

Abstract
Heterogametic sex chromosomes have evolved many times independently, and in many cases, the loss of functional genes from the sex-limited Y or W chromosome leaves only one functional gene copy on the corresponding X or Z chromosome in the heterogametic sex. Because gene dose often correlates with gene expression level, this difference in gene dose between males and females for X- or Z-linked genes in some cases has selected for chromosome-wide transcriptional dosage compensation mechanisms to counteract any reduction in expression in the heterogametic sex. These mechanisms are thought to restore the balance between sex-linked loci and the autosomal genes they interact with, and this also typically results in equal expression between the sexes. However, dosage compensation in many other species is incomplete, and in the case of birds average expression from males (ZZ) remains higher than in females (ZW). Interestingly, recent reports in chickens and related species have shown that the Z chromosome is expressed less in males than would be expected from two copies of the chromosome, and recent data from cell-based approaches on 11 loci in chicken have suggested that one Z chromosome is partially inactivated in males, in a mechanism thought to be homologous to X inactivation in therian mammals. In the present study, we use controlled crosses in three tissues to test for the presence of Z inactivation in males, which would be expected to bias transcription to the active gene copy (allele-specific expression). We show that for the vast majority of genes on the chicken Z chromosome, males express both parental alleles at statistically similar levels, indicating no Z chromosome inactivation. For those Z chromosome loci with detectable ASE in males, we show that the most likely cause is cis-regulatory variation, rather than Z chromosome inactivation. Taken together, our results indicate that unlike the X chromosome in mammals, Z inactivation does not affect an appreciable number of loci in chicken.
Keywords: dosage compensation, allele-specific expression, transcription
Introduction
In diploid species, sex chromosomes occur either as female (females ZW, males ZZ) or male (females XX, males XY) heterogamety. Distinguishable sex chromosomes arise after recombination is halted between the emerging X and Y or Z and W chromosomes. Suppressing recombination between the sex chromosomes results in a loss of recombination for the sex-limited Y or W chromosome, which can lead to the widespread loss of gene activity for Y- or W-linked genes [reviewed in Bachtrog (2013)]. As gene activity on the Y or W chromosome is lost, the heterogametic sex (XY males or ZW females) experiences half the gene dose relative to that in the homogametic sex (XX females or ZZ males).
For many loci, gene expression correlates with gene dose, and therefore reduced gene dose results in reduced expression (Muller 1925; Birchler et al. 2005; Malone et al. 2012). Many genes in the genome are dosage sensitive, and to counteract the deleterious effects of reduced expression for these dosage sensitive genes, chromosome-wide mechanisms of dosage compensation have evolved in several systems (Julien et al. 2012; Mullon et al. 2015). However, the exact status of dosage compensation in some systems remains controversial. In therian mammals, females (XX) inactivate one X chromosome (Lyon 1999), which balances expression to the single X chromosome in males (XY). Female X inactivation has often been proposed to result from selection in males to hyper-express their single X chromosome (Ohno 1967). Because gene expression is often highly correlated between males and females, this could lead to hyper-expression in females, which could be problematic if expression exceeds optimal levels or if stoichiometric balance with interacting autosomal loci is disrupted. Theoretically, female inactivation would counter this over-expression and restore ancestral diploid expression levels.
However, recent observations have cast doubt on this hypothesis. Although expression of some X-linked loci in both females and males are upregulated to restore ancestral diploid expression (Nguyen and Disteche 2006; Lin et al. 2007; Johnston et al. 2008; Moreira de Mello et al. 2010; Pessia et al. 2012), average expression of X-linked genes in females is less than the diploid autosomal average, which is often taken as a proxy for expected expression in the homogametic sex (Julien et al. 2012). Conversely, recent studies found that mammalian expression of X-linked genes is not significantly different from that in species where the X is autosomal (Julien et al. 2012; Lin et al. 2012), suggesting that the lower expression in females for X-linked loci is not due to X-inactivation per se, and causing some controversy over the mechanism and evolutionary consequences of mammalian dosage compensation (Chen and Zhang 2016; He and Zhang 2016).
Importantly, complete mechanisms of dosage compensation are not ubiquitous in systems with heteromorphic sex chromosomes, and many species exhibit incomplete dosage compensation. In systems with incomplete dosage compensation, although some genes are compensated, presumably because they are dosage-sensitive, the heterogametic sex tolerates reduced expression for many X- or Z-linked genes (Itoh et al. 2007). It is unclear why some sex chromosome systems are accompanied by complex whole-chromosome complete dosage compensation mechanisms when many other species persist with incomplete compensation [reviewed in Mank (2009, 2013)], although it may be related to the degree of correlation between male and female expression and strength of selection on expression in the heterogametic sex (Mullon et al 2015).
Incomplete dosage compensation has been most often studied in the chicken, and in this system, female average expression from the single Z chromosome is less than Z expression in males as well as less than the average expression of the diploid autosomes (Ellegren et al. 2007; Itoh et al. 2007; Mank and Ellegren 2009; Julien et al. 2012; Zimmer et al. 2016). More interestingly, expression of the Z chromosome in males, the homogametic sex, is less than what would be expected from two copies, and is less than the average expression in males from autosomal loci (Julien et al. 2012; Zimmer et al. 2016). Studies using cell assays for transcription of a limited number of Z-linked loci have suggested that this may be due to partial Z inactivation in male chickens (Livernois et al. 2013; Graves 2014), resulting from a mechanism that is homologous to, but evolutionarily independent from, X inactivation in mammals. However, ancestral reconstruction of expression across amniotes has suggested that the reduced expression in males from the Z chromosome is due to ancestrally low average expression of loci on the syntenic block that formed the Z (Julien et al. 2012).
X inactivation in therian mammals, although random with respect to parent of origin for many loci, exhibits clear parent of origin effects in some tissues and for loci along the chromosome, leading to allele-specific expression (ASE), or a bias for expression from one parent (typically the mother) over the other (Okamoto et al. 2004; Gregg et al. 2010a; Wang et al. 2014). In theory, if the Z is inactivated, or partially inactivated, there should be strong signatures of ASE from Z-linked loci unless inactivation is perfectly balanced between the paternal and maternal Z chromosome. The possibility of Z inactivation has been difficult to test with RNA-Seq data due to the paucity of SNP variation on avian Z chromosomes, and previous work has been limited to a small proportion of Z-linked loci and has not been designed to test for parent-of-origin effects (Zimmer et al. 2016). In order to detect parent-of-origin allele specific expression, and to assess the potential for Z inactivation, we designed an inter-breed cross to maximize the number of Z-linked SNPs and tracked their transmission and expression in the F1 generation. Our results show that there is little evidence of even partial Z inactivation in chickens, and that allele specific expression detected in a limited number of Z-linked genes is due to cis-regulation rather than inactivation.
Materials and Methods
Reciprocal Cross Experiment and Sampling
We used two inbred chicken strains, Cornish Game (GC) and White Leghorn (WL), to generate reciprocal cross progeny (supplementary fig. S1, Supplementary Material online). We chose six offspring (three males and three females) from each cross, with the exception of the CG × WL cross, where we only obtained two daughters. We also collected tissue from three males and three females from each of the purebred parental breeds for expression analysis. Three tissues (brain, liver, and pectoralis muscle) of the 23 chicks we collected 1 day after hatching, and total RNA extracted for transcriptome sequencing (RNA-Seq). One muscle sample in a male from the CG × WL was removed because its expression pattern was a significant outlier. At the same time, blood of the four parental birds was collected for DNA extraction and whole genome re-sequencing.
DNA was extracted using standard phenol–chloroform protocols, and total RNA was extracted by Trizol. Both DNA and RNA were sequenced with 100bp paired-end reads, with 300 bp insert size, In total, we obtained 82.5 Gb of re-sequencing data for the four parents, corresponding to 20× average coverage for each individual, and 246.3 Gb of RNA-Seq data, corresponding to an average of 3.6 million mappable reads per sample. All experiments were approved by the Animal Care and Use Committee of China Agricultural University (Approval ID: XXCB-20090209). All the animals were fed and handled according to the regulations and guidelines established by this committee, and all efforts were made to minimize suffering.
Parental Genome Reconstruction
In order to ensure the accuracy of subsequent RNA-Seq data alignment, we reconstructed each of the four parental genomes. We first mapped re-sequencing data to the chicken reference genome (Gallus_gallus-4.0, http://hgdownload.soe.ucsc.edu/goldenPath/galGal4/bigZips/) with the Burrows-Wheeler Aligner (BWA) (Li and Durbin 2010), and then sorted the resulting bam files and removed duplicated reads with the Picard toolkit (http://broadinstitute.github.io/picard/). We used the Genome Analysis Toolkit (GAKT) (McKenna et al. 2010) for SNP calling. Bases in the reference genome were substituted by breed variants if breed variants were supported by more reads, based on output from Vcftools (Danecek et al. 2011).
We filtered SNPs in our parental breeds to identify those SNPs that were reciprocally fixed in each parental breed and supported by >10 reads (relaxed to >4 reads in females for Z loci to account for lower expression levels). These conditions were chosen to eliminate potential sources of bias (Gregg et al. 2010b; DeVeale et al. 2012).
RNA-Seq Data Analysis
Parental genomes were used to replace the reference genome in the RNA-Seq data alignment for progeny of both inter-breed crosses using Tophat (Trapnell et al. 2009), with max-deletion-length = 1, intron-length between 3 and 25,000, read-realign-edit-dist as 0, and microexon-search. RNA-Seq data of purebred Cornish Game and White Leghorn were aligned to the chicken reference genome (Gallus_gallus-4.0) using the same parameters. Duplicate reads were removed with Samtools (Li et al. 2009) in order to remove potential sources of bias (Dozmorov et al. 2015). We converted the resulting bam files to vcf files using the mpileup function of Samtools in order to call SNPs. For the progeny of both crosses, we only assessed those loci where the parental strains had fixed alternative SNPSs.
Cufflinks (Trapnell et al. 2012) was used estimate transcript abundance. We calculated log2 transformed measure of fragments per kilobase of exon per million fragments mapped [log2(FPKM + 1)] in order to estimate transcript abundance, and used FPKM for normalization and filtering, using previously implemented thresholds (Zimmer et al. 2016) to facilitate comparisons. We also used FPKM to calculate the pairwise correlation coefficient (Pearson correlation test) of gene expression between all samples in order to identify potential outliers, and to calculate the Z:A expression ratio for both males and females.
In order to assess allele-specific expression, we aligned RNA-Seq reads from the progeny of reciprocal crosses to the genome of both parents, and SNPs with >10 normalized reads in every sample were retained. We calculated the ratio of paternal:maternal allelic expression, and tested the difference between maternal and paternal expression with paired t-tests.
To identify whether the SNPs with significant levels of ASE on the Z chromosome are the result of Z inactivation or cis-acting elements inherited from the parental breeds, we assessed normalized read counts of the two parental alleles, and compared them to the parental expression levels. Ratios were transformed by log2, and correlation coefficients were calculated by Pearson correlation test.
Results
We designed two reciprocal crosses between Cornish Game and White Leghorn breeds, and in each cross, we resequenced the parental genomes and measured F1 transcription with RNA-Seq in three males and three females. We also measured transcription in our parental strains in three males and three females for comparison. Our study design maximizes the number of Z-linked SNPs, which has hampered previous studies, and allows us to parse parent-of-origin allele-specific expression from cis-regulatory variation.
After trimming, we had on average 22.8, 17.8, and 21.3 million mappable reads per individual in brain, liver, and muscle, respectively. After normalization, within-treatment pairwise correlations for gene expression were very high (Pearson’s r2 > 0.90, supplementary fig. S2, Supplementary Material online). Gene expression may vary considerably between the sexes for any single locus (Mank 2017), as well as extensively among loci within each sex, therefore many studies of sex chromosome expression compare average expression across chromosomal categories (Julien et al. 2012; Hough et al. 2014; Zimmer et al 2016) rather than for any single locus. Using this approach, we observe that male expression of the Z chromosome is roughly 1.3- to 1.5-fold greater than female average Z expression, consistent with the doubled dose of Z-linked genes in males, consistent with previous reports (Ellegren et al. 2007; Mank and Ellegren 2009; Itoh et al. 2010). More importantly, average expression of the Z chromosome in both males and females is significantly less than average autosomal expression (fig. 1A). In males, the expression ratios of Z:A with the average of all crossbred individuals were 0.91, 0.93, and 0.89 in brain, liver, and muscle, respectively, whereas in female, the ratios were 0.76, 0.82, and 0.79 (fig. 1B).
Fig. 1.—
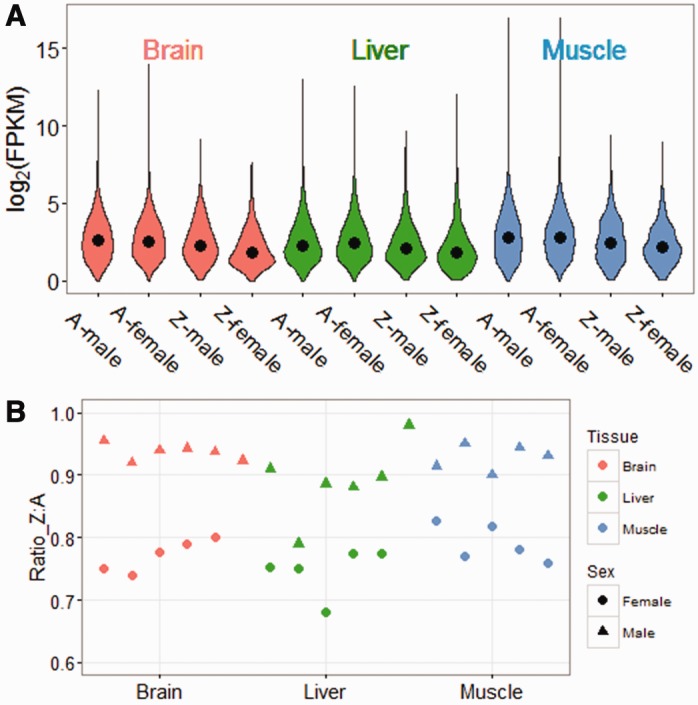
Expression status of Z chromosome and Autosome in three tissues. (A) Violin plots show log2(FPKM) for all the genes with significant expression after filtering. For each tissue, we calculated average FPKM of all females or all males from the two interbreed crosses for each gene. The black points in violins refer to median value of all genes. (B) Z:Autosome expression ratio of three tissues of each crossbred individual. Triangles represent females, circles represent males.
We next assembled the genomes of the four parents in order to eliminate mapping bias of breed-specific SNPs that differ from the Chicken Genome Assembly (galGal4) (Fresard et al. 2014). We recovered nearly 5 million SNPs in each parental genome, and of those, 463,926 SNPs were unique to either the Cornish Game or White Leghorn breeds in our cross. These SNPs covered 7,575 genes, including 61,365 SNPs located in 453 Z-linked loci, which is more than half of the 884 loci currently annotated to the chicken Z chromosome. We only assessed allele-specific expression for SNPs with sufficient reads coverage (>10) in each of the three replicates per group, resulting in SNP coverage for 988 autosomal genes (covered by 2,564 SNP) and 81 Z-linked genes (covered by 312 SNPS) in brain, 679 autosomal genes (covered by 1,758 SNPs) and 60 Z-linked genes (covered by 235 SNPS) in liver, and 961 autosomal genes (covered by 2,677 SNPs) and 76 Z-linked genes (covered by 248 SNPs) in muscle.
We then assessed allele-specific expression in replicate F1 males and females from each cross. Alignment results indicate that the proportion of reads transcribed from each parental allele follows a standard normal distribution, except for the SNPs on Z chromosome in females, which transcribed entirely from their father (fig. 2, table 1). Our data indicate that males express both parental alleles for the vast majority of Z-linked genes (fig. 3). For the Z chromosomes, we identified significant levels (chi-square test, P < 0.05) of ASE in 18 SNPs in the brain (corresponding to 13 genes), 2 SNPs in the liver (2 genes), and 3 SNPs in muscle (3 genes) in the reciprocal crosses. For autosomal loci, we identified significant levels of ASE in 166 SNPs in the brain (corresponding to 113 genes), 120 SNPs in the liver (73 genes), and 120 SNPs in muscle (76 genes). The proportion of Z-linked genes with significant ASE was not significantly different than the proportion of autosomal loci in any of our three tissues.
Fig. 2.—

Overview of RNA-Seq reads aligned to parental genomes. Data from three tissues were taken together. The X axis represents the proportion of reads mapped to paternal allele, Y axis represents SNP count.
Table 1.
Expression of Parental Alleles
| Tissue | Cross | Autosomes |
Z Chromosome |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Male ASE (paternal/maternal)a | Female ASE (paternal/maternal)b | Male/Female Expressionc | Male ASE (paternal/maternal)a | P-valued | Male/Female Expressionc | Number of Z SNPs | Number of Z Genes | ||
| Brain | WL×CG | 1.033 | 1.015 | 0.992 | 1.027 | 0.519 | 1.438 | 625 | 115 |
| CG×WL | 1.000 | 1.002 | 0.914 | 1.047 | 0.059 | 1.284 | 676 | 125 | |
| Liver | WL×CG | 1.014 | 1.000 | 1.039 | 1.108 | 0.343 | 1.493 | 360 | 75 |
| CG×WL | 1.008 | 1.006 | 1.018 | 1.109 | 0.087 | 1.537 | 401 | 82 | |
| Muscle | WL×CG | 0.999 | 1.037 | 1.034 | 1.077 | 0.054 | 1.386 | 539 | 116 |
| CG×WL | 0.989 | 0.994 | 0.951 | 0.993 | 0.017 | 1.302 | 607 | 124 | |
The ratio of normalized reads count mapped to paternal genome versus maternal genome in male progeny.
The ratio of normalized reads count mapped to paternal genome versus maternal genome in in female progeny.
Expression ratio of male to female (FPKM).
P-value of paired t-test based on reads count mapped to paternal alleles and maternal alleles of Z linked SNPs in males.
Fig. 3.—

Expression level of male relative to female on Z chromosome in three tissues. The Y axis shows male expression levels (include male_maternal, male_paternal, and male) relative to females. Red points refer to reads transcribed from the maternal allele in males, green points refer to reads transcribed from the paternal allele in males, and blue points refer to all male reads. Each point represents one SNP.
To identify the relationship between ASE gene expression patterns and their parental expression level, and to differentiate ASE from partial Z inactivation from cis-regulatory variation, we performed correlation test on the expression level ratio of the parental breeds (CG/WL) and ratio of two allele (CG/WL) in our two reciprocal crosses. The ratio of CG/WL alleles was significantly correlated with parental expression (Cross WL × CG: r2 = 0.92401, P-value < 0.0005; Cross CG × WL: r2 = 0.64690, P-value < 0.05, table 2). Slopes of their regression line were close to 1, and intercepts were approximately equal to 0 (fig. 4), even after removing the two loci with strong allele-specific effects. This suggests that the genes on the Z-chromosome with ASE are primarily influenced by cis-regulatory factors inherited from their parents rather than inactivation.
Table 2.
Expression Pattern of ASE Genes
| Tissue | Pos. | Gene | Cross WL×CG |
Cross CG×WL |
Purebred |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| WLa | CG | log2(C/W) | CG | WL | log2(C/W) | CG | WL | log2(C/W) | |||
| Brain | 479598 | WDR7 | 114.00 | 67.93 | −0.747 | 35.19 | 76.64 | −1.123 | 134.88 | 96.06 | 0.490 |
| Brain | 6926664 | KIAA1328 | 6.82 | 20.73 | 1.603 | 12.64 | 3.13 | 2.014 | 11.19 | 7.33 | 0.611 |
| Brain | 8939108 | —b | 49.12 | 87.83 | 0.838 | 90.28 | 55.98 | 0.690 | 168.81 | 78.18 | 1.110 |
| Brain | 37189505 | —c | 38.27 | 16.51 | −1.213 | 14.86 | 37.12 | −1.321 | 31.50 | 41.61 | −0.402 |
| Brain | 37799432 | CEP78 | 3.49 | 12.31 | 1.819 | 12.74 | 1.10 | 3.532 | 22.15 | 8.46 | 1.388 |
| Brain | 37799454 | CEP78 | 3.49 | 12.31 | 1.820 | 12.74 | 1.10 | 3.533 | 20.17 | 8.43 | 1.258 |
| Brain | 37836738 | PSAT1 | 116.20 | 254.64 | 1.132 | 349.22 | 171.90 | 1.023 | 559.59 | 209.44 | 1.418 |
| Brain | 46766148 | MAN2A1 | 8.73 | 29.58 | 1.760 | 45.00 | 13.90 | 1.695 | 52.85 | 28.97 | 0.868 |
| Brain | 52189031 | CPLX1 | 141.82 | 11.09 | −3.676 | 70.83 | 191.96 | −1.438 | 32.56 | 371.94 | −3.514 |
| Brain | 60325035 | —d | 20.61 | 64.95 | 1.656 | 44.71 | 23.54 | 0.926 | 94.39 | 32.95 | 1.518 |
| Liver | 10438089 | DNAJC21 | 6.07 | 15.14 | 1.318 | 25.44 | 10.29 | 1.306 | 21.69 | 22.68 | −0.0640 |
| Muscle | 37836020 | PSAT1 | 16.29 | 3.07 | −2.408 | 4.78 | 14.50 | −1.600 | 15.43 | 17.90 | −0.214 |
| Muscle | 41269793 | CTSL2 | 48.01 | 25.02 | −0.940 | 19.25 | 88.40 | −2.200 | 60.28 | 37.69 | 0.677 |
| Correlation coefficient (r2) | 0.92401 | 0.64690 | |||||||||
| P-valuee | 0.000133 | 0.0432 | |||||||||
WL and CG in interbreed crosses represent the reads number mapped to the White Leghorn allele and Cornish allele, respectively.
ENSGALG00000002386.
ENSGALG00000027927.
gga-mir-9-2-TMEM161B.
Correlation coefficient and P-value in table refer to correlation of log2(CG/WL) compared with the ratio of purebred log2(CG/WL), which was tested by Pearson correlation test.
Fig. 4.—
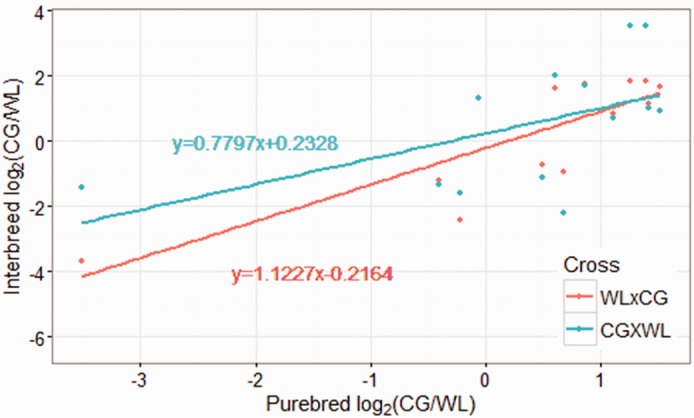
Correlation of ASE genes expression pattern with their parental expression in brain. The X axis shows the expression ratio of purebred CG to purebred WL. The y axis shows the expression ratio of the CG allele to the WL allele in the WL × CG (red) and CG × WL (blue) interbreed crosses.
Discussion
Our data confirms previous reports (Ellegren et al. 2007; Itoh et al. 2007; Mank and Ellegren 2009; Julien et al. 2012; Zimmer et al. 2016) of incomplete Z chromosome dosage compensation in females, where average expression on the Z chromosomes in females is less than the autosomal average. This is consistent with the fact that although females have two copies of all autosomal genes, they only have one copy of Z-linked loci. Our results are also consistent with previous studies that recovered average male:female expression ratios on the Z chromosome ranging from 1.2 to 1.5 (Mank and Ellegren 2009; Itoh et al. 2010). Importantly, the Z:A expression ratio in males in our data is consistent with previous findings (Julien et al. 2012; Zimmer et al. 2016), showing that although males have two copies of both Z-linked and autosomal genes, the average expression from the Z chromosome is less than that on for the autosomes.
Recent work has suggested that although birds lack complete Z chromosome dosage compensation, dosage sensitive genes are effectively compensated on a gene-by-gene basis (Wright et al. 2015b; Zimmer et al. 2016). This is in sharp contrast to the eutherian X chromosome, where X inactivation in females, controlled by the X-inactivation centre (Xic) (Okamoto and Heard 2009), inactivates nearly all genes on the X chromosomes, despite the fact that only a minority are thought to be dosage sensitive. However, Livernois et al. (2013), using RNA-fluorescent in situ hybridization (RNA-FISH) on 11 genes of the chicken Z chromosome, found evidence of one active Z chromosome and one partially inactive Z chromosome in the cells of male chickens. Their data was consistent with partial Z inactivation in males, suggesting convergent mechanisms in therian mammals and birds to achieve dosage parity between the sexes. This was somewhat at odds with other work (Julien et al. 2012) that found the ancestral expression of Z-linked genes was simply lower than average. However, both results achieved by Zimmer et al. (2016) and Livernois et al. (2013) were very limited in the number of loci they could assess, and neither we able to examine parent-of-origin effects.
Even partial inactivation of the Z chromosome is likely to leave a signature of allele-specific expression unless inactivation is perfectly balanced between the paternal and maternal copy. In marsupials, the paternal X is preferentially inactivated (Wang et al. 2014). In eutherian mammals, which have a more random form of X inactivation, the maternal copy is nonetheless more often transcribed than the paternal copy (Okamoto et al. 2004; Gregg et al. 2010a). This suggests that if the Z is even partially inactivated in males, we would expect greater allele-specific expression compared with autosomal loci. However, testing the possibility of Z inactivation in chicken on a broader scale using allele-specific expression has been difficult due to the low amount of sequence variation on this chromosome (Sundstrom et al. 2004; Wright et al. 2015a).
In order to maximize the number of informative Z-linked SNPs, we performed reciprocal crosses of two chicken breeds. This design allows us to not only assess allele-specific expression across a far larger number of Z-linked loci than previously assayed (Zimmer et al. 2016), but to also test for any parent-of-origin effects. Using this approach, which assesses the largest number of Z-linked genes to date, we found that both parental alleles of most Z-linked genes are transcribed equally in males, which excludes the possibility of even partial parent-of-origin inactivation. Although we cannot rule out the possibility of random inactivation entirely, it is unlikely to be the case as it would have to be perfectly balanced between both maternal and paternal copies for the length of the Z chromosome. Although we did detect a few instances of significant ASE in Z-linked genes, the proportion of Z-linked genes with ASE was not statistically different than that of the autosomes. Moreover, when we compared expression of these genes with the parental expression levels, our results indicate that the instances of ASE on the Z chromosome are likely due to cis-acting regulatory variation, rather than partial chromosomal inactivation.
Our study is somewhat limited by the presence and location of SNPs in our parental breeds, and we did not recover SNPs for all genes within the chicken genome. We were therefore unable to measure ASE in genes for which we did not recover informative SNPs. However, our study is based on a far broader number of genes than previous cell-based assays (Livernois et al. 2013), and therefore represents the most comprehensive study in Z inactivation to date.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Acknowledgments
This work was supported by the earmarked fund for the Beijing Innovation Team of the Modern Agro-industry Technology Research System (CARSPSTP), National Scientific Supporting Projects of China (2015BAD03B03), and a European Research Council grant to J.E.M. (agreement 680951). We gratefully acknowledge our colleagues in the Poultry Team at the National Engineering Laboratory for Animal Breeding of China Agricultural University, for their assistance on sample collection and helpful comments on the manuscript.
Literature Cited
- Bachtrog D. 2013. Y-chromosome evolution: emerging insights into processes of Y-chromosome degeneration. Nat Rev Genet. 14:113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Riddle NC, Auger DL, Veitia RA. 2005. Dosage balance in gene regulation: biological implications. Trends Genet. 21:219–226 [DOI] [PubMed] [Google Scholar]
- Chen X, Zhang J. 2016. The X to autosome expression ratio in haploid and diploid human embryonic stem cells. Mol Biol Evol. 33:3104–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, et al. 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVeale B, van der Kooy D, Babak T. 2012. Critical evaluation of imprinted gene expression by RNA-Seq: a new perspective. PLoS Genet. 8:e1002600.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dozmorov MG, et al. 2015. Detrimental effects of duplicate reads and low complexity regions on RNA- and ChIP-seq data. BMC Bioinformatics 16 Suppl 13:S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, et al. 2007. Faced with inequality: chicken do not have a general dosage compensation of sex-linked genes. BMC Biol. 5:40.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresard L, et al. 2014. Transcriptome-wide investigation of genomic imprinting in chicken. Nucleic Acids Res. 42:3768–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves JA. 2014. Avian sex, sex chromosomes, and dosage compensation in the age of genomics. Chromosome Res. 22:45–57. [DOI] [PubMed] [Google Scholar]
- Gregg C, Zhang J, Butler JE, Haig D, Dulac C. 2010a. Sex-specific parent-of-origin allelic expression in the mouse brain. Science 329:682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg C, et al. 2010b. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 329:643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Zhang J. 2016. X-Chromosome dosage compensation In: eLS. Chichester: John Wiley & Sons, Ltd; p. 1–7. [Google Scholar]
- Hough J, Hollister JD, Wang W, Barrett SC, Wright SI. 2014. Genetic degeneration of old and young Y chromosomes in the flowering plant Rumex hastatulus. Proc Natl Acad Sci U S A. 111:7713–7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, et al. 2007. Dosage compensation is less effective in birds than in mammals. J Biol. 6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, et al. 2010. Sex bias and dosage compensation in the zebra finch versus chicken genomes: general and specialized patterns among birds. Genome Res. 20:512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston CM, et al. 2008. Large-scale population study of human cell lines indicates that dosage compensation is virtually complete. PLoS Genet. 4:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien P, et al. 2012. Mechanisms and evolutionary patterns of mammalian and avian dosage compensation. PLoS Biol. 10:e1001328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Xing K, Zhang J, He X. 2012. Expression reduction in mammalian X chromosome evolution refutes Ohno's hypothesis of dosage compensation. Proc Natl Acad Sci U S A. 109:11752–11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, et al. 2007. Dosage compensation in the mouse balances up-regulation and silencing of X-linked genes. PLoS Biol. 5:e326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livernois AM, Waters SA, Deakin JE, Marshall Graves JA, Waters PD. 2013. Independent evolution of transcriptional inactivation on sex chromosomes in birds and mammals. PLoS Genet. 9:e1003635.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF. 1999. X-chromosome inactivation. Curr Biol. 9:R235–R237. [DOI] [PubMed] [Google Scholar]
- Malone JH, et al. 2012. Mediation of Drosophila autosomal dosage effects and compensation by network interactions. Genome Biol. 13:r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank JE. 2017. The transcriptional architecture of phenotypic dimorphism. Nat Ecol Evol. 1:0006.. [DOI] [PubMed] [Google Scholar]
- Mank JE. 2013. Sex chromosome dosage compensation: definitely not for everyone. Trends Genet. 29:677–683. [DOI] [PubMed] [Google Scholar]
- Mank JE. 2009. The W, X, Y and Z of sex-chromosome dosage compensation. Trends Genet. 25:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank JE, Ellegren H. 2009. All dosage compensation is local: gene-by-gene regulation of sex-biased expression on the chicken Z chromosome. Heredity (Edinburgh) 102:312–320. [DOI] [PubMed] [Google Scholar]
- McKenna A, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira de Mello JC, et al. 2010. Random X inactivation and extensive mosaicism in human placenta revealed by analysis of allele-specific gene expression along the X chromosome. PLoS One 5:e10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. 1925. Why polyploidy is rare in animals than in plants. Am Nat. 59:346–353. [Google Scholar]
- Mullon C, Wright AE, Reuter M, Pomiankowski A, Mank JE. 2015. Evolution of dosage compensation under sexual selection differs between X and Z chromosomes. Nat Commun. 6:7720.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DK, Disteche CM. 2006. Dosage compensation of the active X chromosome in mammals. Nat Genet. 38:47–53. [DOI] [PubMed] [Google Scholar]
- Ohno S. 1967. Sex chromosomes and sex-linked genes. Berlin: Springer-Verlag. [Google Scholar]
- Okamoto I, Heard E. 2009. Lessons from comparative analysis of X-chromosome inactivation in mammals. Chromosome Res. 17:659–669. [DOI] [PubMed] [Google Scholar]
- Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E. 2004. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 303:644–649. [DOI] [PubMed] [Google Scholar]
- Pessia E, Makino T, Bailly-Bechet M, McLysaght A, Marais GA. 2012. Mammalian X chromosome inactivation evolved as a dosage-compensation mechanism for dosage-sensitive genes on the X chromosome. Proc Natl Acad Sci U S A. 109:5346–5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundstrom H, Webster MT, Ellegren H. 2004. Reduced variation on the chicken Z chromosome. Genetics 167:377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Douglas KC, Vandeberg JL, Clark AG, Samollow PB. 2014. Chromosome-wide profiling of X-chromosome inactivation and epigenetic states in fetal brain and placenta of the opossum, Monodelphis domestica. Genome Res. 24:70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AE, et al. 2015a. Variation in promiscuity and sexual selection drives avian rate of Faster-Z evolution. Mol Ecol. 24:1218–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AE, Zimmer F, Harrison PW, Mank JE. 2015b. Conservation of regional variation in sex-specific sex chromosome regulation. Genetics 201:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer F, Harrison PW, Dessimoz C, Mank JE. 2016. Compensation of dosage-sensitive genes on the chicken Z chromosome. Genome Biol Evol. 8:1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.