

Abstract
Rickettsiae are obligate intracellular bacteria that have small genomes as a result of reductive evolution. Many Rickettsia species of the spotted fever group (SFG) cause tick-borne diseases known as “spotted fevers”. The life cycle of SFG rickettsiae is closely associated with that of the tick, which is generally thought to act as a bacterial vector and reservoir that maintains the bacterium through transstadial and transovarial transmission. Each SFG member is thought to have adapted to a specific tick species, thus restricting the bacterial distribution to a relatively limited geographic region. These unique features of SFG rickettsiae allow investigation of how the genomes of such biologically and ecologically specialized bacteria evolve after genome reduction and the types of population structures that are generated. Here, we performed a nationwide, high-resolution phylogenetic analysis of Rickettsia japonica, an etiological agent of Japanese spotted fever that is distributed in Japan and Korea. The comparison of complete or nearly complete sequences obtained from 31 R. japonica strains isolated from various sources in Japan over the past 30 years demonstrated an extremely low level of genomic diversity. In particular, only 34 single nucleotide polymorphisms were identified among the 27 strains of the major lineage containing all clinical isolates and tick isolates from the three tick species. Our data provide novel insights into the biology and genome evolution of R. japonica, including the possibilities of recent clonal expansion and a long generation time in nature due to the long dormant phase associated with tick life cycles.
Keywords: rickettsia, intracellular bacteria, intra-species genomic diversity, genome evolution, generation time
Introduction
Rickettsiae are Gram-negative bacteria that together with the genus Orientia constitute the family Rickettsiaceae in the order Rickettsiales (Dumler et al. 2001). Rickettsiae have an obligate intracellular life cycle and a small genome (1.1–1.6 Mb) as a result of the reductive evolution that occurred after their specialization to an intracellular niche (Blanc et al. 2007). Members of the genus Rickettsia are divided into four groups: the spotted fever group (SFG), the typhus group (TG), the R. belli group, and the R. canadensis group. The former two groups cause various arthropod-borne diseases in humans (Parola et al. 2005). The SFG consists of more than 25 validated species, and more new species or subspecies are being proposed. Among these species/subspecies, at least 24 are known or suspected to be pathogenic in humans, causing tick-borne diseases, or “spotted fevers” such as Rocky Mountain spotted fever, which is caused by R. rickettsii (Parola et al. 2013). The most notable biological and ecological aspect of SFG rickettsiae is their life cycle, which is closely associated with that of ticks. Ticks of the family Ixodidae (known as “hard” ticks) are generally thought to act as both vectors and reservoirs for most SFG rickettsiae, because the bacteria are maintained in ticks through transstadial (transfer of bacteria from stage to stage in the tick life cycle) and transovarial transmission (Burgdorfer and Brinton 1975; Socolovschi et al. 2009). However, the mode of association with ticks varies among species. Some are probably beneficial symbionts, whereas others have undetermined benefit to hosts despite transstadial and transovarial maintenance for generations. Others appear to be harmful to arthropod hosts. Moreover, soft ticks (Argasidae) are increasingly known to harbor SFG rickettsiae (Kawabata et al. 2006; Reeves et al. 2006; Duh et al. 2010; Lafri et al. 2015). Rickettsia akari and R. felis are associated with mites and fleas, respectively (Merhej and Raoult 2011).
Although the life cycle of each species is not yet fully understood, each SFG member is thought to have adapted to a specific tick species, thus restricting the bacterial distribution to a relatively limited geographic region and causing a region-specific “spotted fever”. These unique features of SFG rickettsiae allow investigation of how the genomes of such biologically and ecologically specialized bacteria evolve after genome reduction and the types of population structures generated. Owing to improvements in sequencing technology and the small genome sizes of rickettsiae, over 17 species of SFG rickettsiae have been sequenced. Comparative genome analyses using these sequences have demonstrated that the genomes of SFG rickettsia are very similar in size, mostly ranging from 1.2 to 1.3 Mb, with some exceptions (Ogata et al. 2005). Although relatively well-conserved genome synteny has been observed in most genomes, significant differences exist in the level of gene loss/decay (Merhej and Raoult 2011). Remarkable genomic rearrangement, mostly due to mobile genetic elements, have been described for R. felis and R. peacockii (Ogata et al. 2005; Felsheim et al. 2010; Gillespie et al. 2014). The presence of plasmids and conjugation genes have also been observed in several species (Merhej and Raoult 2011; El Karkouri et al. 2016). These inter-species genomic differences may translate to clinical and biological differences among species (Merhej and Raoult 2011). However, to the best of our knowledge, a large-scale intra-species genomic comparison at the whole genome level has not been conducted for any of the SFG rickettsiae. Only a few reports have analyzed intra-species genomic diversity, but these have been small-scale genomic comparisons using only a few strains (Ellison et al. 2008; Massung et al. 2011; Gillespie et al. 2014; Clark et al. 2015). Although multiple genomes have been sequenced for several species, the work was often performed by different laboratories using different sequencing platforms. Large-scale analyses of genetic diversity and population structure have been conducted in several SFG species, but these studies were based on the sequence diversity in several selected genomic regions (Fournier et al. 2004; Karpathy et al. 2007; Eremeeva and Dasch 2009; Fournier et al. 2009; Massung et al. 2011; Paddock et al. 2014).
Rickettsia japonica is an etiological agent of Japanese (or Oriental) spotted fever (referred to as JSF in this article) that was first discovered in 1984 in Japan (Mahara et al. 1985; Uchida et al. 1986, 1992). JSF patients have been identified in southwestern and central Japan (Mahara 2006) and in Korea (Chung et al. 2006). Although the details of host and reservoir association for R. japonica have not yet been analyzed, the bacterium has been isolated from or detected by PCR in the following eight tick species: Dermacentor taiwanensis, Haemaphysalis hystericis, H. cornigera, H. flava, H. longicornis, H. fromosensis, H. megaspinosa, and Ixodes ovatus (Ando and Fujita 2013). Rickettsia japonica has also been detected in H. longicornis collected in Korea (Lee et al. 2003). A human pathogenic SFG rickettsiae closely related to R. japonica appears to exist in Thailand (Gaywee et al. 2007; Takada et al. 2009). Because R. japonica was isolated from the unfed larvae of D. taiwanensis and H. hystericis (strains DT-1 and HH-1; both analyzed in this study; see Takada et al. 1994 for the reference of DT-1), transovarial transmission occurs in at least these two tick species.
Here, we report the results of a nation-wide phylogenomic analysis of R. japonica. We performed a high-resolution phylogenetic analysis of high-quality genome sequences of 31 strains (11 complete sequences and 20 nearly complete sequences containing a single gap in a tandem repeat-containing region) isolated from various regions and sources in Japan over the past 30 years. The analysis revealed an extremely low level of genomic diversity for R. japonica, thereby providing novel insights into the biology and genomic evolution of R. japonica, including the possibilities of recent clonal expansion or a long generation time in nature, due to the long dormant phase associated with tick life cycles.
Materials and Methods
Bacterial Strains, Cultivation, and Genomic DNA Preparation
The R. japonica strains analyzed in this study are listed in supplementary table S1, Supplementary Material online, in which detailed strain information for each isolate (location and year of isolation, number of in vitro passages, etc.) is also presented. Bacterial cells were inoculated onto confluent monolayers of L929 or Vero cells and incubated at 34 °C for 4–7 days in 5% CO2. After cultivation, the infected cells from two 25-cm3 culture bottles were mechanically exfoliated and disrupted with glass beads, and the cell suspension was separated by centrifugation at 200 × g for 5 min. The supernatant was transferred to a 15-ml tube and kept on ice. The cell pellet was resuspended in a small volume of supernatant, homogenized gently with a glass Dounce homogenizer (GPE Scientific, Bedfordshire, UK), and separated by centrifugation at 200 × g for 5 min. The resulting supernatant was mixed with the first supernatant. Before extraction of the bacterial genomic DNA, the mixture was incubated with DNase I (Invitrogen, Carlsbad, CA) at a final concentration of 1,000 U/ml for 1 h at 37 °C to digest host DNA. DNase I was inactivated by heating at 65 °C for 5 min, and bacterial genomic DNA was then extracted using the DNeasy Blood and Tissue Kit (Qiagen, Tokyo, Japan) according to the manufacturer’s instructions.
Genome Sequencing and Genome Annotation
Two strains (YH_M and MZ08014) were sequenced using the Roche 454 FLX titanium platform. We produced 287,137 and 298,014 shotgun reads of 400-500 bp in length. The sequence reads were assembled with GS Assembler ver. 2.3, and gaps were filled with sequencing PCR products that spanned the gaps, by using an ABI3730 capillary sequencer. The remaining 29 strains were sequenced on the Illumina MiSeq platform. Libraries for paired-end sequencing (average fragment sizes 250–300 bp) were generated using a Nextera XT DNA sample prep kit (Illumina) according to the manufacturer’s instructions, and paired-end sequences (250 bp from each end) were obtained. After trimming of low-quality sequences (quality score; <10), reads were mapped onto the C57BL/6J mouse genome sequence (accession nos. NC_000067 to NC_000087) using the Burrows–Wheeler Alignment tool (BWA) software (Li and Durbin, 2009) to remove the reads derived from the mouse L929 cells used for R. japonica propagation. The remaining Illumina reads (final coverage ranged from 150× to 800×) were assembled using Platanus version 1.1.4 (Kajitani et al. 2014), which usually produced four contigs per strain. Three of the four gaps were filled by sequencing PCR products. To fill the remaining gap derived from the tandem repeat-containing region of the rompA gene, low-coverage PacBio sequences were generated with an RS II system for nine of the 29 strains to determine the numbers of tandem repeats in the rompA gene by using long PacBio sequences spanning the repeat region in the gene. The final sequences of the rompA gene in the nine strains were determined by correcting the PacBio sequences by mapping Illumina reads. The gap sequences in the rompA gene of 20 strains were not determined.
Automated annotation was performed with the Microbial Genome Annotation Pipeline (MiGAP) (www.migap.org/). Manual curation of the annotated data was performed using in silico Molecular Cloning software (in silico biology, inc., Tokyo, Japan). All sequence data obtained in this study (assembled sequences and raw read sequence data) have been deposited in the GenBank/EMBL/DDBJ database. Their accession numbers are shown in supplementary table S1, Supplementary Material online.
Identification and Characterization of SNPs and InDels and Phylogenetic Analysis
A multiple sequence alignment of 31 R. japonica genomes was constructed using GENETYX ver. 16.0, and the alignment was manually curated. The sequence of strain HH-13 was used as a reference for SNP and InDel calling. On the basis of the identified SNPs, phylogenetic trees were constructed using an in-house program (fig. 1) or RAxML (Stamatakis 2014) with 1,000-fold bootstrapping (other figures). The R version 3.2.4 package was used for statistical analysis. The evolutionary analysis of R. rickettsii strains was conducted in MEGA7 (Kumar et al. 2015). The numbers of all possible synonymous and non-synonymous mutations were calculated by computationally changing each nucleotide in all CDSs to the other three bases along the entire YH_M genome by using the Biopython package (Cock et al. 2009).
Fig. 1.—

Geographic distribution of Rickettsia japonica analyzed in this study and associated phylogenetic relationships. Isolation locations for the 31 analyzed strains are shown (see Supplementary fig. S2, Supplementary Material online, for more precise information of the strains isolated in Miyazaki prefecture). Clinical and tick isolates are indicated by red and blue circles, respectively. Larger circles that are divided into two or three parts indicate two or three strains isolated from ticks collected in the same location (within a few kilometers) on the same day. The names of the prefecture where the strains were isolated are indicated. The abbreviations of each prefecture are shown in parentheses. In the box, a phylogenetic tree of the 31 R. japonica strains is shown. The tree was constructed by using 112 SNP sites that were identified among the 31 strains. Clinical and tick isolates are indicated by red and blue circles, respectively. Larger circles that are divided into two, three, or four parts represent 2-, 3-, or 4-member strain sets with identical genome sequences in terms of SNPs, respectively. Small dots represent hypothetical intermediates, and the dot-to-dot distance corresponds to one SNP difference. The years and locations of strain isolation are shown in parentheses. Among the three lineages identified, Lineage II contains strains HH-16/17/18, and Lineage III contains one strain (OHH-1). Lineage I contains the other 27 strains. Among the 112 SNPs, we found no homoplasic SNP (the same mutation that occurred in different lineages independently or in parallel).
Strain Set and Genome Sequencing
We analyzed a total of 31 R. japonica isolates collected from various regions of Japan between 1985 and 2014 (fig. 1 and supplementary table S1, Supplementary Material online). The oldest strain, YH, is the first-identified isolate of R. japonica (Uchida et al. 1992). Although this strain was previously sequenced (Dong et al. 2012; Matsutani et al. 2013), we re-sequenced it to obtain a highly accurate genome sequence that was comparable to those of the other 30 strains analyzed in this study (referred to here as YH_M to distinguish this sequence from the published genome). Although the regions of isolation were highly biased toward southwestern and central Japan (fig. 1), this distribution exactly reflected the occurrence of JSF cases in Japan. The strain set included 17 clinical isolates and 14 tick-derived isolates from three different tick species (12 from Haemaphysalis hystricis, one from H. longicornis, and one from Dermacentor taiwanensis). One tick strain was isolated from H. longicornis attached to a JSF patient, but other strains were from ticks collected in the environment. Three sets of strains were isolated from ticks (all from H. hystricis) collected in the same region (within a few kilometers) on the same day. One set was composed of three strains (HH06125/HH06154/HH06116) isolated in Uwajima city, Ehime, on Shikoku island. The second set was composed of two strains (HH-12/HH-13) isolated in Onomichi city, Hiroshima, on the main island, and the third was composed of three strains (HH-16/HH-17/HH-18) isolated in the Amami-oshima island close to the Okinawa islands located at the very southern end of Japan.
We sequenced these strains completely (11 strains) or nearly completely (20 strains with one gap in a complicated tandem repeat-containing region of the rompA gene) using Roche 454 or Illumina MiSeq followed by gap filling using capillary sequencing of the gap-covering PCR products. Sequence information from low-coverage PacBio sequencing was also used to obtain the complete sequence of the rompA gene. In addition, all sequence variations, single nucleotide polymorphisms (SNP), and insertion/deletions (InDel) that were identified in the 31 genomes were confirmed either by PCR amplification and sequencing of the respective regions (10 InDel sites) or by manual inspection of Illumina read mapping (other sites).
Results
General Features of R. j aponica Genomes
All R. japonica strain sequences contained a single circular chromosome, and no plasmid was found in any strain. The chromosomes were very similar in size (1,283,227–1,284,037 bp in the 11 completely sequenced strains; see supplementary table S1, Supplementary Material online) with almost identical G + C content (32.4%). Consistently with results from previous reports (Dong et al. 2012; Matsutani et al. 2013), the R. japonica genomes contained one set of rRNA genes (not forming an operon), 33 tRNA genes, and three non-coding RNA genes. A total of 1,186 protein-coding regions (CDSs), including 63 split or truncated genes, were identified in the fully annotated YH_M genome. The difference in the number of CDSs compared with those previously reported was due to differences in the completeness and accuracy of the genome sequences and in the method and criterion used for annotation. In addition, compared with the YH_M sequence obtained in this study, the previously reported full genome sequence of strain YH (Matsutani et al. 2013) contained 75 SNPs, 41 InDels, and translocations of two small segments in the rompA gene. However, these variations were found in only the YH genome and not in any of the other 30 strains analyzed in this study, except for two InDels that were derived from copy number differences in the tandem repeats. Thus, all or almost all of the variations were probably derived from sequence errors and misassembly in the YH genome. All sequence differences found between the YH_M and YH sequences are shown in supplementary table S2, Supplementary Material online. The YH sequence of Dong et al. (2012) is a draft sequence obtained by low-coverage 454 sequencing, and it contained more than 200 SNPs/InDels as compared with our YH_M sequences. Therefore, we did not perform a fine sequence comparison with our YH_M sequence.
Phylogenetic Analysis of R. j aponica Strains
We identified only 112 SNP sites and 44 InDels among the 31 R. japonica genomes (tables 1 and 2; see supplementary tables S3 and S4, Supplementary Material online listing all identified SNPs and InDels, respectively). On the basis of these SNPs, a phylogenetic tree of the 31 strains was constructed (fig. 1). Notably, we found seven groups of strains that did not contain SNPs within each group. Additionally, no InDels were detected in each group, with a few exceptions. Three of the seven groups were the aforementioned strain sets, which were composed of the strains isolated from ticks collected in the same place on the same day, thus suggesting that ticks inhabiting the same location carry a single R. japonica clone, although we cannot fully exclude the possibility that some of these ticks originated from the progeny of the same female. Other groups were composed of strains isolated in different years or locations or from different sources (fig. 1).
Table 1.
Summary of the SNP Analysis of the 31 Rickettsia japonica Genomes
| Number Of SNPs | Transition:Transversion | Intragenic:Intergenic | Synonymous:Nonsynonymous | |
|---|---|---|---|---|
| Total | 112 | 87:25 | 71:41 | 32:39 |
| Lineage I-specific | 34 | 24:10 | 23:11 | 8:15 |
| (Specific to a single strain) | (14) | (10:4) | (10:4) | (4:6) |
| (Shared by 2 or more strains) | (20) | (14:6) | (13:7) | (4:9) |
| Lineages II & III-specific | 13 | 11:2 | 9:4 | 6:3 |
| Lineage II-specific | 19 | 15:4 | 13:6 | 8:5 |
| Lineage III-specific | 46 | 37:9 | 26:20 | 10:16 |
Table 2.
Summary of the InDel Analysis of the 31 Rickettsia japonica Genomes
| Number of InDels | Intergenic:Intragenic | PolyA Or PolyT | Tandem repeats | Others | |
|---|---|---|---|---|---|
| Total | 43 | 23:20 | 16 | 11 | 16 |
| Only in Lineages II and III | 25 | 17:8 | 11 | 2 | 12 |
| (Common to Lineages II & III) | (3) | (0:3) | (0) | (1) | (2) |
| (Specific to Lineage II) | (10) | (8:2) | (5) | (0) | (5) |
| (Specific to Lineage III) | (12) | (9:3) | (6) | (1) | (5) |
| Other distribution patterns | 18 | 6:12 | 5 | 9 | 4 |
Despite such a low genomic diversity, the 31 strains were separated into three distinct lineages—one major and two minor. The major lineage (referred to as Lineage I) contained 27 strains, including all clinical strains isolated in various regions and 10 tick strains isolated from three different species collected in various regions. Strain YH_M was also included in this lineage. Lineage I was extremely homogenous, with a total of only 34 SNPs and a maximum difference of 14 SNPs between the strains, whereas several sublineages were distinguished (fig. 1).
Of the two minor lineages, Lineage II was composed of three tick strains (HH-16/17/18) isolated from the Amami-oshima island, which is geographically distant from the collection locations of the other strains. However, a strain of Lineage III (composed of the single strain OHH-1) was isolated from the main island. Thus, distributions of genetically distant strains are not limited to remote islands.
Characterization of SNPs
Most of the 112 SNPs identified (78%) were generated by transition (table 1; see also supplementary fig. S1, Supplementary Material online, for their genomic locations). Of the 71 intragenic SNPs, 39 were non-synonymous substitutions (table 1, fig. 2, and supplementary table S3, Supplementary Material online). Among the Lineage I-specific SNPs, the proportion of non-synonymous SNPs (15 out of 23) was higher than that in other categories (table 1). Importantly, a statistically significant difference was not detected between this proportion and that expected by counting all possible non-synonymous mutations (P = 0.1218 by a two-sided binomial test), thus suggesting that many of these non-synonymous substitutions are neutral or that insufficient time passed for accumulating enough numbers of substitutions to make it possible to detect selection. The 112 SNPs were distributed across the entire genome, and most were sporadic (fig. 2). However, we found two SNP clusters, one specific to Lineage II (at nucleotide 273,160–273,161) and one specific to Lineage III (at 686,049–686,060). All SNPs in these clusters were generated by transversion (supplementary table S3, Supplementary Material online). The clustering of these SNPs suggests that these two clusters were introduced by recombination.
Fig. 2.—

Distribution of SNPs and InDels in the 31 Rickettsia japonica genomes. The top line indicates the full-length R. japonica genome. Genomes from patients and ticks are indicated by red and blue horizontal lines, respectively. Genomic locations of all SNPs and InDels that were identified in each genome are indicated by short vertical lines and triangles, respectively. Synonymous, non-synonymous and intergenic SNPs are indicated by blue, red, and black lines, respectively. Intragenic and intergenic InDels are indicated by equilateral and isosceles triangles, respectively. The dotted rectangles represent the sequence gaps in the rompA gene. SNP clusters found in Lineages II and III are indicated by asterisks. In this figure, HH-13 was used as a reference, and the genomes are ordered according to the maximum likelihood phylogenetic tree shown on the left-hand side.
Lineage I was highly homogeneous but contained various strains isolated from different locations in different years. Therefore, we analyzed the correlations of pair-wise SNP distances among the Lineage I strains and their geographical and temporal distances (fig. 3). Whereas no correlation was detected between the SNP distances and the temporal distances, the SNP distances exhibited a low but positive correlation with the geographic distances. In this regard, the genetic relationship of the 10 strains isolated in the Miyazaki prefecture on Kyushu island may be noteworthy. These strains were separated into three sublineages (M00021/MZ08014/M13010, M99023/M08024/HH07124/M99015, and M99123/M14012/M114024 in fig. 1) and exhibited some geographic separation according to lineage (supplementary fig. S2, Supplementary Material online). However, in each sublineage, only zero to two SNPs were generated over the 10–15 years period, thus again highlighting the limited heterogeneity detected for R. japonica genomes.
Fig. 3.—
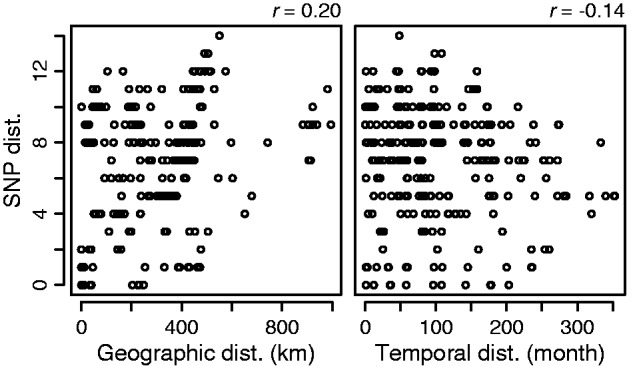
Correlations of SNP distances and geographical or temporal distances among the Lineage I strains. Pair-wise SNP distances and geographical and temporal distances among the Lineage I strains were determined, and the correlations of SNP distances and geographical (left panel) or temporal (right panel) distances were investigated. Lineage I contains two sets of strains (HH-12/13 and HH-16/17/19) that were isolated from ticks that were collected in the same location on the same day. As no SNPs were found in each set of strains, only one strain from each set was included in this analysis. Pearson’s correlation coefficients were calculated using the R package.
Characterization of InDels
All 44 identified InDels were small (up to 429 bp; supplementary table S4, Supplementary Material online) and distributed throughout the genomes (fig. 2). More than half of the InDels (25 InDels) were found in only Lineage II or III (table 2), and many were derived from polyA or polyT sequences or tandem repeats (16 and 11 InDels, respectively). Notably, eight of the 11 tandem repeat-associated InDels were found in four genes encoding cell-surface proteins (rOmpA, rOmpB, Sca1, and Sca2 proteins), and one was found in a gene encoding an ankyrin-repeat containing protein (supplementary fig. S3, Supplementary Material online). The occurrence of these InDels does not always follow the strain phylogeny as expected from their association with tandem-repeats; thus, they cannot be used in a phylogenetic analysis.
Discussion
In this study, we collected 31 R. japonica strains isolated over the past 30 years from various sources and locations in Japan, and we obtained 11 complete and 20 nearly complete genome sequences. These high-quality sequences allowed us to perform a nation-wide, high-resolution phylogenetic analysis of R. japonica. Although three lineages of R. japonica (Lineages I, II, and III) were distinguished, only 112 SNPs and 44 InDels were detected among the 31 strains, thus demonstrating a very low level of genomic diversity for R. japonica inhabiting Japan (fig. 1 and tables 1 and 2). In particular, the major lineage (Lineage I) was extremely homogeneous, and only 34 SNPs were detected among the 27 strains of this lineage. We detected a weak correlation of SNP distances and geographic distances among the Lineage I strains (fig. 3) and some geographical separation of sublineages. However, no correlation was found between SNP distances and temporal distances among the strains even though they were isolated over 30 years, thus indicating a very low SNP accumulation rate. In fact, none or only two SNPs were generated in the local populations inhabiting the Miyazaki prefecture over 10 years (supplementary fig. S2, Supplementary Material online). Because an accelerated rate of sequence evolution is well known in endosymbionts such as Buchnera aphidicola (Moran 1996; Itoh et al. 2002; Moran et al. 2009), the observed low genomic diversity of R. japonica was an unexpected result. However, evolutionary rates can vary among endosymbiont lineages (Bennett et al. 2014), and a similarly low level of sequence diversity has been observed within each clade of B. aphidicola from a single aphid host species inhabiting North America (Moran et al. 2009).
How has such a low level of genomic diversity been maintained in R. japonica, particularly in Lineage I? One plausible explanation is the recent geographic expansion of the Lineage I clone. This hypothesis is supported by the finding that the proportion of nonsynonymous SNPs in the Lineage I-specific SNPs was not significantly different from that expected by counting all possible mutations. If this explanation is true, Lineage I may be progressing toward northern Japan, where no victims of JSF have yet been reported even though four of the eight potential tick host species of R. japonica (H. longicornis, H. flava, H. megaspinosa, and Ixodes ovatus) are located there (Yamaguti et al. 1971; Ando and Fujita 2013). Several Japanese research groups have repeatedly performed surveys of rickettsia-infected ticks at several locations in the northern part of the main island and in Hokkaido over the past 20 years, but no tick carrying R. japonica has ever been detected (Fujita 2008; Ando et al., unpublished data). The lack of JSF patients may be due to many factors; the idea of ongoing expansion of Lineage I is just one possibility. However, this hypothesis is of great public health importance and is worthy of verification by a long-term, systematic survey of JSF patients and R. japonica-infected ticks in these regions. In addition, the genomes of Korean R. japonica strains and an R. japonica-like agent detected in Thailand should be analyzed and should provide valuable information concerning this issue.
Another possible explanation may be a long generation time for R. japonica in nature. SFG rickettsiae, including R. japonica, have a unique life cycle that is closely linked to that of ticks (Socolovschi et al. 2009). After hatching from eggs, ticks develop into larvae and then into nymphs and adults. The ticks must consume a blood meal at each stage of this cycle (three times total). In the case of ticks inhabiting temperate regions, two or more years are required to complete the life cycle because inter-stadial periods generally last more than one month and ticks enter diapause in the winter (Saito 1960; Balashov 1972; Randolph 2008). Moreover, unfed nymph and adult ticks of many species can survive for a year or more before they have a chance to consume a blood meal by eking out the limited energy from their previous meal. Some argasid species can survive as long as 10 or 11 years without a blood meal (Balashov 1972). During any period of diapause or quiescence, rickettsia cells in ticks probably remain in a dormant state. If so, rickettsia cells replicate fewer times in a single tick’s life cycle. In addition, only rickettsia cells that invade oocytes during active oogenesis, which occurs after nymphal and adult meals, can be passed on to the next tick generation (Socolovschi et al. 2009). Therefore, the actual generation time of tick-associated rickettsia such as R. japonica in nature may be very long, although the estimated doubling time of the R. japonica strain YH in L929 cells is approximately six hours (Ando, unpublished).
In addition to these two possibilities, it may be necessary to consider other mechanisms for the generation and maintenance of the observed low level of sequence diversity of R. japonica. These possibilities include the presence of a highly efficient proofreading system and a high degree of selection against variants. Regarding the generation time in nature, the rate of autophagic destruction of rickettsial cells should be considered. The difference in the replication rate among various tissues, particularly ovaries and eggs, may also be an important factor, because rickettsia cells probably replicate more in these issues, and these cells are the population inherited by the tick progenies. The experimental verification of these possibilities may be technically difficult but worthwhile. In addition, fine within-species phylogenetic analyses should also be conducted in other SFG members to examine whether similar genomic features are present. In such studies, considerable attention should be paid to the quality of genome sequences, because the level of sequence diversity among rickettsia strains may be within the range of sequencing errors. If a long generation time truly occurs in nature, similar genomic features may be observed in other SFG rickettsiae.
Another notable finding was that all clinical isolates belonged to Lineage I, a result that may simply reflect a higher prevalence of ticks infected with this lineage or may be due to a difference in pathogenicity in humans between this lineage and the other two minor lineages. We observed no significant differences in the growth of these lineages in cultured cells. However, the 13 SNPs that are shared by Lineage I strains but are not present in Lineages II and III may be noteworthy. Among the 13 SNPs, nine were intragenic, and three of the nine were non-synonymous (table 1). These non-synonymous SNPs were found in the genes for the rOmpB protein, a two-domain glycosyltransferase and a predicted transcriptional activator. rOmpB is known to play important roles in adhesion and invasion into host cells (Hackstadt et al. 1992; Uchiyama 2003). Although the functions of the latter two proteins are unknown, if the potential virulence of Lineage I is higher than that of other lineages, some of these three SNPs may be associated with increased virulence potential. Additionally, three InDels that were found in Lineages II/III but not in Lineage I may also be of note. Two of these InDels occurred in the gene for rOmpB and in a gene encoding the actin polymerization protein RickA, which mediates the intracellular motility of rickettsia (Gouin et al. 2004). However, to determine the potential virulence of Lineage I in humans, the lineages of additional clinical isolates should be searched for; these Lineage II/III-specific mutations may also have deleterious effects on the survival or persistence of R. japonica. The delineating SNPs and InDels for each lineage should contribute to this surveillance.
Finally, from the perspective of host adaptation or coevolution with host ticks, it may be noteworthy that Lineage I contained tick strains from three different tick species. This finding suggests that the transmission of R. japonica among different tick species may occur relatively frequently in nature and that such a host switch may not affect the genetic diversity and population structure of R. japonica. This frequent transmission may be related to the broad tick host range of R. japonica, which is somewhat unusual among SFG rickettsiae (Parola et al. 2013). A recent analysis of R. rickettsii, which also has a broad tick host range, has suggested that its phylogeography is probably determined by ecological and environmental factors that exist independently of the distribution of a particular tick vector (Paddock et al. 2014). However, in this study, we analyzed only one strain each from H. longicornis and D. taiwanensis. Therefore, we cannot exclude the possibility that these ticks acquired R. japonica from H. hystricis before sampling by mechanisms such as co-feeding and that these R. japonica strains thus may not have adapted to new tick species yet. This issue requires further investigation of more R. japonica strains from different species and investigations in other SFG rickettsia species that have narrow or broad tick host ranges.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the Ministry of Health, Labour and Welfare, Japan [grant number H24-Shinkou-Ippan-008]; and the Research Program on Emerging and Reemerging Infectious Diseases from Japan Agency for Medical Research and Development, AMED [grant number; not assigned]. We thank N. Kawano, A. Yoshida, Y. Takeshita, H. Iguchi, and N. Fuji for their technical assistance. A. Akter and S. I. Mondal were supported by scholarships from the Japan Student Services Organization and from the Japanese Ministry of Education, Culture, Sports, Science and Technology, respectively. The authors declare no conflict of interest.
Literature Cited
- Ando S, Fujita H. 2013. Diversity between spotted fever group rickettsia and ticks as vector (in Japanese; an English abstract available). Med Entomol Zool 64:5–7. [Google Scholar]
- Balashov YS. 1972. Bloodsucking ticks (Ixodoidea)—vectors of diseases of man and animals. Miscellaneous Publications of the Entomological Society of America 8:159–376. [Google Scholar]
- Bennett GM, McCutcheon JP, MacDonald BR, Romanovicz D, Moran NA. 2014. Differential genome evolution between companion symbionts in an insect-bacterial symbiosis. MBio 5:e01697-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G, et al. 2007. Reductive genome evolution from the mother of Rickettsia . PLoS Genet. 3:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorfer W, Brinton LP. 1975. Mechanisms of transovarial infection of spotted fever Rickettsiae in ticks. Ann N Y Acad Sci. 266:61–72. [DOI] [PubMed] [Google Scholar]
- Chung MH, et al. 2006. Japanese spotted fever, South Korea. Emerg Infect Dis 12:1122–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark TR, et al. 2015. Comparative genome sequencing of Rickettsia rickettsii strains that differ in virulence. Infect Immun 83:1568–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJ, et al. 2009. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25:1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, El Karkouri K, Robert C, Raoult D, Fournier PE. 2012. Genomic analysis of Rickettsia japonica strain YHT . J Bacteriol 194:6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duh D, et al. 2010. Rickettsia hoogstraalii sp. nov., isolated from hard- and soft-bodied ticks. Int J Syst Evol Microbiol. 60:977–984. [DOI] [PubMed] [Google Scholar]
- Dumler JS, et al. 2001. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and ‘HGE agent’ as subjective synonyms of Ehrlichia phagocytophila . Int J Syst Evol Microbiol. 51:2145–2165. [DOI] [PubMed] [Google Scholar]
- El Karkouri K, Pontarotti P, Raoult D, Fournier PE. 2016. Origin and evolution of rickettsial plasmids. PLoS One 11:e0147492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison DW, et al. 2008. Genomic comparison of virulent Rickettsia rickettsii Sheila Smith and avirulent Rickettsia rickettsii Iowa. Infect Immun 76:542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eremeeva ME, Dasch GA. 2009. Closing the gaps between genotype and phenotype in Rickettsia rickettsii. Ann N Y Acad Sci. 1166:12–26. [DOI] [PubMed] [Google Scholar]
- Felsheim RF, Kurtti TJ, Munderloh UG. 2010. Genome sequence of the endosymbiont Rickettsia peacockii and comparison with virulent Rickettsia rickettsii: identification of virulence factors. PLoS One 4:e8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier PE, Zhu Y, Ogata H, Raoult D. 2004. Use of highly variable intergenic spacer sequences for multispacer typing of Rickettsia conorii strains. J Clin Microbiol. 42:5757–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier PE, et al. 2009. Analysis of the Rickettsia africae genome reveals that virulence acquisition in Rickettsia species may be explained by genome reduction. BMC Genomics 10:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H. 2008. Cell culture system for isolation of disease agents: 15 years of experience in Ohara Research Laboratory (in Japanese; an English abstract available). Ann Rep Ohara Hosp 48:21–42. [Google Scholar]
- Gaywee J, et al. 2007. Human infection with Rickettsia sp. related to R. japonica. Thailand. Emerg Infect Dis 13:671–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JJ, et al. 2014. Genomic diversification in strains of Rickettsia felis Isolated from different arthropods. Genome Biol Evol. 7:35–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouin E, et al. 2004. The RickA protein of Rickettsia conorii activates the Arp2/3 complex. Nature 427:457–461. [DOI] [PubMed] [Google Scholar]
- Hackstadt T, Messer R, Cieplak W, Peacock MG. 1992. Evidence for proteolytic cleavage of the 120-kilodalton outer membrane protein of rickettsiae: identification of an avirulent mutant deficient in processing. Infect Immun 60:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Martin W, Nei M. 2002. Acceleration of genomic evolution caused by enhanced mutation rate in endocellular symbionts. Proc Natl Acad Sci U S A. 99:12944–12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajitani R, et al. 2014. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24:1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpathy SE, Dasch GA, Eremeeva ME. 2007. Molecular typing of isolates of Rickettsia rickettsii by use of DNA sequencing of variable intergenic regions. J Clin Microbiol. 45:2545–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata H, et al. 2006. First detection of Rickettsia in soft-bodied ticks associated with seabirds, Japan. Microbiol Immunol 50:403–406. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K. 2015. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafri I, et al. 2015. Detection of a novel Rickettsia sp. in soft ticks (Acari: Argasidae) in Algeri. Microbes Infect 17:859–861. [DOI] [PubMed] [Google Scholar]
- Lee JH, et al. 2003. Identification of the spotted fever group rickettsiae detected from Haemaphysalis longicornis in Korea. Microbiol Immunol 47:301–304. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahara F, et al. 1985. The first report of rickettsial infections of spotted fever group in Japan three clinical cases (in Japanese). J Jpn Assoc Infect Dis 59:1165–1172. [DOI] [PubMed] [Google Scholar]
- Mahara F. 2006. Rickettsioses in Japan and the Far East. Ann N Y Acad Sci. 1078:60–73. [DOI] [PubMed] [Google Scholar]
- Massung RF, Dasch GA, Eremeeva ME. 2011. Rickettsia and Coxiella. In: Budowle B, Schutzer SE, Breeze RG, Keim PS, Morse SA. editors. Microbial Forensics, 2nd Edition. Elsevier. p. 277–296. [Google Scholar]
- Matsutani M, et al. 2013. Complete genomic DNA sequence of the East Asian spotted fever disease agent Rickettsia japonica . PLoS One 8:e71861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merhej V, Raoult D. 2011. Rickettsial evolution in the light of comparative genomics. Biol Rev Camb Philos Soc 86:379–405. [DOI] [PubMed] [Google Scholar]
- Moran NA. 1996. Accelerated evolution and Muller's rachet in endosymbiotic bacteria. Proc Natl Acad Sci U S A. 93:2873–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, McLaughlin HJ, Sorek R. 2009. The dynamics and time scale of ongoing genomic erosion in symbiotic bacteria. Science 323:379–382. [DOI] [PubMed] [Google Scholar]
- Ogata H, et al. 2005. The genome sequence of Rickettsia felis identifies the first putative conjugative plasmid in an obligate intracellular parasite. PLoS Biol. 3:e248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddock CD, et al. 2014. Phylogeography of Rickettsia rickettsii genotypes associated with fatal Rocky Mountain spotted fever. Am J Trop Med Hyg 91:589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parola P, Paddock CD, Raoult D. 2005. Tick-borne rickettsioses around the world: Diseases challenging old concepts. Clin Microbiol Rev 18:716–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parola P, et al. 2013. Update on tick-borne rickettsioses around the world: a geographic approach. Clin Microbiol Rev 26:657–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph SE. 2008. The impact of tick ecology on pathogen transmission dynamics In: Bowman AS, Nuttall P, editors. Ticks: biology, diseases, and control. New York: Cambridge University Press; p. 40–72. [Google Scholar]
- Reeves WK, et al. 2006. Borrelia, Coxiella, and Rickettsia in Carios capensis (Acari: Argasidae) from a brown pelican (Pelecanus occidentalis) rookery in South Carolina. USA Exp Appl Acarol 39:321e9. [DOI] [PubMed] [Google Scholar]
- Saito Y. 1960. Studies on Ixodid ticks. Part II. On the rearing and life history of the three tick species (Haemaphysalis hystricis, Ixodes japonensis, and Ixodes persulcasus persulcatus) in Japan. Acta Med Biol. 7:303–321. [Google Scholar]
- Socolovschi C, Mediannikov O, Raoult D, Parola P. 2009. The relationship between spotted fever group Rickettsiae and ixodid ticks. Vet Res. 40:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada N, et al. 2009. Spotted fever group Rickettsia sp. closely related to Rickettsia japonica, Thailand. Emerg Infect Dis 15:610–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada N, Fujita H, Yano Y, Tsuboi Y, Mahara F. 1994. First isolation of a Rickettsia closely related to Japanese spotted fever pathogen from a tick in Japan. J Med Entomol 31:183–185. [DOI] [PubMed] [Google Scholar]
- Uchida T, Tashiro F, Funato T, Kitamura Y. 1986. Isolation of a spotted fever group Rickettsia from a patient with febrile exanthematous illness in Shikoku, Japan. Microbiol Immunol 30:1323–1326. [DOI] [PubMed] [Google Scholar]
- Uchida T, Uchiyama T, Kumano K, Walker DH. 1992. Rickettsia japonica sp. nov., the etiological agent of spotted fever group rickettsiosis in Japan. Int J Syst Bacteriol 42:303–305. [DOI] [PubMed] [Google Scholar]
- Uchiyama T. 2003. Adherence to and invasion of Vero cells by recombinant Escherichia coli expressing the outer membrane protein rOmpB of Rickettsia japonica . Ann N Y Acad Sci. 990:585–590. [DOI] [PubMed] [Google Scholar]
- Yamaguti N, Tipton VJ, Keegan HL, Toshioka S. 1971. Ticks of Japan, Korea, and Ryukyu Islands. Brigham Young Univ Sci Bull Biol Ser 15:1–266. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.