

Abstract
The vascular smooth muscle cell (VSMC) phenotypic switch is a key pathophysiological change in various cardiovascular diseases, such as aortic dissection (AD), with a high morbidity. Polycystin-1 (PC1) is significantly downregulated in the VSMCs of AD patients. PC1 is an integral membrane glycoprotein and kinase that regulates different biological processes, including cell proliferation, apoptosis, and cell polarity. However, the role of PC1 in intracellular signaling pathways remains poorly understood. In this study, PC1 downregulation in VSMCs promoted the expression of SM22, ACTA2 and calponin 1 (CNN1) proteins. Furthermore, PC1 downregulation in VSMCs upregulated phospho-MEK, phospho-ERK and myc, but did not change phospho-JNK and phospho-p38. These findings suggest that the MEK/ERK/myc signaling pathway is involved in PC1-mediated human VSMC phenotypic switch. Opposite results were observed when an ERK inhibitor was used in VSMCs downregulated by PC1. When the C-terminal domain of PC1 (PC1 C-tail) was overexpressed in VSMCs, the expression levels of phosphor-ERK, myc, SM22, ACTA2 and CNN1 proteins were downregulated. The group with the overexpressed mutant protein (S4166A) in the PC1 C-tail showed similar results to the group with the downregulated PC1 in VSMCs. These results suggest that the Ser at the 4166 site in PC1 is crucial in the PC1 mediated MEK/ERK/myc signaling pathway, which might be the key pathophysiological cause of AD.
Key words: Aortic dissection, polycystin-1, ERK, VSMC, phenotypic switch
Introduction
Aortic dissection (AD) is a fatal condition involving the aorta. In recent years, the prevalence of AD has increased worldwide, which led to the high demand for medical resources and social wealth.1 Vascular smooth muscle cells (VSMCs), the main components of aorta media, exhibit two phenotypes, namely, contractile and synthetic.2,3 Contractile VSMCs display weak proliferation and migration abilities, and exhibit a limited ability to synthesize extracellular matrices. By contrast, synthetic VSMCs feature strong proliferation and migration abilities, hypertrophic appearance, and “hill and valley” growth. Synthetic VSMCs show a strong ability to synthesize extracellular matrices (e.g., collagen, elastin, and proteoglycans).4 Most mature and healthy VSMCs are of the contractile phenotype and exhibit a relatively high contractile capability and a low proliferation ability.5 However, different physiological and pathological stimuli may cause the VSMC phenotypic switch, which leads to the proliferation, migration, and synthesis of excessive extracellular matrices.6 These effects result in pathological changes to tissues and organs. The morphological changes of AD primarily occur in aorta media.7 Under normal conditions, contractile VSMCs can maintain the vascular tone and homeostasis of the blood vessel wall. However, the number and proportion of synthetic VSMCs in AD patients are significantly increased, which reduce aortic elasticity and promote rupture.6,8,9 These results suggest that the phenotypic switch is an important factor in the occurrence of AD.6
Polycystin-1 (PC1), a 4302-amino acid (aa) cell-surface protein encoded by the polycystic kidney disease 1 gene (PKD1), is distributed in numerous tissues and involved in cell-matrix interactions and cell signaling.10 This protein contains a short cytoplasmic C terminal (PC1 C-tail, 197 aa), 11 transmembrane receptor-like glycoproteins, and a large N terminal extracellular region.11 Mutations in PC1 are indicators of vascular diseases and therefore of prognostic importance.12 Furthermore, the intracellular C-terminal domain of PC1 comprises specific tyrosine and serine sites for phosphorylation by c-src, focal adhesion kinase, and protein kinase A (PKA), as well as an RRSSR consensus sequence for putative S4166 phosphorylation.13 PC1 has also been implicated in various downstream signaling events; furthermore, it can be cleaved and localized to the nucleus via a nuclear localization signal present within its sequence.14 Overexpression of the C-terminal domain in ventricular myocytes increases [3H]Leucine incorporation by nearly twofold.15 These results suggest that the C-terminal domain of PC1 is an important factor in vascular diseases.
In the present study, we hypothesized that PC1 is an important factor in the VSMC phenotypic switch and in AD occurrence. To test this hypothesis, we determined whether the expression level of PC1 in AD patients is different from that of normal vessel walls and whether PC1 can promote the VSMC phenotypic switch from contractile to synthetic. Then, we explored the mechanism of the PC1 C-terminal domain on several kinases in VSMCs. Furthermore, we confirmed the role of PC1(S4166) on VSMC phenotypic switch.
Materials and Methods
Preparation of aortic tissues
Paraffin sections of aortic tissues were collected from AD patients and normal participants from the First Affiliated Hospital of Anhui Medical University. The study was approved by the Association of Medical Ethics of the hospital. A written informed consent was obtained from all participants.
Cell culture
Human aortic VSMCs were obtained from American Type Culture Collection (Rockville, MD, USA) and were cultured in F12K Kaighn’s modification medium supplemented with 10% fetal bovine serum (FBS) and 1 × Antibiotic-Antimycotic Solution at 37°C, humidified atmosphere containing 5% CO2. The growth medium was replaced every 3-4 day and the cells were seeded onto Petri dishes or multi-well plates at a ratio of 1 to 4 upon 80% confluence, and were starved for 24 h in serum-free medium prior to use.16,17
VSMC proliferation assay and cell cycle analysis
Cell counting kit-8 kits (CCK-8, Beyotime Institute of Biotechnology, Shanghai, China) were used for evaluation VSMCs proliferation. Cell growth was arrested by incubation of the cells in serum-free medium for 24 h prior to be used according to the manufacturer’s instructions. Then, VSMCs were seeded onto a 96-well cell culture plate at a density of 2×103 cells/well for 24 h at 37°C, and treated with different concentrations of salusin-β for 24, 48 or 72 h. Finally, 10 μL of CCK-8 solution was added into each well, and incubated for 2 h at 37°C. The absorbance was conducted at 450 nm using a microplate reader (ELX800, BioTek Instruments, Inc., Vermont, USA). The distribution of various phases of the cell cycle was used to evaluate the VSMCs proliferation with flow cytometry.
Constructs and production of lentivirus
Recombinant lentivirus not expressing PC1 was designed by RNA interference. And recombinant lentivirus expressing GFP was used as a control. The sequence of PC1-shRNA1, sense: 5’-GATCCCGACAAGCAGTCCCTGACCTTCAAGAGAGGTCAGGGACTGCTTGTCGTTTTTTG-3’; The negative control shRNA, sense: 5’-CGCGTCCCCCACCTTTCGGCACTCTCCCTTCAAGAGGGGAGAGTGCCGAAAGGTGTTTTTGGAAAT-3’. Oligo DNA was annealed and subcloned to pLVX-shRNA2 vector, and then the connected compound was transfected with competent cell DH5. The recombinant plasmid was sequenced to verify whether insert sequences were consistent with oligo DNA sequences. A packaging plasmid system pLVX-shRNA2, psPAX2 and pMD2G were co-transfected into 293T cells via Lipofectamine 2000. The transfected cells were incubated for 6 h, and then the complete medium was replaced. The viral particles were subsequently harvested at 48 h after transfection, 0.45-μm cellulose acetate filter was used to remove the debris, under centrifugation at 50,000 rpm (4°C) for 2 h and resolved in formulation buffer. The titer of virions was determined through the transduction of HEK293 cells with serial dilutions of the vector and GFP expression assessment by fluorescent cells counting under fluorescence microscopy after 96 h. Viral stocks were aliquoted and stored until they were used.
Haematoxylin eosin (HE) and Masson’s trichrome staining
The thoracic aorta were fixed in 4% paraformaldehyde in PBS, embedded in paraffin, sectioned into 5-μm thick, and stained with Haematoxylin-Eosin staining or Masson’s trichrome staining according to the instructions. Vascular remodeling was analyzed under light microscopy (BX 45, Olympus Co., Tokyo, Japan).
Immunohistochemistry
The thoracic aorta were fixed in 4% paraformaldehyde in PBS, embedded in paraffin, sectioned into 5-μm thick slices, blocked with BSA, the sections were incubated with primary antibody against PC1 (ABT128, Millipore, Billerica, MA, USA) overnight at 4°C and then biotinylated secondary antibody for 60 min at room temperature. Vascular remodeling was analyzed under light microscopy (BX 45, Olympus Corporation, Tokyo, Japan).
RNA extraction and real-time quantitative PCR analysis
The mRNA of PC1, SM22, ACTA2, CNN1, Myc and CyclinD1 were analyzed by real-time quantitative PCR. Total RNA was isolated with Trizol reagent according to the manufacturer’s instructions. RNA concentration and quality were checked by spectrophotometry. cDNA was synthesized from RNA by reverse transcription. Real-time quantitative PCR was performed on an ABI PRISM 7500 sequence detection system. The 25 μL reaction mixture included 12.5 μL 2× SYBR Green PCR Mix, 5 μL diluted cDNA (1:10), and 0.5 μM sense and antisense primers. Amplification was carried out under the following conditions: initial denaturation for 10 min at 95°C, denaturation for 10 s at 95°C, annealing for 30 s at an optimal temperature for each cDNA, and extension for 30 s at 72°C. The mRNA levels of target genes were calculated after normalization to tubulin mRNA.
Western blot
The content of PC1, SM22, ACTA2, CNN1, p38, ERK, JNK, PI3K and Myc, CyclinD1, and the phosphorylation of p38, ERK, JNK and PI3K were determined by Western blot. Preparation of whole cell lysates and tissue homogenates, and the immunoblotting assay were performed according to previously described procedures.18 The primary antibodies were obtained from the following sources: anti-Polycystin-1 (ABT128; Millipore); anti-SM22 alpha (ab14106; Abcam, Cambridge, UK); anti-alpha smooth muscle Actin (ab5694; Abcam); anti-Calponin (ab46794; Abcam); anti-alpha Tubulin (ab52866; Abcam); anti-p38 (phospho Y182) (ab47363; Abcam); Anti-p38 (ab7952; Abcam); Anti-ERK1 (pT202/pY204) + ERK2 (pT185/pY187) (ab50011; Abcam); Anti-ERK1 + ERK2(ab17942; Abcam); Anti-JNK1+JNK2 (phospho T183 + Y185) antibody (ab4821); Anti-JNK1+JNK2 (ab37228; Abcam); Anti-PI3K p85 (phospho Y607); Anti-PI3K p85 (ab189403; Abcam); anti-myc (ab32072; Abcam) and anti-Cyclin D1 (ab134175; Abcam).
Transwell invasion assay
Culture inserts of 8-μm pore size (Transwell; Costar; Sigma-Aldrich, St. Louis, MO, USA) were coated with Matrigel (BD Biosciences, San Jose, CA, USA) (100 μg/well) and placed into the wells of 24-well culture plates. In the lower chamber, 500 μL of medium containing 10% FBS was added, and 1×105 cells were seeded to the upper chamber. After 36 h of incubation at 37°C with 5% CO2, the number of cells that had migrated through the pores were fixed with 10% formalin and stained with 0.05% crystal violet. Data are the mean ± SD of three independent experiments.
Statistical analysis
All data are expressed as the mean ± standard deviation (SD). Statistical analysis were performed using SPSS version 18.0 software. Comparisons between groups were made using one-way analysis of variance. P<0.05 was considered to be statistically significant.
Results
Downregulation of PC1 remodels the aorta of AD patients
HE staining and Masson’s trichrome staining revealed the remodeling of arteries of the AD patients. Furthermore, HE-stained aortic sections showed that the normal aortic wall was composed of an inner layer, a middle layer, and an outer layer. The VSMCs in the middle layer were neatly arranged, and few inflammatory cells were scattered. The middle layer of the aortic wall from each AD patient became thinner and was accompanied by the loss of SMCs and accumulation of the outer layer and inflammatory cells. Masson’s trichrome staining showed that the collagen fibers from AD tissue were thick and disorganized, and their collagen accumulation increased in the aortic media compared with the controls (Figure 1A). These results showed that aorta from an AD patient shows fractures (red arrows) compared with that from a normal person. Moreover, immunohistochemical results showed that the expression level of PC1 was lower in the AD patient than in the controls. Using an antibody against the C-terminal region of PC1, the PC1 C-tail was detected in the nucleus (Figure 1B).
Figure 1.

PC1 was downregulated in the aorta of AD. A) HE staining and Masson’s trichrome staining of aortas from AD patients and normal individuals (ctrl); red arrows indicate aortic fractures. B) PC1 immunohistochemistry of aortas from AD patients and normal individuals (ctrl). Scale bars: 100 μm.
The PC1 mRNA and protein levels in AD patients and normal persons were also detected by Q-PCR and Western blot. PC1 mRNA was significantly lower in the aortic vascular smooth muscle of the AD patients than in the controls (Figure 2A). Similarly, the PC1 protein level was significantly lower in the AD patients than in the controls (Figure 2 B,C).
Figure 2.

PC1 mRNA and protein levels in AD patients and normal persons. A) Expression of PC1 mRNA in the control and AD; AD patient has a significantly lower expression of PC1 than the control. B) Western blot of four cases of AD and control. C) Normalization of the PC1 protein of the AD and control groups; AD patient has a significantly lower expression of PC1 than the control. Each experimental condition was performed in triplicate and applied in three independent experiments. Values are expressed as mean ± SD; *P<0.01.
Genetic inhibition of PC1 promotes the proliferation and migration of VSMCs
The Q-PCR and Western blot were used to detect the efficiency of LV-PC1-kd. As shown in Figure 3 A,B, the PC1 was significantly downregulated by LV-PC1-kd. The proliferation of VSMCs was detected through cell counting kit-8 (CCK8) assay. Results revealed that the proliferation of VSMCs in the group with knock-downed PC1 was enhanced, especially after 72 h (Figure 3C). The mean OD450 values at 24 h were 0.61 and 0.73 in the control and PC1 knockdown groups, respectively (P=0.088). The mean OD450 values increased to 0.73 and 0.99 in the control and PC1 knockdown groups, respectively (P=0.023). Furthermore, the mean value significantly increased to 0.96 and 1.41 in the control and PC1 knockdown groups after 72 h (P=0.008). This results indicates the significant difference between the two groups. PC1 downregulation in the VSMCs reduced the cell ratio in the G0/G1 phase and increased that in the G2/M phase compared with the vehicle treatment in VSMCs (Figure 3D). The percentages of cells in the G0/G1 phase was 60.8 and 46.4 in the control and PC1 knockdown groups, respectively (P=0.010). The percentages of cells in the S phase were 33.1 and 38.8 in the control and PC1 knockdown groups, correspondingly (P=0.168). The percentages of cells in the G2/M phase was 6.02 and 14.8 in the control and PC1 knockdown groups, respectively (P=0.036) (Figure 3E). These data suggests the significant influence of PC1 in cellular proliferation. Moreover, in the transwell assay, the VSMCs with PC1 knockdown showed a strong migration (Figure 3 F,G). These results suggest that PC1 influences the proliferation, cell cycle distribution, and migration of VSMCs.
Figure 3.
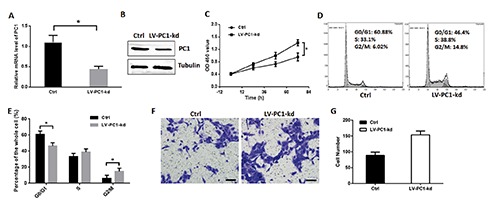
PC1 knockdown promoted VSMC proliferation. A) VSMC was infected by LV-PC1-kd for 72 h, and Q-PCR was used to detect the efficiency. B) Western blot was used to detect the efficiency of LV-PC1-kd. C) Cell proliferation was detected through CCK8 kit and valuated by an absorbance at 450 nm; two groups of VSMCs were detected: one served as control, and the other was infected with lentivirus containing PC1-shRNA that can knockdown PC1. D) Cell cycle of two groups of VSMCs detected through flow cytometry in a representative experiment. E) Cell ratios in the G0/G1, S, and G2/M phases of the two VSMC groups. F) Transwell assay of PC1-kd VSMCs in a representative experiment. The VSMCs migrated through transwell chamber were colored purple by 0.05% crystal violet; scale bars: 50 μm. G) The number of migration cells in the transwell assay of each VSMC group. Each experimental condition was performed in triplicate and applied in three independent experiments. Values are expressed as mean ± SD; *P<0.01.
Genetic inhibition of PC1 promotes the differentiation of VSMCs
SM22, ACTA2 and CNN1 are biomarkers of the VSMCs phenotypic switch from contractile to synthetic; these biomarkers exhibit lower expression levels in the synthetic phenotype than in the contractile phenotype.19,20 In the present study, we determined the expression levels of these three genes to monitor the differentiation of VSMCs. Real-time polymerase chain reaction (PCR) showed that PC1 downregulation decreased the mRNA expression levels of SM22, ACTA2 and CNN1 in VSMCs (Figure 4A). Similarly, Western blot results showed that PC1 knockdown decreased the protein concentrations of SM22, ACTA2 and CNN1 in VSMCs (Figure 4 B,C). Overall, these results suggest that PC1 knockdown promotes VSMC differentiation.
Figure 4.
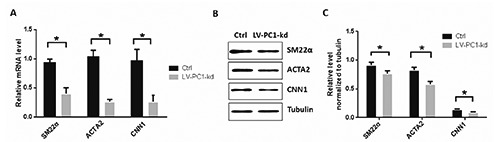
PC1 knockdown promoted the VSMC phenotypic switch. A) mRNA expression levels of SM22α, ACTA2, and CNN1 were detected through RT-PCR; two groups of VSMCs were detected: one served as control, and the other was infected with lentivirus containing PC1-shRNA that can knockdown PC1. B) Protein levels of SM22α, ACTA2, and CNN1 detected through Western blot. C) Relative levels of SM22α, ACTA2, and CNN1 were normalized to tubulin through grayscale scanning. Each experimental condition was performed in triplicate and applied in three independent experiments. Values are expressed as mean ± SD; *P<0.01.
Regulation of ERK phosphorylation in VSMCs
Many signaling pathways regulate the VSMC phenotypic switch.21-23 To determine signalling pathway involved in this process, we identified the phosphorylation levels of p38, JNK and PI3K in VSMCs. PC1 downregulation promoted the phosphorylation of ERK and MEK, but exerted no significant effect on the phosphorylation of p38, JNK and PI3K in VSMCs (Figure 5 A,B). The expression levels of myc and cyclinD1, which can be regulated by ERK were also detected. PC1 knockdown clearly increased the mRNA and protein expression levels of myc and cyclinD1 (Figure 5 C,D). These results indicate that the MEK/ERK/myc signaling pathway plays an important role in the proliferation and differentiation of VSMCs.
Figure 5.

Effect of PC1 on MAPK pathways. A) Phosphorylated p38, ERK, JNK, PI3K and MEK were detected through Western blot; two VSMCs groups were detected: one served as the control, and the other was infected with lentivirus containing PC1-shRNA that can knockdown PC1. B) Relative level of phosphorylation was normalized to total specific protein, such as phosphor-ERK/ERK, through gray scale scanning. C) Protein expression levels of myc and cylinD1 were detected through Western blot. D) Relative mRNA levels of myc and cyclinD1 in the two VSMC groups were detected through RT-PCR. Each experimental condition was performed in triplicate and applied in three independent experiments. Values are expressed as mean ± SD; *P<0.01.
ERK phosphorylation in PC1-downregulated VSMCs is inhibited by SCH772984
An ERK inhibitor was used to determine the MEK/ERK/myc signaling pathway involved in the proliferation and differentiation of VSMCs. SCH772984, an inhibitor of ERK1 and ERK2, can inhibit the proliferation of VSMCs. The mean OD450 values of the control, PC1 knockdown, and SCH772984 groups were 0.91, 1.40, and 1.05, respectively, with a significant difference between the PC1 knockdown and SCH772984 groups (P<0.001) (Figure 6A). Flow cytometry results showed that the mean percentage of cells in the G2/M phase of the control, PC1 knockdown, and SCH772984 groups were 10.2, 15.1, and 8.8, respectively, with a significant difference between the PC1 knockdown and SCH772984 groups (P<0.001) (Figure 6 B,C). SCH772984 addition decreased the proliferation of VSMCs with PC1 knockdown. Interestingly, it did not significantly change the protein expression levels of ACTA2, SM22, and cyclinD1 (Figure 6D).
Figure 6.

ERK inhibitor SCH772984 can inhibit the effects of PC1 knockdown on VSMCs. A) Cell proliferation was detected through a CCK8 kit and evaluated by absorbance at 450 nm; three groups were detected: VSMC control, without lentivirus and SCH772984; LV-PC1-kd + DMSO, VSMCs with lentivirus with PC1-shRNA and DMSO as solvent control; LV-PC1-kd + Comp., VSMCs containing lentivirus with PC1-shRNA and ERK inhibitor SCH772984. B) Cell cycles of the three VSMC groups were detected through flow cytometry; the result is from a representative experiment. C) Cell ratios of the G0/G1, S, G2/M phases in the three VSMC groups. D) Protein expression levels of ACTA2, SM22α and cyclinD1 were detected through Western blot. Each experimental condition was performed in triplicate and applied in three independent experiments. Values are expressed as mean ± SD; *P<0.01.
Cytoplasmic tail mutation of PC1 promoted ERK phosphorylation
Considering that PC1 downregulation can promote the proliferation of VSMCs, we hypothesized that altering the expression of wild PC1 affects VSMCs proliferation. Thus, we created a PC1 (pS4166A) mutant to examine the phosphorylation of ERK and the expression of SM22, ACTA2, CNN1, cyclinD1 and myc. Western blot results showed that the protein expression levels of phospho-ERK, ERK, SM22, ACTA2, CNN1, cyclinD1 and myc in the PC1(S4166A) mutant were similar to those in the PC1 knockdown cells (Figure 7 A,B). These results suggest that PC1 C-tail can increase the expression of SM22, ACTA2, and CNN1 which are markers of the contractile phenotype of VSMCs, thereby promoting the phenotypic switch to synthetic phenotype and the proliferation of VSMCs. Furthermore, S4166 is a crucial factor in the function of PC1 C-tail (Figure 7C).
Figure 7.

Function of PC1 C-tail and the mutant PC1 C-tail formed at S4166A. A) Diagrams of PC1, PC1 C-tail and S4166A mutant site; blue strip represents the extracellular region, the green strip represents the helical structure, and red strip represents the cytoplasmic region. B) Protein levels of phospho-ERK, ERK, ACTA2, SM22α, CNN1, cyclinD1, and myc in four VSMC groups were detected: Ctrl, VSMCs without any transfection; LV-PC1-kd, VSMCs infected with lentivirus with PC1-shRNA that can knockdown PC1; PC1, VSMCs infected by lentivirus with PC1 C-tail coding sequence that can promote PC1 C-tail expression; and PC1(S4166A), VSMCs infected by lentivirus with mutant PC1 C-tail at the S4166A coding sequence that can express the mutant form of the PC1 C-tail. C) Diagram of signaling pathway in VSMC phenotypic switch induced by PC1.
On the basis of these results, we hypothesized that PC1 can regulate the MEK/ERK/myc pathway and induce the VSMC phenotypic switch. However, the mechanism by which PC1 regulates CyclinD1 is still unknown. The PC1 C-tail, especially S4166, is the key factor in the entire process.
Discussion
In the current study, PC1 was significantly downregulated in the VSMCs of AD patients. PC1 was involved in MEK/ERK/myc pathway and PC1 downregulation promoted VSMC proliferation and phenotypic switch. The PC1 C-tail was also a key factor in the ERK pathway. The effect of overexpressed S4166 mutant in the VSMCs, was similar to that of the downregulated PC1. There results suggest that the PC1 C-tail is the key pathophysiological cause of AD, and is involved in the MEK/ERK/myc signaling pathway. The aorta is composed of three layers, particularly, the tunica adventitia, tunica media, and tunica intima. The tunica media is composed of elastic fibers and VSMCs. The pathology of AD is medial degeneration caused by the VSMC phenotypic switch.24-26 Our findings suggest that the intervention of PC1 can potentially prevent excessive vascular proliferation and medial degeneration, thereby reducing the risk of AD.
PC1 is a transmembrane molecule that plays an essential role in renal tubular morphogenesis. PC1 dysfunction causes human autosomal dominant polycystic kidney disease (ADPKD). Previous studies showed that AD is related to ADPKD, hypertension and atherosclerosis. PC1 downregulation induces the proliferation of human VSMCs. In this regard, CCK8 assay is a convenient colorimetric assay for testing VSMC proliferation. In the current study, we confirmed that PC1 knockdown can stimulate the proliferation of VSMCs in a time-dependent manner. Furthermore, PC1 downregulation increased the ratio of cells in the S and G2/M phases, and promoted VSMC migration. ACTA2 encodes the VSMC-specific α-actin, a component of the contractile complex, SM22 is a 22 kDa lineage-restricted protein of smooth muscle cells (SMC) that is physically associated with cytoskeletal actin filament bundles in contractile VSMCs, CNN1 can bind to actin and calmodulin and can regulate and modulate smooth muscle contraction. Thus, ACTA2, SM22, and CNN1 can reflect VSMC proliferation. The mRNA and protein levels decreased when PC1 knocked down in VSMCs. This effect was also observe in AD patients. These data indicate that PC1 downregulation of PC1 is an important triggering factor of AD formation.
The pathways involved in the pathogenesis of AD includes the TGF-β, Smad3, and angiotensin II signaling pathways and the VSMC pathway.6,27,28 In the current study, we measured the phosphorylation of p38, ERK, JNK, and PI3K and found that ERK phosphorylation significantly increased. The ERK signaling pathway, which involves MEK, myc, and cyclinD, is important in the cell proliferation and migration. By transducing the stimulatory extracellular signals to the nucleus, the MEK/ERK/myc signaling pathway can trigger a series of biological reactions, such as cell proliferation, differentiation, and apoptosis, and plays an important regulatory role in these processes. Recent studies have shown that the MEK/ERK/myc signaling pathway is related to the contractile function of vascular smooth muscle, and contributes to VSMC dysfunction, vasoconstriction, and vascular remodeling.15 PC1 is involved in the regulation of the VSMC phenotypic switch.15 In our previous study, the mutant PC1 was detected in the vessel tissue of patients (unpublished data).
Gene mutations and environmental factors, such as hypertension, can increase the vascular pressure and activate the AD-related pathways.29,30 PC1 can be phosphorylated at S4166 in the C-terminal domain by protein kinase X (PRKX).13 PRKX is a cAMP-dependent CREB kinase that can promote cell migration and proliferation. PRKX shows similar roles to PC1 in VSMCs.31 Mutations in the S4166 of the PC1 C-tail cause ERK phosphorylation, increase the expression levels of cyclinD1 and myc; and decrease ACTA2, SM22, and CNN1. These effects were also observed in VSMCs with PC1 knockdown. These results indicate that the PC1 C-tail plays an important role in the VSMC phenotypic switch.
In conclusion, these findings demonstrate that PC1 regulates AD by modulating the phenotype of VSMCs. PC1 downregulation may induce AD formation, which involves the MEK/ERK/myc signaling pathway. The PC1 C-tail, especially the S4166 site, plays the main role in this process.
ACKNOWledgments
the authors would like to thank Dr. Jian Cui, Peking Union Medical College for helping with cellular experiments.
References
- 1.Nienaber CA, Clough RE. Management of acute aortic dissection. Lancet, 2015;385:800-811 [DOI] [PubMed] [Google Scholar]
- 2.Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol, 2012; 74:13-40 [DOI] [PubMed] [Google Scholar]
- 3.Branchetti E, Poggio P, Sainger R, Shang E, Grau JB, Jackson BM, et al. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc Res, 2013; 100:316-324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chistiakov DA, Orekhov AN, Bobryshev YV. Vascular smooth muscle cell in atherosclerosis. Acta Physiol (Oxf) 2015;214:33-50. [DOI] [PubMed] [Google Scholar]
- 5.Uryga AK, Bennett MR. Ageing induced vascular smooth muscle cell senescence in atherosclerosis. J Physiol 2016;594:2115-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu SB, Zhu J, Zhou ZZ, Xi EP, Wang RP, Zhang Y. TGF-beta1 induces human aortic vascular smooth muscle cell phenotype switch through PI3K/AKT/ID2 signaling. Am J Transl Res 2015;7:2764-74. [PMC free article] [PubMed] [Google Scholar]
- 7.Kudryavtseva O, Aalkjaer C, Matchkov VV. Vascular smooth muscle cell phenotype is defined by Ca2+-dependent transcription factors. FEBS J 2013;280:5488-99. [DOI] [PubMed] [Google Scholar]
- 8.El-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol 2009;6:771-86. [DOI] [PubMed] [Google Scholar]
- 9.Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res 2012;95:194-204. [DOI] [PubMed] [Google Scholar]
- 10.Trudel M, Yao Q, Qian F. The Role of g-protein-coupled receptor proteolysis site cleavage of Polycystin-1 in renal physiology and polycystic kidney disease. Cells 2016;5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 1995;10:151-60. [DOI] [PubMed] [Google Scholar]
- 12.Rossetti S, Chauveau D, Kubly V, Slezak JM, Saggar-Malik AK, Pei Y, et al. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet 2003;361:2196-201. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Burrow CR, Polgar K, Hyink DP, Gusella GL, Wilson PD. Protein kinase X (PRKX) can rescue the effects of polycystic kidney disease-1 gene (PKD1) deficiency. Biochim Biophys Acta, 2008;1782:1-9. [DOI] [PubMed] [Google Scholar]
- 14.Chapin HC, Caplan MJ. The cell biology of polycystic kidney disease. J Cell Biol 2010;191:701-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pedrozo Z, Criollo A, Battiprolu PK, Morales CR, Contreras-Ferrat A, Fernandez C, et al. Polycystin-1 is a cardiomyocyte mechanosensor that governs l-type Ca2+ channel protein stability. Circulation 2015;131:2131-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto K, Ohishi M, Ho C, Kurtz TW, Rakugi H. Telmisartan-induced inhibition of vascular cell proliferation beyond angiotensin receptor blockade and peroxisome proliferator-activated receptor-gamma activation. Hypertension 2009;54:1353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 2008;117:1161-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Liu Y. Blockage of tubular epithelial to myofibroblast transition by hepatocyte growth factor prevents renal interstitial fibrosis. J Am Soc Nephrol 2002;13:96-107. [DOI] [PubMed] [Google Scholar]
- 19.Dong LH, Li L, Song Y, Duan ZL, Sun SG, Lin YL, et al. TRAF6-mediated SM22alpha K21 ubiquitination promotes G6PD activation and NADPH production, contributing to GSH homeostasis and VSMC survival in vitro and in vivo. Circ Res 2015;117:684-94. [DOI] [PubMed] [Google Scholar]
- 20.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res 2016;118:692-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding Y, Zhang M, Zhang W, Lu Q, Cai Z, Song P, et al. AMP-activated protein kinase alpha 2 deletion induces VSMC phenotypic switching and reduces features of atherosclerotic rosclerotic plaque stability. Circ Res 2016;119:718-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagayama K, Kyotani Y, Zhao J, Ito S, Ozawa K, Bolstad FA, et al. Exendin-4 prevents vascular smooth muscle cell proliferation and migration by angiotensin II via 26. the inhibition of ERK1/2 and JNK signaling pathways. PLoS One 2015;10:e0137960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kay AM, Simpson CL, Stewart JA., Jr. The 27. role of AGE/RAGE signaling in diabetes-mediated vascular calcification. J Diabetes Res 2016;2016:6809703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugiyama S, Kugiyama K, Nakamura S, Kataoka K, Aikawa M, Shimizu K, et al. Characterization of smooth muscle-like cells in circulating human peripheral 28. blood. Atherosclerosis 2006; 87:351-62. [DOI] [PubMed] [Google Scholar]
- 25.Wang N, Yang J, Yu X, Hu J, Xing C, Ju X, et al. Radial artery calcification in endstage renal disease patients is associated with deposition of osteopontin and diminished expression of alpha-smooth muscle actin. Nephrology (Carlton) 2008; 3:367-75. [DOI] [PubMed] [Google Scholar]
- 26.Mehrotra R. Disordered mineral metabolism and vascular calcification in nondialyzed chronic kidney disease patients. J Ren Nutr 2006;16:100-18. [DOI] [PubMed] [Google Scholar]
- 27.Hilhorst-Hofstee Y, Scholte AJ, Rijlaarsdam ME, van Haeringen A, Kroft LJ, Reijnierse M, et al. An unanticipated copy number variant of chromosome 15 disrupting SMAD3 reveals a three-generation family at serious risk for aortic dissection. Clin Genet 2013;83:337-44. [DOI] [PubMed] [Google Scholar]
- 28.Fry JL, Shiraishi Y, Turcotte R, Yu X, Gao YZ, Akiki R, et al. Vascular smooth muscle sirtuin-1 protects against aortic dissection during angiotensin II-induced hypertension. J Am Heart Assoc 2015;4:e002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pannu H, Avidan N, Tran-Fadulu V, Milewicz DM. Genetic basis of thoracic aortic aneurysms and dissections: potential relevance to abdominal aortic aneurysms. Ann N Y Acad Sci 2006;1085:242-55. [DOI] [PubMed] [Google Scholar]
- 30.Guo D, Hasham S, Kuang SQ, Vaughan CJ, Boerwinkle E, Chen H, et al. Familial thoracic aortic aneurysms and dissections: genetic heterogeneity with a major locus mapping to 5q13-14. Circulation 2001;103:2461-8. [DOI] [PubMed] [Google Scholar]
- 31.Li X, Li HP, Amsler K, Hyink D, Wilson PD, Burrow CR. PRKX, a phylogenetically and functionally distinct cAMP-dependent protein kinase, activates renal epithelial cell migration and morphogenesis. Proc Natl Acad Sci USA 2002;99:9260-5. [DOI] [PMC free article] [PubMed] [Google Scholar]