenopiles
Introduction
Small, noncoding RNA molecules have been shown to have important regulatory roles in diverse eukaryotic lineages (Carthew and Sontheimer 2009). One particularly important class of small RNA is the microRNAs (miRNAs). These molecules are produced by digestion of double stranded hairpin regions of RNA transcripts by RNAseIII endonucleases such as Dicer and Drosha. The small RNAs produced, which are usually 20–24 nucleotides long but can be up to 26 nucleotides (Kozomara and Griffiths-Jones 2011), then bind to Argonaute proteins within RNA-induced silencing complexes (RISCs). The miRNA is used as a guide by the RISC complex to recognize specific RNA targets and effect an action such as slicing (cutting) the RNA strand at the binding site or preventing it from being processed by other complexes such as the ribosome (inhibition of translation).
miRNAs are thought to have evolved from more ancient small RNA regulatory systems such as the small interfering RNA (siRNA) pathway (Ghildiyal and Zamore 2009). This scenario has been suggested to explain why the core protein components of the miRNA biogenesis pathway, such as Dicer and Argonaute, appear to be very ancient and are found throughout the diverse lineages of the eukaryotic tree (Cerutti and Casas-Mollano 2006; Mukherjee et al. 2013), while miRNAs themselves have only been definitively identified in a relatively small number of lineages (Tarver et al. 2012).
To date, there is convincing evidence for miRNAs in at least five eukaryotic lineages: animals (Tarver et al. 2012, 2013), land plants (Taylor et al. 2014), chlorophyte green algae (Chlamydomonas reinhardtii; Molnar et al. 2007; Zhao et al. 2007), social amoebae (Dicytostelium discoideum; Hinas et al. 2007; Avesson et al. 2012) and brown algae (Ectocarpus sp.; Cock et al. 2010; Tarver et al. 2015). In addition, a large collection of 102 miRNAs was reported recently for the dinoflagellate Symbiodinium kawagutii (Lin et al. 2015) and a small number of miRNA-like molecules have been identified in the fungus Neurospora crassa (Lee et al. 2010), but confirmation of the candidate miRNAs in these latter two species awaits the publication of detailed small RNA read mapping information.
There is no evidence of conservation of miRNAs between the phylogenetic groups, indicating that miRNA systems evolved independently in each lineage (Grimson et al. 2008; Ghildiyal and Zamore 2009; Axtell et al. 2011; Kozomara and Griffiths-Jones 2011). The absence of miRNAs in many other eukaryotic lineages supports this conclusion (Tarver et al. 2012). Note that, based on the absence of similarity between the miRNAs of demosponges and those of other animal groups, it has been proposed that miRNAs may actually have evolved twice in the animal lineage (Sperling et al. 2010). However, if recent evidence for miRNAs in unicellular relatives of metazoans (Brate et al. 2016) is confirmed, a single origin for all animal miRNAs may be more likely.
The brown algae are members of the stramenopile lineage and, to date, are the only group within this lineage that have been shown convincingly to possess miRNAs. Indeed detailed analyses of the genomes of three different diatoms (another stramenopile group) failed to detect any canonical miRNAs (Lopez-Gomollon et al. 2014; Rogato et al. 2014). Based on this observation, it has been proposed that the evolution of miRNAs in the brown algae may have been important for the emergence of complex multicellularity in this group (Cock and Collén 2015; Tarver et al. 2015), a suggestion that has been made for other multicellular lineages that possess miRNAs (Mattick 2004; Peterson et al. 2009).
In this study, we have searched for miRNA loci in a second brown algal species, the kelp S. japonica. We show that, like the filamentous brown alga Ectocarpus sp., this species possesses a large and diverse miRNA repertoire: 117 miRNA loci that fall into 98 different miRNA families. Surprisingly, although the S. japonica and Ectocarpus sp. miRNAs shared a number of common structural features, they did not share any sequence similarity. In other lineages, such as animals and land plants, a subset of miRNAs are conserved over long periods of evolutionary time (Bartel 2009; Nozawa et al. 2010; Tarver et al. 2013; Taylor et al. 2014). The absence of sequence similarity between the S. japonica and Ectocarpus sp. miRNAs suggests that brown algae do not exhibit this phenomenon of long-term conservation of miRNA loci.
Materials and Methods
Small RNA Sequencing
The generation of 11,363,272 and 8,901,516 sRNA reads, respectively, for heat shocked (20 °C) and control (10 °C) samples of young sporophytes of S. japonica variety “Huangguan No.1” has been described previously (Liu et al. 2015). In addition, 14,747,252 and 15,486,821 sRNA reads were generated for sporophytes subjected to either blue light or grown under dark conditions, respectively. For these latter samples, juvenile sporophytes (∼3.5–4.0 cm in width and 35–40 cm in length) of S. japonica variety “Zhongke” were collected from a cultivation raft and washed several times with cold, sterilized seawater to eliminate epiphytes. The sporophytes were then maintained in filtered sterilized seawater at 10 °C under 35 ± 5 mol m−2 s−1 light with a 10-h light:14-h dark period. After 3 days of incubation under these conditions, five individuals each were transferred to either blue light conditions or to the dark for 2 h before harvesting by wrapping in aluminium paper in the dark and immediately storing at −80 °C for RNA extraction. The four collections of sRNA reads (raw reads) were pooled and Cutadapt (Martin 2011) was used to remove 5′ and 3′ adapter sequences and to select reads of between 18 and 28 nucleotides. Read quality was evaluated using Fastqc (Andrews 2016). This process generated a total of 47,689,097 clean reads. The sRNA sequence data is accessible from either the SRA Knowledge Base (accession number SRR4343884) for the heat shock and control samples or from the Gene Expression Omnibus database (accession number GSE36704) for the blue light and dark treatments (supplementary table S1, Supplementary Material online).
Identification of S. japonica miRNAs
Candidate miRNA loci were identified using both miRDeep2 (Friedlander et al. 2012), which uses criteria based on the characteristics of animal miRNAs, and miRDeep-p (Yang and Li 2011), adapted for the identification of plant miRNAs, which usually have longer precursor sequences. The 47,689,097 pooled, clean reads were provided as input for each program. BLAST was used to compare the precursors or the candidate miRNAs identified by the two programs and candidates identified by miRDeep-p were only retained if they were not already among the candidates identified by miRDeep2. The candidate miRNA loci generated by this process were analysed manually using a set of criteria based on highly conserved features of experimentally characterized miRNAs for both plants and animals (see results section for details; Tarver et al. 2012, 2013; Taylor et al. 2014).
A custom script employing Bowtie (Langmead et al. 2009) was used to map and align all of the sRNA sequence reads onto the candidate precursor miRNA loci with no mismatches allowed. The script returned the absolute numbers of reads for each unique sRNA species mapped to each pre-miRNA and also calculated the percentage of each of these sRNA species with respect to the total sum of reads mapped to each pre-miRNA locus. For each miRNA locus, the script then queried the most abundant mapped sRNA species (corresponding to the miRNA) against miRBase V21 using BLAST and if matches were detected this was recorded in the output file together with the e-value for the match. Folding of the pre-miRNA sequences was carried out using Vienna RNAfold (Lorenz et al. 2011) using standard settings and the structure, folding energy and complementarity were also recorded in the output file.
Foldback length was calculated by removing all sequence at and distal to the first occurrence of at least three unpaired nucleotides or of any additional secondary structure and then measuring the length of the RNA strand constituting the stem-loop.
Identification of miRNA Families and Clusters
miRNA families were identified using a combination of two approaches. First, ungapped alignments of the miRNA seed regions (nucleotides two to eight; Bartel 2009) were generated with Muscle (Edgar 2004) and pairwise sequence identity calculated with MEGA7 (Tamura et al. 2011). miRNA loci with identical seed regions were grouped into families. Second, pairwise comparisons of all the S. japonica pre-miRNA sequences were carried out using BLASTn with a threshold E-value of 10−4 to group pre-miRNAs into families. The final list of families was generated by merging the results of these two analyses.
miRNAs located at a distance of <5 kb from each other on the chromosome were defined as belonging to the same cluster (Nozawa et al. 2012).
Evaluation of miRNA Divergence Rates
To evaluate the rates of divergence of brown algal miRNAs, the Ectocarpus sp. miRNAs (Tarver et al. 2015) were compared with homologous loci in a draft genome assembly for another Ectocarpus species, Ectocarpus fasciculatus. Illumina HiSeq2500 125-bp paired-end sequence data (a total of 92,225,940 reads) was generated for female and male E. fasciculatus strains Ec846 and Ec847 (supplementary table S1, Supplementary Material online) and assembled with SOAPdenovo (Luo et al. 2012) to generate 709,900 sequence scaffolds. Regions homologous to Ectocarpus sp. pre-miRNA sequences were identified by BLASTn (cut-off <e−10) and the potential of the homologous E. fasciculatus genomic regions to form hairpins was evaluated using Vienna RNAfold (Lorenz et al. 2011). Control genomic regions were obtained by extracting regions the same size as each of the Ectocarpus sp. pre-miRNAs but located 1-kb upstream and 1-kb downstream of their chromosomal positions. After elimination of any extracted region that contained protein-coding exon sequence, the remaining sequences were compared with the E. fasciculatus draft genome scaffolds with BLASTn (cut-off <e−10) to identify the homologous regions in E. fasciculatus. Only pairs of homologous control regions with a BLASTn e-value of <e−10 were retained for further analysis. Percentage identities between homologous pre-miRNA sequences and between homologous control sequences were calculated using EMBOSS Needle (Li et al. 2015). The rate of evolution of very young Drosophila miRNAs was calculated in the same manner by comparing the pre-miRNA sequences of the eight miRNAs corresponding to branch 6 in figure 2 of Nozawa et al. (2010) between D. melanogaster and D. simulans and between D. melanogaster and D. sechellia.
Fig. 2.—
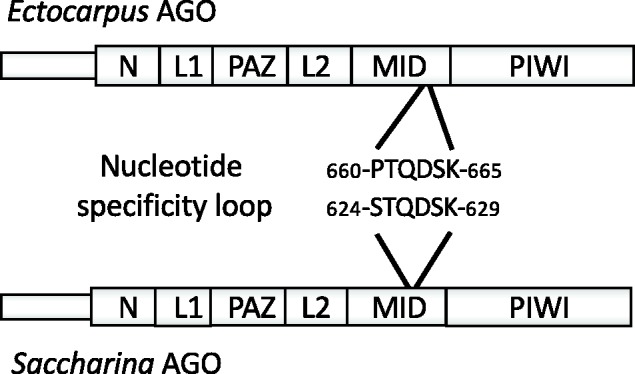
Schematic representation of the Argonaute proteins from Ectocarpus sp. (Ec-01_009010.1) and Saccharina japonica (SJ13826) showing the dissimilar nucleotide specificity loops.
Comparisons of miRNA Loci with Other Genome Features to Infer Evolutionary Origin
To search for evidence that the S. japonica miRNAs may have been derived from repeated regions of the genome or from protein-coding genes, the following analyses were carried out: 1) RepeatMasker (http://repeatmasker.org; last accessed March 11, 2017) was used to compare the S. japonica pre-miRNA sequences with Repbase to detect the presence of repeated elements and 2) BLASTn comparison of the S. japonica pre-miRNAs with the S. japonica genome sequence was used to search for similarity to protein-coding regions.
Comparison of the S. japonica miRNAs with Those of Species from Other Eukaryotic Lineages
To compare general structural features and genomic characteristics of the S. japonica miRNAs with those of the miRNA complements of species from other eukaryotic lineages, we used sets of miRNA loci that had been previously obtained (Tarver et al. 2015) by recovering the complete inventory of miRNA entries for each species from miRBase (Kozomara and Griffiths-Jones 2011) and then filtering based on the criteria proposed by Tarver et al. (2013) and Taylor et al. (2014). The miRBase accession numbers of the miRNAs obtained after filtering are provided in supplementary table S2, Supplementary Material online. The species used were Drosphila melanogaster (animals), Dictyostelium discoideum (slime molds), Arabidopsis thaliana (land plants) and Chlamydomonas reinhardtii (chlorophyte green algae).
Expression of S. japonica miRNAs
The expression level of S. japonica miRNAs was calculated as the number of sRNA reads corresponding to the miRNA per million reads mapped to the genome (RPM). The miRNA was defined as the most strongly expressed sRNA produced by each miRNA locus (Berezikov et al. 2011).
Identification of Potential Targets of S. japonica miRNAs
TAPIR (Bonnet et al. 2010) was used in precise mode with the default options to identify both potential target genes of S. japonica miRNAs and potential target mimics. Comparison of the sets of predicted protein-coding target genes of S. japonica and Ectocarpus sp. miRNAs was carried out based on a list of 9,773 genes that were determined to be orthologous between the two species based on one-to-one reciprocal best BLASTp matches with an e-value maximum cut off of 0.0001. The S. japonica genome annotation includes a total of 17,529 protein-coding genes (Ye et al. 2015) and therefore 55.8% of these genes have a one-to-one orthologue in Ectocarpus sp.
Results
Identification of S. japonica miRNAs
To identify S. japonica miRNA loci, the genome sequence (Ye et al. 2015), together with mapping information for 47 million small RNA reads from four different libraries, were analysed using the algorithms miRDeep2 (Friedlander et al. 2012) and miRDeep-p (Yang and Li 2011), identifying 1,837 and 2,081 candidate miRNAs, respectively. The combined set of candidate miRNAs were then screened manually by applying a set of stringent criteria based on highly conserved features common to both land plant and animal miRNA loci (Tarver et al. 2012, 2013; Taylor et al. 2014) that had been used previously to identify miRNA loci in Ectocarpus sp. Candidate miRNAs were rejected 1) if less than 15 nucleotides of the miRNA paired with the opposite arm of the hairpin, 2) if there was no evidence for expression (mapped sRNA-seq reads) of both the miRNA and the miRNA* (with at least four sRNA-seq reads for the miRNA), 3) if there were not two-nucleotide 3′ overhangs at both ends of the miRNA/miRNA* hybrid, 4) if the cleavage of the miRNA at its 5′ end was not precise (<66% of miRNA reads starting at the same nucleotide), 5) if the terminal loop from the miRNA to the mRNA* was less than eight nucleotides long, 6) if there was a mismatch of more than one nucleotide at either end of the miRNA/miRNA* hybrid and 7) if there were additional loops in the miRNA hairpin. We also verified that the genomic region did not form an extended hairpin with additional small RNA species on either the miRNA or miRNA* arm that were more abundantly expressed than the miRNA or miRNA* themselves, respectively. This latter test was carried out because many candidate miRNAs in Ectocarpus sp. were shown to be located on extended hairpin regions (Tarver et al. 2015). Application of these criteria led to the retention of 117 of the candidate miRNAs (fig. 1;supplementary fig. S1 and table S3, Supplementary Material online).
Fig. 1.—

Four representative Saccharina japonica miRNAs. The miRNA and miRNA* are indicated in blue and red, respectively. See supplementary figure S1, Supplementary Material online, for similar diagrams for the full set of 117 S. japonica miRNAs.
Saccharina japonica and Ectocarpus sp. miRNAs Do Not Share Sequence Similarity but Have Similar General Characteristics
Comparison of the S. japonica and Ectocarpus sp. miRNAs and pre-miRNAs using BLAST detected no loci that shared significant sequence similarity between the two species. However, despite this absence of sequence similarity, the miRNA complements of S. japonica and Ectocarpus sp. shared numerous general characteristics. For example, the Ectocarpus sp. miRNAs are highly diverse compared with the miRNA complements of plants and animals; the 64 Ectocarpus sp. miRNAs fall into 62 different families (Tarver et al. 2015). A high level of diversity was also noted for the S. japonica miRNAs; the 117 miRNAs retained after application of the selection criteria fell into 98 different miRNA families using the same criteria (supplementary table S3, Supplementary Material online). Moreover, the S. japonica miRNA families were small, 17 having only two members and one having three members.
Similarly, as in Ectocarpus sp., the S. japonica miRNAs tend not to occur in clusters in the genome. Only, six small miRNA clusters were identified in S. japonica, and all but one of these clusters consisted of only two genes (supplementary table S3, Supplementary Material online). Only one of the six clusters included two unrelated miRNAs, indicating that the majority of the clusters arose through local miRNA gene duplications. The identification of a large number of families and a small number of clusters indicated that, as was concluded for Ectocarpus sp., only a relatively small proportion of the S. japonica miRNA loci are likely to have been generated by gene duplication. This situation contrasts markedly with that in land plants and animals, where miRNA gene clusters are common (Hertel et al. 2006; Nozawa et al. 2010, 2012; Marco et al. 2012) and both gene and genome duplication events appear to have made an important contribution to the expansion of the miRNA families (Hertel et al. 2006; Li and Mao 2007; Heimberg et al. 2008; Nozawa et al. 2012).
An analysis of the S. japonica pre-miRNA sequences with RepeatMasker did not detect any significant similarities with transposon sequences, while BLAST comparisons between the pre-miRNA sequences and the genome sequence did not detect any evidence of similarity to protein-coding genes. It therefore seems unlikely that evolution of miRNA loci from repeat elements (Piriyapongsa and Jordan 2007, 2008; Avesson et al. 2012) or from duplicated copies of protein-coding genes (Allen et al. 2004; Rajagopalan et al. 2006) are important mechanisms for generating miRNA loci in S. japonica. Taken together, these observations suggest that the majority of the S. japonica miRNAs have probably evolved from hairpin regions of the genome (Felippes et al. 2008; Nozawa et al. 2010; Campo-Paysaa et al. 2011), the remainder arising as a result of local gene duplications. Evolution of miRNAs from genomic hairpins has been proposed to be the most important mechanism of miRNA evolution in the animal lineage (Nozawa et al. 2012), and this also appears to have been the case for Ectocarpus sp. (Tarver et al. 2015).
Another common feature of the S. japonica and Ectocarpus sp. miRNAs is that the same proportion of miRNAs (75%) are located within transcribed genes in both species (table 1; Tarver et al. 2015), with most of these miRNAs being located in introns (97% and 93%, respectively, for the two species). In this respect, brown algal miRNA resemble animal miRNAs (Nozawa et al. 2010; Campo-Paysaa et al. 2011) more than those of land plants (Nozawa et al. 2012). None of these intron-located S. japonica miRNAs were mirtrons (Curtis et al. 2012). As was observed for Ectocarpus sp., the majority of the S. japonica miRNAs that were located in genes were transcribed from the same strand as the host gene mRNA (94% and 90% for two species, respectively, supplementary table S3, Supplementary Material online; Tarver et al. 2015).
Table 1.
Characteristics of miRNAs from the Brown Algae Saccharina japonica and Ectocarpus sp. and from Representative Species for Four Other Eukaryotic Lineages
| Species | Saccharina | Ectocarpus | Drosophila | Dictyostelium | Arabidopsis | Chlamydomonas |
|---|---|---|---|---|---|---|
| Total number of miRNA loci | 117 | 64 | 110 | 11 | 64 | 10 |
| Total number of miRNA families | 105 | 62 | 94 | 11 | 40 | 10 |
| Percent of miRNA loci in genes | 75.2 | 75.0 | 55.5 | 9.1 | 9.4 | 10.0 |
| Mean foldback length (nt) | 183 | 170 | 83 | 132 | 136 | 140 |
| Percent 21 nt miRNAs | 88.9 | 84.4 | 17.3 | 90.9 | 73.4 | 80.0 |
| Percent U at first nt of miRNA | 63.3 | 92.2 | 73.6 | 27.3 | 76.6 | 80.0 |
| Percent U at first nt of miRNA* | 44.4 | 34.4 | 22.7 | 27.3 | 21.88 | 20.0 |
| Percent miRNAs at 3p position | 55.6 | 65.6 | 60.0 | 72.7 | 48.4 | 60.0 |
| Mean expression ratio miRNA/miRNA* | 207 | 446 | 82 | 18 | 225 | 425 |
| Median expression ratio miRNA/miRNA* | 7 | 35 | 21 | 10 | 18 | 34 |
Note.—nt, nucleotide; U, uracil; 3p, three prime.
The S. japonica and Ectocarpus sp. miRNAs also shared several structural features, independent of the selection criteria (table 1). For example, the average length of a miRNA foldback loop in Ectocarpus sp. (170 nt) is longer than the average lengths that have been calculated for other eukaryotic groups, where average lengths vary between 82 and 140 nt (Tarver et al. 2015). The foldback loops of S. japonica miRNAs were also exceptionally long on average (183 nt). Comparison of the mature miRNAs revealed that there is a strong tendency in both of the brown algal species for these molecules to be 21 nucleotides in length (89% for S. japonica and 84% for Ectocarpus sp.). Among the other eukaryotic lineages, only Dictyostelium exhibited a stronger bias towards 21 nucleotide miRNAs (but note that the data for this amoeba is based on a small complement of only 11 miRNAs; table 1). Also, on average, in the brown algae the miRNA was markedly more abundant than the miRNA* (average miRNA/miRNA* RPM ratios of 207 and 446 for S. japonica and Ectocarpus sp., respectively).
In contrast, although there was a preference for a U residue at the first position of the miRNA in S. japonica (63% of miRNAs), the preference was not as marked as in Ectocarpus sp., where 92% of miRNAs begin with a U residue (Tarver et al. 2015). Analyses of the structures of human and Arabidopsis Argonaute proteins have shown that the nucleotide specificity loop within the middle (mid) domain of these proteins play an important role in determining whether these proteins have a preference for miRNAs with specific 5′ nucleotides (Frank et al. 2010, 2012). Comparison of the Ectocarpus sp. and S. japonica Argonaute proteins indicated that the nucleotide specificity loops are slightly different (fig. 2). It is possible that this difference underlies the observed difference in preference for a U at the first position of the miRNA. In neither brown algal species was there a strong preference for U at the first position of the miRNA*.
It has been demonstrated in both animal and plant systems that some pre-miRNA hairpins can be processed to produce additional miRNA-like molecules, from regions of the hairpin upstream or downstream of the miRNA/miRNA* hybrid and that these additional molecules can have biological functions (Langenberger et al. 2009; Shi et al. 2009; Zhang et al. 2010; Bortoluzzi et al. 2011). When these molecules are in phase with the miRNA/miRNA* pair, they are called miRNA–offset RNAs (moRNAs). moRNAs are not a common feature of Ectocarpus sp. miRNA loci, although one potential moRNA was detected (Tarver et al. 2015). In S. japonica, although additional small RNA products were detected for some of the miRNA loci (SjapMIR91, SjapMIR100, SjapMIR102), we did not detect any moRNA/moRNA* pairs that satisfied the structural criteria we applied for the miRNAs.
Expression Patterns of the S. japonica miRNAs
As was observed for Ectocarpus sp. (Tarver et al. 2015), the S. japonica miRNAs exhibited a broad range of abundances, from 0.1 to 18157.6 RPM (mean 365.1 RPM). On average, the miRNA was considerably more abundant than the miRNA* (mean ratio: 207), a feature that was also observed in Ectocarpus sp. and is more typical of plant than of animal miRNAs.
Divergence Rates of Brown Algal miRNA Loci
The shared features of the S. japonica and Ectocarpus sp. miRNAs described earlier indicated that they were likely to be derived from a common, ancestral miRNA system but the absence of sequence similarity between the two miRNA complements precluded calculation of divergence rates. We therefore compared the Ectocarpus sp. miRNAs with the corresponding loci in a more closely related species, Ectocarpus fasciculatus, in order to obtain an estimate of the rate at which miRNA loci evolve in brown algal genomes. We identified 21 sequences in a draft assembly of the Ectocarpus fasciculatus genome that were both homologous (best BLASTn match, <e-10) to an Ectocarpus sp. pre-miRNA and which were predicted to fold into a hairpin loop (supplementary fig. S2, Supplementary Material online). As no estimation is currently available for the time of emergence of the genus Ectocarpus, we based our calculations of gene divergence rates on an estimated maximum divergence time between the genus Ectocarpus and the sister genus Kuckuckia of 25 Myr (Kawai et al. 2015). Using this value, we obtained a minimum estimation of the mean divergence rate for the Ectocarpus pre-miRNAs of 7.7×10−9 substitutions/site/year. This minimum mean divergence rate, which is likely to considerably underestimate the actual mean divergence rate, is comparable with values calculated for very young miRNA loci (mean divergence rate 2.6×10−8 substitutions/site/year, see Materials and Methods) in Drosophila (Nozawa et al. 2010). Detailed analysis of the birth and death of miRNA loci within the genus Drosophila have shown that the majority of new miRNA loci that arise in this genus degenerate very rapidly and are lost from the genome but the genome also contains a small proportion of miRNAs that have been conserved over much longer evolutionary times, in some cases hundreds of millions of years (Lu et al. 2008; Nozawa et al. 2010; Fromm et al. 2015). Taken together, the data presented here suggest that, in contrast, the brown algae do not possess a subset of miRNAs that is conserved over long evolutionary times.
The comparison between Ectocarpus sp. and E. fasciculatus indicated a rapid rate of evolution of miRNA loci in these brown algae. Note, however, that rates of divergence of the miRNA loci were significantly lower than for a set of control, homologous, genomic regions (Kruskal–Wallis test, P = 1.13×10−8; supplementary fig. S3, Supplementary Material online) indicating that the brown algal miRNA loci are nonetheless evolving under constraint.
Prediction of Target Genes and Target Mimics for the S. japonica miRNAs
At present, very little is known about how brown algal miRNAs function at the molecular level. In particular, no miRNA target genes have been confirmed experimentally and it is not known whether brown algal miRNAs recognize their putative targets with a high degree of complementarity (as tends to be the case in plants and green algae; Brodersen et al. 2008) or whether complementarity is low (as tends to be the case for animal miRNAs; Bartel 2009). To establish a set of potential target genes for the S. japonica miRNAs, we therefore carried out a screen for potential targets based on the assumption that complementarity with the miRNA is high, as this facilitated target identification. The screen was carried out using TAPIR (Bonnet et al. 2010), which not only allows the prediction of potential target genes but can also predict potential target mimics, that is, sequences that could potentially form a duplex with a miRNA that has a large bulge in the regions of positions 10 and 11 preventing cleavage of the target by the argonaute protein (Franco-Zorrilla et al. 2007). The search was applied to both the full set of S. japonica protein-coding (mRNA) transcripts (Ye et al. 2015) and to a recently published set of S. japonica long noncoding RNAs (lncRNAs) (Cormier et al. 2016). A total of 368 potential target sites were identified in the transcripts of 317 genes (243 corresponding to mRNA genes and 74 corresponding to lncRNAs; supplementary table S4, Supplementary Material online), plus 594 potential target mimic sites in the transcripts of 495 genes (447 mRNA, 48 lncRNA; supplementary table S5, Supplementary Material online). Experimental analysis will be necessary to validate these candidate target genes as bona fide miRNA targets.
The number of target genes (mRNA and lncRNA) predicted for each individual miRNA varied from 0 (44 miRNAs) to 29 (supplementary table S4, Supplementary Material online). Very few of the predicted mRNA target genes were predicted to be targeted by more than one miRNA and when this did occur (23 genes, each recognized by two miRNAs) it was due to two identical miRNAs (encoded by different genetic loci) recognizing the same target site. This observation is consistent with the high sequence diversity of the S. japonica miRNAs.
The majority of the predicted target genes were of unknown function, but putative functions could be assigned to 79 genes based on BLAST matches to the public databases (supplementary table S4, Supplementary Material online). The most common functional groups represented within this set of genes were metabolic processes (12 genes), cellular regulation and signal transduction (9 genes), life cycle (8 genes), membrane transporters (6 genes) and chromatin regulation (5 genes). The predicted target genes represented a broad range of functional categories, as was previously observed for Ectocarpus sp. (table 2; Tarver et al. 2015). However, none of the predicted protein-coding miRNA target genes in S. japonica were orthologous to predicted protein-coding miRNA target genes in Ectocarpus sp. (Tarver et al. 2015), indicating that the miRNA complements of the two species target completely different sets of genes. Such a large difference between the sets of predicted miRNA target genes for the two species might be expected if brown algal miRNAs preferentially target recently evolved genes. However, this does not appear to be the case because 51.8% of the predicted S. japonica target genes had a one-to-one orthologue in the Ectocarpus sp. genome, a value that is similar to the proportion of one-to-one orthologues when the two complete predicted proteomes are compared (55.8%; see the methods section for details).
Table 2.
Comparison of the Sets of Predicted miRNA Target Genes in Saccharina and Ectocarpus, Grouped by Functional Category
| Functional Category | Saccharina Target Genes | Ectocarpus Target Genes |
|---|---|---|
| Cell wall | 2 | 2 |
| Cellular regulation | 9 | 10 |
| Cytoskeleton | 4 | 4 |
| Defence | 0 | 10 |
| DNA or chromatin modification | 5 | 6 |
| Electron transport | 2 | 0 |
| Inserted viral gene | 0 | 1 |
| Life cycle | 8 | 0 |
| Membrane trafficking | 3 | 2 |
| Membrane transporter | 6 | 10 |
| Metabolism | 12 | 12 |
| Mitosis | 0 | 1 |
| Photosynthesis | 0 | 2 |
| Protein synthesis | 0 | 3 |
| Protein-binding | 0 | 18 |
| Proteolysis | 2 | 11 |
| RNA metabolism | 4 | 4 |
| Stress response | 4 | 1 |
| Transcription | 4 | 5 |
| Unknown function | 178 | 58 |
| TOTAL | 243 | 160 |
Discussion
This study identified 117 miRNA loci in the S. japonica genome using stringent selection criteria, and these miRNAs fell into 98 different families. Surprisingly, none of these miRNAs are conserved in the Ectocarpus sp. genome and none of the 64 Ectocarpus sp. miRNAs (Tarver et al. 2015) are conserved in S. japonica. Despite the absence of sequence similarity between miRNAs from the two species, S. japonica and Ectocarpus sp. miRNAs share a number of emergent features (i.e., independent of the selection criteria used to identify them), indicating that they have been generated by similar evolutionary processes and have been under similar evolutionary constraints. These shared features include a strong preference for 21 nucleotide mature miRNAs, exceptionally long foldback loops, high miRNA/miRNA expression ratios and high sequence diversity, the latter resulting in a large number of miRNA families. In both species there is also a strong tendency for miRNA loci to be included within genes rather than in intergenic regions and for the miRNAs not to be clustered in the genome. Finally, in both cases, miRNA loci appear to have arisen predominantly from genomic hairpins, rather than being derived from transposons or fragments of protein-coding genes or by miRNA duplication. Note, however, that miRNA duplication appears to have been more frequent in S. japonica than in Ectocarpus sp., presumably reflecting the larger size of the former’s genome (537 Mb [Ye et al. 2015] compared with 214 Mb for Ectocarpus sp. [Cock et al. 2010]).
Importantly, miRNA complements from species belonging to other eukaryotic lineages often exhibited marked differences for many of the parameters listed earlier (table 1). This indicates that the shared features of the brown algal miRNAs are not due to universal constraints on miRNA evolution but to constraints specific to the brown algal lineage.
Taken together, these observations suggest that the S. japonica and Ectocarpus sp. miRNAs are likely to be derived from a common, ancestral miRNA system. We cannot formerly rule out the possibility that miRNA systems evolved independently in the Ectocarpales and the Laminariales, but this hypothesis would seem to be less likely given the similarities between the miRNA complements in the two species analysed and the rarity of independently evolved miRNA systems across the eukaryotic tree. If the S. japonica and Ectocarpus sp. miRNAs are derived from a common, ancestral miRNA system, then this implies that individual miRNA loci are not maintained over long periods of evolutionary time in brown algal genomes (the last common ancestor of S. japonica and Ectocarpus sp. is thought to have existed between 80 and 110 Ma; Silberfeld et al. 2010; Kawai et al. 2015). This contrasts with the situation in the land plant and animal lineages, where some miRNAs have been strongly conserved over evolutionary time, in some cases for >600 Myr (Berezikov 2011; Tarver et al. 2013; Taylor et al. 2014).
The lack of sequence similarity between S. japonica and Ectocarpus sp. miRNAs could be due to the ancestral miRNAs having diverged to the point that they now share no sequence similarity or to the ancestral loci having been replaced by new miRNA loci that evolved after the divergence of the Ectocarpales and the Laminariales (or a combination of these two processes). Comparison of the Ectocarpus sp. miRNA loci with the corresponding regions of the E. fasciculatus genome indicated that miRNA sequences have been diverging rapidly within the genus Ectocarpus. Sequence divergence is therefore likely to be at least one of the process that underlie long-term changes in miRNA complements within the brown algae.
Although brown algal miRNAs do not appear to be conserved over long time periods, we did find evidence that they are evolving under constraint. Within the genus Ectocarpus, for example, miRNA hairpin loop regions were significantly more strongly conserved than neighbouring regions of noncoding genomic sequence. Moreover, the structural similarities between the S. japonica and Ectocarpus sp. miRNAs also indicate constrained sequence evolution. These observations are consistent with the brown algal miRNAs representing functional loci but further work will be needed to confirm this experimentally and to explore their specific functions.
Prediction of potential target genes using TAPIR suggested that the miRNA complements of S. japonica and Ectocarpus sp. target distinct sets of protein-coding genes in the two species. This suggests that, not only are the miRNAs complements of these two species unrelated at the sequence level, but the two miRNA complements are likely to regulate different sets of miRNA targets. Taken together, these observations suggest that miRNAs function differently in brown algae to their counterparts in the animal and land plant lineages. Rather than being key, evolutionarily conserved regulatory components, it is possible that brown algal miRNAs play a less central role in gene regulation, perhaps only mediating subtle adjustments to gene expression levels. Again, functional analysis of individual brown algal miRNAs will be necessary to explore this hypothesis and to extend comparisons with the miRNA systems of other lineages. It is also interesting to bear in mind that the brown algae are the most recently evolved of the eukaryotic groups that exhibit complex multicellularity (Cock et al. 2010), the brown algal lineage having emerged only ∼260 Ma (Kawai et al. 2015). If, as we have suggested, the evolution of miRNAs in this lineage was linked to the transition to multicellularity (Tarver et al. 2015), it is possible that the miRNA system may still be in a relatively primitive state compared with the miRNA systems of long-established multicellular groups such as the animals and land plants.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We would like to thank Masafumi Nozawa for providing us with information about the Drosophila miRNAs. This work was supported by the Centre National de la Recherche Scientifique, the University Pierre and Marie Curie, the European Research Council grant “Sexsea” (638240), the Central Public-interest Scientific Institution Basal Research Fund, CAFS (NO. 2016PT03), the National Natural Science Foundation of China (31302188, 31272660) and the Qingdao National Laboratory for Marine Science and Technology (2015ASKJ02).
Literature Cited
- Allen E, et al. 2004. Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet. 36:1282–1290. [DOI] [PubMed] [Google Scholar]
- Andrews S. 2016. FastQC a quality control tool for high throughput sequence data. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Avesson L, Reimegard J, Wagner EG, Soderbom F. 2012. MicroRNAs in Amoebozoa: deep sequencing of the small RNA population in the social amoeba Dictyostelium discoideum reveals developmentally regulated microRNAs. RNA 18:1771–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axtell MJ, Westholm JO, Lai EC. 2011. Vive la difference: biogenesis and evolution of microRNAs in plants and animals. Genome Biol. 12:221.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, et al. 2011. Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res. 21:203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E. 2011. Evolution of microRNA diversity and regulation in animals. Nat Rev Genet. 12:846–860. [DOI] [PubMed] [Google Scholar]
- Bonnet E, He Y, Billiau K, Van de Peer Y. 2010. TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 26:1566–1568. [DOI] [PubMed] [Google Scholar]
- Bortoluzzi S, Biasiolo M, Bisognin A. 2011. MicroRNA-offset RNAs (moRNAs): by-product spectators or functional players?. Trends Mol Med. 17:473–474. [DOI] [PubMed] [Google Scholar]
- Brate J, et al. 2016. Pre-metazoan origin of animal miRNAs. bioRxiv 076190.
- Brodersen P, et al. 2008. Widespread translational inhibition by plant miRNAs and siRNAs. Science 320:1185–1190. [DOI] [PubMed] [Google Scholar]
- Campo-Paysaa F, Semon M, Cameron RA, Peterson KJ, Schubert M. 2011. microRNA complements in deuterostomes: origin and evolution of microRNAs. Evol Dev. 13:15–27. [DOI] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. 2009. Origins and Mechanisms of miRNAs and siRNAs. Cell 136:642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti H, Casas-Mollano JA. 2006. On the origin and functions of RNA-mediated silencing: from protists to man. Curr Genet. 50:81–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock JM, Collén J. 2015. Independent emergence of complex multicellularity in the brown and red algae In: Ruiz-Trillo I, Nedelcu AM, editors. Evolutionary transitions to multicellular life. Advances in marine genomics. Springer Verlag, Berlin. p. 335–361. [Google Scholar]
- Cock JM, et al. 2010. The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature 465:617–621. [DOI] [PubMed] [Google Scholar]
- Cormier A, et al. 2017. Re-annotation, improved large-scale assembly and establishment of a catalogue of noncoding loci for the genome of the model brown alga Ectocarpus. New Phytol. 214:219–232. [DOI] [PubMed] [Google Scholar]
- Curtis HJ, Sibley CR, Wood MJA. 2012. Mirtrons, an emerging class of atypical miRNA. Wiley Interdiscip Rev RNA 3:617–632. [DOI] [PubMed] [Google Scholar]
- Edgar R. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felippes FF, Schneeberger K, Dezulian T, Huson DH, Weigel D. 2008. Evolution of Arabidopsis thaliana microRNAs from random sequences. RNA 14:2455–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Zorrilla JM, et al. 2007. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet. 39:1033–1037. [DOI] [PubMed] [Google Scholar]
- Frank F, Hauver J, Sonenberg N, Nagar B. 2012. Arabidopsis Argonaute MID domains use their nucleotide specificity loop to sort small RNAs. EMBO J. 31:3588–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank F, Sonenberg N, Nagar B. 2010. Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature 465:818–822. [DOI] [PubMed] [Google Scholar]
- Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N. 2012. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm B, et al. 2015. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu Rev Genet. 49:213–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M, Zamore PD. 2009. Small silencing RNAs: an expanding universe. Nat Rev Genet. 10:94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, et al. 2008. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature 455:1193–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimberg AM, Sempere LF, Moy VN, Donoghue PC, Peterson KJ. 2008. MicroRNAs and the advent of vertebrate morphological complexity. Proc Natl Acad Sci U S A. 105:2946–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel J, et al. 2006. The expansion of the metazoan microRNA repertoire. BMC Genomics 7:25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinas A, et al. 2007. The small RNA repertoire of Dictyostelium discoideum and its regulation by components of the RNAi pathway. Nucleic Acids Res. 35:6714–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai H, Hanyuda T, Draisma SGA, Wilce RT, Andersen RA. 2015. Molecular phylogeny of two unusual brown algae, Phaeostrophion irregulare and Platysiphon glacialis, proposal of the Stschapoviales ord. nov. and Platysiphonaceae fam. nov., and a re-examination of divergence times for brown algal orders. J Phycol. 51:918–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. 2011. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39:D152–D157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenberger D, et al. 2009. Evidence for human microRNA-offset RNAs in small RNA sequencing data. Bioinformatics 25:2298–2301. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, et al. 2010. Diverse pathways generate microRNA-like RNAs and Dicer-independent small interfering RNAs in fungi. Mol Cell 38:803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Mao L. 2007. Evolution of plant microRNA gene families. Cell Res. 17:212–218. [DOI] [PubMed] [Google Scholar]
- Li W, et al. 2015. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 43:W580–W584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, et al. 2015. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 350:691–694. [DOI] [PubMed] [Google Scholar]
- Liu F, Wang W, Sun X, Liang Z, Wang F. 2015. Conserved and novel heat stress-responsive microRNAs were identified by deep sequencing in Saccharina japonica (Laminariales, Phaeophyta). Plant Cell Environ. 38:1357–1367. [DOI] [PubMed] [Google Scholar]
- Lopez-Gomollon S, et al. 2014. Global discovery and characterization of small non-coding RNAs in marine microalgae. BMC Genomics 15:697.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, et al. 2011. ViennaRNA package 2.0. Algorithms Mol Biol. 6:26.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, et al. 2008. The birth and death of microRNA genes in Drosophila. Nat Genet. 40:351–355. [DOI] [PubMed] [Google Scholar]
- Luo R, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1:18.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Hooks K, Griffiths-Jones S. 2012. Evolution and function of the extended miR-2 microRNA family. RNA Biol. 9:242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17 Available from: http://journal.embnet.org/index.php/embnetjournal/article/view/200/479. [Google Scholar]
- Mattick JS. 2004. RNA regulation: a new genetics?. Nat Rev Genet. 5:316–323. [DOI] [PubMed] [Google Scholar]
- Molnar A, Schwach F, Studholme DJ, Thuenemann EC, Baulcombe DC. 2007. miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature 447:1126–1129. [DOI] [PubMed] [Google Scholar]
- Mukherjee K, Campos H, Kolaczkowski B. 2013. Evolution of animal and plant dicers: early parallel duplications and recurrent adaptation of antiviral RNA binding in plants. Mol Biol Evol. 30:627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa M, Miura S, Nei M. 2010. Origins and evolution of microRNA genes in Drosophila species. Genome Biol Evol. 2:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa M, Miura S, Nei M. 2012. Origins and evolution of microRNA genes in plant species. Genome Biol Evol. 4:230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson KJ, Dietrich MR, McPeek MA. 2009. MicroRNAs and metazoan macroevolution: insights into canalization, complexity, and the Cambrian explosion. Bioessays 31:736–747. [DOI] [PubMed] [Google Scholar]
- Piriyapongsa J, Jordan IK. 2007. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One 2:e203.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piriyapongsa J, Jordan IK. 2008. Dual coding of siRNAs and miRNAs by plant transposable elements. RNA 14:814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. 2006. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 20:3407–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogato A, et al. 2014. The diversity of small non-coding RNAs in the diatom Phaeodactylum tricornutum. BMC Genomics 15:698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Hendrix D, Levine M, Haley B. 2009. A distinct class of small RNAs arises from pre-miRNA-proximal regions in a simple chordate. Nat Struct Mol Biol. 16:183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberfeld T, et al. 2010. A multi-locus time-calibrated phylogeny of the brown algae (Heterokonta, Ochrophyta, Phaeophyceae): investigating the evolutionary nature of the ‘brown algal crown radiation’. Mol Phylogenet Evol. 56:659–674. [DOI] [PubMed] [Google Scholar]
- Sperling EA, Robinson JM, Pisani D, Peterson KJ. 2010. Where’s the glass? Biomarkers, molecular clocks, and microRNAs suggest a 200-Myr missing Precambrian fossil record of siliceous sponge spicules. Geobiology 8:24–36. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28:2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarver JE, Donoghue PC, Peterson KJ. 2012. Do miRNAs have a deep evolutionary history?. Bioessays 34:857–866. [DOI] [PubMed] [Google Scholar]
- Tarver JE, et al. 2013. miRNAs: small genes with big potential in metazoan phylogenetics. Mol Biol Evol. 30:2369–2382. [DOI] [PubMed] [Google Scholar]
- Tarver JE, et al. 2015. microRNAs and the evolution of complex multicellularity: identification of a large, diverse complement of microRNAs in the brown alga Ectocarpus. Nucl Acids Res. 43:6384–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RS, Tarver JE, Hiscock SJ, Donoghue PC. 2014. Evolutionary history of plant microRNAs. Trends Plant Sci. 19:175–182. [DOI] [PubMed] [Google Scholar]
- Yang X, Li L. 2011. miRDeep-P: a computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 27:2614–2615. [DOI] [PubMed] [Google Scholar]
- Ye N, et al. 2015. Saccharina genomes provide novel insight into kelp biology. Nat Commun. 6:6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, et al. 2010. Multiple distinct small RNAs originate from the same microRNA precursors. Genome Biol. 11:R81.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, et al. 2007. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev. 21:1190–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.