

Abstract
Mycobacterium tuberculosis is divided into several distinct lineages, and various genetic markers such as IS-elements, VNTR, and SNPs are used for lineage identification. We propose an M. tuberculosis classification approach based on functional polymorphisms in virulence genes. An M. tuberculosis virulence genes catalog has been established, including 319 genes from various protein groups, such as proteases, cell wall proteins, fatty acid and lipid metabolism proteins, sigma factors, toxin–antitoxin systems. Another catalog of 1,573 M. tuberculosis isolates of different lineages has been developed. The developed SNP-calling program has identified 3,563 nonsynonymous SNPs. The constructed SNP-based phylogeny reflected the evolutionary relationship between lineages and detected new sublineages. SNP analysis of sublineage F15/LAM4/KZN revealed four lineage-specific mutations in cyp125, mce3B, vapC25, and vapB34. The Ural lineage has been divided into two geographical clusters based on different SNPs in virulence genes. A new sublineage, B0/N-90, was detected inside the Beijing-B0/W-148 by SNPs in irtB, mce3F and vapC46. We have found 27 members of B0/N-90 among the 227 available genomes of the Beijing-B0/W-148 sublineage. Whole-genome sequencing of strain B9741, isolated from an HIV-positive patient, was demonstrated to belong to the new B0/N-90 group. A primer set for PCR detection of B0/N-90 lineage-specific mutations has been developed. The prospective use of mce3 mutant genes as genetically engineered vaccine is discussed.
Keywords: Mycobacterium tuberculosis, virulence genes, lineage-specific SNP, phylogenetic analysis
Introduction
Genetically heterogeneous Mycobacterium tuberculosis (MT) species are divided into several groups, so-called lineages or genotypes, in which they are characterized by specific variations that gradually accumulate during the course of evolution (Filliol et al. 2006; Gagneux et al. 2006; Gagneux and Peter 2007; Prozorov and Danilenko 2011; Jagielski et al. 2014). Lineages can vary by gene copy number, insertions sequences (IS-elements), deletion/insertion of several nucleotides, and point mutations known as single nucleotide polymorphism (SNP). Due to advances in phylogenetic analysis new sublineages are constantly being indentified (Gagneux et al. 2006; Coll et al. 2014; Eldholm et al. 2016).
A wide range of methods have been developed for classifying isolates into lineages, based on different polymorphisms such as regions of difference (RD), variable number tandem repeat (VNTR) loci, double-repetitive (DR) loci, and SNPs (Jagielski et al. 2014). SNPs are considered the most promising marker as it provides high resolution and unambiguous results. SNPs in various gene groups can be used for genotyping, including housekeeping and drug resistance genes (Ford et al. 2013; Coll et al. 2014; Feuerriegel et al. 2014). Synonymous (sSNPs) and nonsynonymous (nsSNP) SNPs are used for genotyping. Some amino acid changes can be neutral, but some can affect protein function. The usage of such functional nsSNPs allows for the detection of new bacterial groups with potentially changed phenotype.
Isolates of different lineages vary by many phenotypes such as the tendency to develop drug resistance, virulence levels, and pathogenicity, which influences the disease severity (Homolka et al. 2012; Ford et al. 2013; Reiling 2013). This diversity is created by mutations affecting gene product structure and function. Aside from mutations in functional genes, clinical tuberculosis severity depends on the balance between the host’s health, genetic background, immune status, diet, environmental status, and microbiota composition (Carding et al. 2015) with the pathogen’s virulence system (Cobat et al. 2013). Immune status is considered the most essential factor for successful mycobacteria eradication. A hereditary inability to initiate steps in immune response leads to genetic susceptibility to tuberculosis (Hill 2001). Many factors lead to immune suppression, such as diseases (HIV, hepatitis, diabetes, and cancer), stress, and long-term use of drugs, or environmental pollution. Hence, the risk of infection and pathogenesis are determined by the interaction between the pathogen and the host’s state.
MT virulence and pathogenicity are conditioned by a range of genes, the participation in pathogenesis of which has been experimentally shown. The MT genome encodes more than 300 virulence genes from various groups, such as serine-threonine proteinkinases, systems toxin–antitoxin (TA systems), sigma factors, and type VII secretion system (Forrellad et al. 2013; Prozorov et al. 2014). Their products play crucial roles in different stages of infection, such as mucosal colonization, cell invasion, avoidance of host immune response, and survival under stress conditions (Forrellad et al. 2013; Prozorov et al. 2014; Tiwari et al. 2015). Mutations in these genes may influence the pathogen phenotype and, subsequently, provide promising data to analyze.
Previously, we were the first group to use functional mutations in structural genes of TA systems that influence the formation of a persistent state, and showed the correlation between particular polymorphism patterns and lineage (Zaychikova et al. 2015). The purpose of this work is to establish a mutation catalog of various virulence genes (including TA systems) and to use it for the identification of new epidemiologically dangerous sublineages that arose in various regions during the last decades.
Materials and Methods
Virulence Genes Catalog Development
The definition of “virulence” is still widely discussed and its defining parameters and conditions are unsettled. Here, by the term “virulence”, we mean the ability of a pathogen to cause disease, overcome the host resistance mechanism via invasion and adhesion to host cells, and adapt to hostile environments, including immune response modulation. To develop am M. tuberculosis virulence gene catalog, we used various reviews and articles on this theme (Zhao and Xie 2011; Burian et al. 2013; Forrellad et al. 2013). The following groups of gene products were analyzed:
Cell Wall Proteins
Five percent of all MT cell wall proteins have been shown to contribute to bacteria virulence (Forrellad et al. 2013). Their function varies from adhesion/invasion to transport proteins (Raynaud et al. 2002; Rengarajan et al. 2005; Stewart et al. 2005). One of the most essential members of this family are Mce (mammalian cell entry) family proteins that are organized in four large operons (Singh et al. 2016). Presumably, these proteins are involved in adhesion and invasion to macrophages on different stages of infection. They play key roles in bacterial survival inside macrophages during the early events of infection. However, their exact action mechanism remains elusive. A high level of polymorphism of the mce operon genes has been reported by comparing drug sensitive isolates to resistant (Pasricha et al. 2011).
Another major component of M. tuberculosis cell wall are lipoproteins, since they are involved in transmembrane transport, adhesion, signal transduction, and protein degradation (Sander et al. 2004).
Fatty Acid and Lipid Metabolism
Cell wall lipids play an important role in M. tuberculosis pathogenesis by modulating the immune response and interaction with the pathogen (Forrellad et al. 2013). MT cell wall contains mycolic acids, sulfolipids, di- and tri-acylated trehaloses and phtiocerol dimycocerosated (PDIM). PDIMs are essential virulence factors, especially on the early stages of the infection (Sirakova et al. 2003). Biosynthesis of these virulence-associated lipids requires material and energy that are provided by M. tuberculosis cholesterol catabolism pathways. Utilization of cholesterol from host cells membranes contributes to the survival of bacteria during infection (Pandey and Sassetti 2008).
Type VII Secretion Systems
The type VII secretion system (T7SS) play key roles in bacterial pathogenesis. The M. tuberculosis secretion system is responsible for virulence factors transport, both to extracellular space and directly into host cells (Das et al. 2011). MT genome encodes five clusters of T7SS: ESX1-5. ESX-1 and ESX-5 have been shown to be involved in virulence (Wards et al. 2000; Forrellad et al. 2013).
Proteins Inhibiting Antimicrobial Responses of the Macrophage
Proteins of this group help M. tuberculosis survive under macrophage stress conditions. Their functions are various, such as increasing resistance to host toxic compounds (reactive oxygen or nitrogen species), phagosome arresting, inhibiting phagosome–lysosome fusion, and apoptosis inhibition (Zahrt and Deretic 2002; Bach et al. 2008; Sun et al. 2010; Behar et al. 2011).
Gene Expression Regulators: Sigma Factors, Two-Component Systems, Serine–Threonine Proteinkinases
Bacteria adapt to changes in the environment using RNA polymerase, which has a protein complex of five core subunits and dissociable sigma factor subunit. Sigma factors are mostly responsible for promoter recognition and, therefore, they mediate differential expression of many genes, including virulence factors (Bashyam and Seyed 2004; Burian et al. 2013).
Two-component systems are the prokaryotic signal transduction system, formed by a sensor protein (transmembrane histidine kinase) that senses a specific extracellular signal and a regulator protein that modulates the expression of target genes. Such systems adapt the bacteria to different aspects of environmental conditions, including the interaction with the host immune system. Today, 11 paired two-component systems have been described and 4 of them are participating in virulence (Parish et al. 2003; He et al. 2006; Walters et al. 2006; Converse et al. 2009).
Serine–threonine kinases are involved in signal transduction and metabolism regulation in response to environmental changes. MT genome includes 11 serine–threonine protein kinases, 3 of them have been shown to have assigned roles in virulence at various stages of infection, like invasion of macrophage, cell growth and division, and apoptosis regulation (Greenstein et al. 2006; Kumar and Narayanan 2012).
Toxin–Antitoxin Systems
TA systems represent a module of two nearby genes, which contribute to bacterial growth and persistent state formation (Maisonneuve et al. 2011). TA systems help bacteria to avoid the host’s immune response effectively (hence, acting as virulence genes) by developing a latent infection, which can later switch back to its active form under favorable conditions (i.e., immune system depression or antibiotic therapy termination) (Fasani and Michael 2013).
Proteases
Proteases play key roles in cellular homeostasis by controlling proteins involved in transcription, regulation, metabolism and virulence. Proteases inactivate the host’s defense mechanisms by modulating the immune response. Some pathogens, including M. tuberculosis, use extracellular proteases as virulence factors that influence tissue destruction (Zhao and Xie 2011).
Metal Transporter Proteins
MT metal transporter systems are essential for survival in macrophages, which iron concentration is 1,000 times lower than normal (Rodriguez and Smith 2006). High metal ion concentration is extremely toxic for microorganisms. Therefore, increasing ion concentration, for instance zinc, plays a crucial role in macrophage antimicrobial responses. As a result, M. tuberculosis bacteria have developed both import and export metal system during evolution (Quadri 2008).
Genome Catalog Development
We used publicly available genomes of MT deposited in GenBank database. All genomes were genotyped using lineage-specific SNPs, proposed by (Homolka et al. 2012; Zaychikova et al. 2015), genomes with satisfactory assembly quality were selected.
In addition to GenBank, the GMTV (Genome-based Mycobacterium Tuberculosis Variation) database (Chernyaeva et al. 2014) was used for analysis. GMTV contains a full list of SNPs in “vcf” format from approximately 1800 genomes, and approximately 1,000 were isolated in Russia. The vast majority of these genomes are contained in GenBank only in SRA format, which makes convenient BLAST-search impossible.
Program for SNP Detection
A Perl program was developed to detect SNPs using the BioPerl package. The program used gene and genome catalogs to build an Excel table with a full list of SNPs in all genes and genomes. The position, nucleotide, and amino acid changes were calculated for every mutation. Sequences were aligned using BLAST v.2.3.0+. Alignments with an identity >98% and coverage >95% were retained for SNP-calling. Only nonsynonymous mutations were analyzed. Insertions and deletions were excluded, as they are often the result of genome assembly errors.
Once the full list of mutations was acquired, a lineage-specific SNP search was carried out. The Pearson correlation coefficient (P) was calculated for every SNP, and mutations with P ≥ 0.95 were considered to be lineage-specific.
The effect of mutations was assessed using three algorithms (MAPP, SIFT and PolyPhen-1) from online consensus classifier PredictSNP1.0 (Bendl et al. 2014). As PredictSNP was designed for genetic disease prediction in human, we chose these algorithms, because they do not use supervised-learning methods and were not trained on human datasets. In context of this research the deleterious SNP prediction means the possible change of protein function that leads to virulence change.
Whole Genome Alignment
Whole genome alignment was made using MUMMER v.3.23 (Kurtz et al. 2004). Each genome was aligned to the H37Rv reference genome (GCA_000195955.2). An in-house Perl script was used to filter only the nonsynonymous SNPs for every genome and calculate the mean values for all lineages. Insertions and deletions were excluded. Mutations in high variable regions (PE/PPE families, pks12, lppA, lppB) were also excluded.
Phylogenetic Analysis
Phylogenetic trees were constructed using concatenated aligned sequences of 128 structural virulence genes (supplementary material S1, Supplementary Material online). The maximum likelihood phylogenetic tree was computed using RAxML v.8.2.9 (Stamatakis 2014), using the GTRCAT model with 1,000 rapid bootstrap inferences and a thorough Maximum Likelihood search. The resulting trees were rooted on M. canettii (GCA_000253375.1) for the global phylogeny tree and on H37Rv (GCA_000195955.2) for trees of lineages LAM, Beijing-B0/W-148, and Ural. Trees were visualized using the interactive tree of life software (iTOL) (Letunic and Bork 2007).
PCR Analysis
PCR amplification was performed using a High Fidelity PCR Enzyme Mix (Fermentas). PCR mixtures contained 20 pmol primers in 100 ml. Primers sequences are described in supplementary material S2, Supplementary Material online. Amplification result analysis was conducted using 1% agarose gel electrophoresis method.
Whole Genome Sequencing
Genome sequencing of strain B9741 was carried out on Roche 454 GS Junior instrument. A total number of 139,538 reads was generated, and reads were assembled to a draft genome using the GS de novo Assembler v.3.0. The resulting genome contained 195 contigs and was deposited at GenBank under accession number LVJJ00000000 (Shur et al. 2016).
Results
We established a broad catalog of 319 virulence genes in more than 14 groups (table 1). The catalog consists of genes that encode proteins that have been shown to contribute to M. tuberculosis virulence and pathogenesis experimentally (Forrellad et al. 2013; Prozorov et al. 2014). The catalog includes both structural and regulatory virulence genes, playing roles in various stages of infection. Genes from the PE/PPE family, which were shown to contribute to the bacterium’s virulence, were excluded from analysis due to a high genetic variability (Fishbein et al. 2015). The full catalog is provided in supplementary material S1, Supplementary Material online.
Table 1.
Distribution of 319 Selected Virulence Genes between Functional Groups of Protein Gene Products
| Group | Number of Genes | Main Functions |
|---|---|---|
| Cell wall proteins | 48 | Transport of virulence factors, adhesion, interaction with host immune system |
| Lipoproteins | 7 | Interaction with macrophages, adhesion, signal transduction |
| Lipid and fatty acid metabolism | 17 | Suppression of immune response, interaction with host cells |
| Mycolic acid synthesis | 12 | Interaction with macrophages |
| Cholesterol catabolism | 6 | Using cholesterol as an energy source, synthesis of virulence-associated PDIM lipids |
| Type VII secretion system | 17 | Export of signal molecules and inhibitors of antimicrobial response, induction of bacteria’s exit from macrophages |
| Proteins inhibiting antimicrobial responses of the macrophage | 15 | Survival in macrophages |
| Sigma factors | 8 | Transcriptional regulation of virulence factors |
| Two-component systems | 12 | Signal transduction, participation in adaptation to hostile environment |
| Serine–threonine proteinkinases | 3 | Apoptose inhibition, inhibition of phagosome–lysosome fusion, reduction of phagocytosis |
| TA systems | 142 | Survival under hostile conditions through switching to the latent state |
| Proteases | 7 | Regulation of virulence factors, host cells degradation, protection from immune response |
| Metal transporter proteins | 7 | Survival in hostile conditions, export of toxic ions—antimicrobial factors |
| Other | 18 | – |
| Total | 319 |
The genome catalog included 1,573 M. tuberculosis genomes of 18 various lineages and sublineages (table 2). The catalog included 27 complete genomes and 1,546 in draft form. Only genomes with unambiguously defined genotypes and reasonable number of mutations (which meant fine assembly quality) were added to the catalog. Supplementary material S3, Supplementary Material online, contains a full list of genomes with the name, lineage and WGS number provided.
Table 2.
Studied Mycobacterium tuberculosis Lineages and Sublineages
| Lineage and Sublineage | Number of Genomes | Main Countries of Isolation |
|---|---|---|
| Beijing | 557 | |
| Beijing-modern | 295 | South Africa, India, Sweden, China |
| Beijing-B0/W-148 | 87(+140)a | Belarus, Iran, Sweden, (Russia)a |
| Beijing-ancestral | 175 | South Africa, South Korea |
| Cameroon | 6 | Mali |
| Delhi/CAS | 102 | India, Belarus, Uganda |
| EAI | 197 | |
| EAI-Manila | 18 | India, Taiwan |
| Other | 179 | India |
| X | 165 | |
| Ghana | 37 | Mali |
| Haarlem | 49 | South Africa, Romania, Belarus |
| Other | 79 | South Africa, Mali |
| LAM: | 337 | |
| LAM1 | 31 | Panama |
| LAM2 | 9 | Panama |
| F15/LAM4/KZN | 150 | South Africa |
| Other | 147 | South Africa, Belarus |
| S | 109 | South Africa, Uganda |
| SMI-049 | 5 | Sweden |
| T-H37Rv-like | 12 | – |
| Ural | 83 | Moldova, Sweden, Belarus |
| Total | 1,573 |
Note.—Beijing lineage is divided into two sublineages: Beijing-modern and Beijing-ancestral. Sublineage Beijing-modern includes Beijing-B0/W-148 sublineage. EAI lineage includes EAI-Manila sublineage. X lineage includes sublineages Ghana and Haarlem. LAM lineage includes sublineages LAM1, LAM2 and F15/LAM4/KZN.
Additionally analyzed genomes from GMTV database.
All studied lineages belong to the lineages 1–4 of MT. Lineages 2, 3 and 4 are considered as “modern”, and lineage 1 as “ancient” (Hershberg et al. 2008). MT also includes ancient lineages 5, 6, and 7 that were not used in the current analysis because they are spread only across Africa and are rarely transmitted to other continents (Gagneux and Peter 2007; Coll et al. 2014). Plylogenetic relationships between the studied lineages are shown in figure 1. A sample of 154 isolates was used for the tree construction in order to bring all lineages and sublineages to a comparative size of around 10 isolates. All lineages and sublineages have bootstrap support >60%.
Fig. 1.—

Global phylogeny of the main Mycobacterium tuberculosis lineages. The tree has two main branches: EAI (so-called Indo-oceanic lineage 1, one of the most ancient MT strains) and the branch with modern lineages — Delhi/CAS (East-African–Indian lineage 3), Beijing (East-Asian lineage 2), and others (Euro-American lineage 4). The tree was rooted on M. canettii. Bootstrap values >50% are shown.
Distribution of nsSNPs in Virulence Genes
A catalog of SNPs in virulence genes has been developed (supplementary material S4, Supplementary Material online). In 624 alignments, genes were found with a percent identity from 80% to 98%, and in 706 alignments genes were found with a percent identity <80% or not found at all. Four TA system genes (mazE9, relG, vapB11, and vapB44) had no nsSNPs in all isolates. In general, TA genes tend to have less SNPs per gene, both synonymous and nonsynonymous (fig. 2a, t-test, P < 0.001). However, normalization by the sum of gene lengths does not show such striking differences (0.012 and 0.013 nsSNPs per bp and 0.0071 and 0.0067 sSNPs per bp for TA and non-TA genes, respectively). Thus, high conservation of mazE9, relG, vapB11 and vapB44 is not likely to reflect some biological unsubstitutability of these genes.
Fig. 2.—

(a) The numbers of synonymous (blue) and nonsynonymous (orange) SNPs per gene in TA and non-TA genes are statistically different (t-test, P < 0.001). Standard errors for every group are shown. (b) Distribution of nsSNPs in virulence genes by the number of genomes in which mutation is located. (c) Comparison of nsSNPs in virulence genes (red squares) and in the whole genome (blue circles). Values of nsSNPs were divided by maximum to make them vary from 0 to 1 and mean values for every lineage were calculated. Standard deviations for every group are shown.
After the elimination of SNPs from partially aligned genes (identity < 98%), 3,563 nonsynonymous mutations have been identified, more than a half of them specific for a single isolate (fig. 2b).
We examined whether lineages with different virulence levels have different number of nsSNPs in virulence genes. For every genome we counted total numbers of nsSNPs in two groups: in virulence genes and in the whole genome. After that the numbers were divided by a group maximum to normalize data, and mean values for every group and lineage were calculated (supplementary material S2, Supplementary Material online). Figure 2c illustrates comparison of normalized nsSNP values in virulence genes and in the whole genome. It shows that the main mutation trends do not differ between virulence genes and the whole genome. For instance, lineages EAI and Beijing are considered to have a lower and higher virulence level, respectively, but inside one lineage nsSNP values for virulence genes and for the whole genome are almost the same. Hence, we assume that lineage-specific changes in virulence are caused by several crucial functional mutations rather than by bulk nonsynonymous mutations.
Detection of nsSNP Set for Genotyping
A total of 123 lineage-specific nsSNPs (P > 0.95) has been detected in all genes groups, and 79 mutations with P = 1 (supplementary material S5, Supplementary Material online). We have selected a set of 15 virulence genes, the nsSNPs of which can classify an isolate into 1 of the 18 studied lineages (table 3). Lineage-specific mutations with the aforementioned criteria for lineages T-H37Rv-like and X were not found due to their close relationship with the reference H37Rv genome. For lineage X, we propose a lineage-specific mutation in pks5 with P = 0.9, which is still considered as a strong positive correlation, and lineage T-H37Rv-like can be identified by the absence of nsSNP in mce1F.
Table 3.
A Set of Lineage-Specific Nonsynonymous SNPs in Virulence Genes Sufficient for the Resolution of the 18 Studied Mycobacterium tuberculosis Groups
| Lineage or Sublineage | Gene | Groups | SNP | P |
|---|---|---|---|---|
| Beijing | mazF8 | TA systems | G122T G-V | 1 |
| Beijing-modern | vapC37 | TA systems | A46G T-A | 1 |
| Beijing-B0/W-148 | mce3B | Cell wall proteins | T145G S-A | 1 |
| Cameroon | pcaA | Mycolic acid synthesis | C691T H-Y | 1 |
| Delhi/CAS | irtA | Metal transporter proteins | C143T A-V | 0.99 |
| EAI | irtA | Metal transporter proteins | G964T V-F | 0.99 |
| EAI-Manila | mbtB | Metal transporter proteins | G2137A D-N | 1 |
| X | pks5 | Lipid and fatty acid metabolism | A3688G T-A | 0.9 |
| Ghana | yrbE3A | Cell wall proteins | G338C G-A | 1 |
| Haarlem | mgtC | Metal transporter proteins | G545A R-H | 1 |
| LAM | mce3F | Cell wall proteins | C992G P-R | 1 |
| LAM1 | eccD1 | Type VII secretion system | C32T T-I | 1 |
| LAM2 | mmaA4 | Mycolic acid synthesis | G166A E-K | 1 |
| F15/LAM4/KZN | mce3B | Cell wall proteins | T44C F-S | 1 |
| S | mazF6 | TA systems | G175A G-S | 1 |
| SMI-049 | pstS-1 | Lipoproteins | C877G P-A | 1 |
| T-H37Rv-like | mce1F | Cell wall proteins | T1109T | 1 |
| Ural | mce3B | Cell wall proteins | C1000G Q-E | 1 |
Note.—For every SNP, its position, nucleotide and acid change are given. Lineage T-H37Rv-like is defined by the absence of mutation in 1,109 nucleotide in mce1F. P, Pearson correlation coefficient.
Lineage-Specific Mutations in Virulence Genes of F15/LAM4/KZN Sublineage
The Latin American–Mediterranean (LAM) lineage is one of the largest and most widespread MT strains and includes various sublineages defined as spoligotyping families LAM1–LAM11. Here, we applied SNP genotyping sets that allowed detection of three sublineages inside LAM: LAM1, LAM2 (Panama) and F15/LAM4/KZN (South Africa) (Gandhi et al. 2013; Lanzas et al. 2013). Sublineage F15/LAM4/KZN has caused a massive tuberculosis outbreak among HIV-positive patients in South Africa with a high level of lethal outcomes. This sublineage demonstrates changes in virulence (Smith et al. 2014). Experiments on various laboratory models including cell cultures A549 THP-1 have shown an increased necrosis of epithelial cells, higher adhesion and invasion (Ashiru et al. 2010; Gandhi et al. 2013; Sarkar et al. 2016). Out of the 150 F15/LAM4/KZN isolates used in this work, more than half were isolated from HIV-positive patients (Cohen et al. 2015).
LAM lineage phylogeny is shown in figure 3a. Three sublineages were separated into distinct branches with high bootstrap values. Sublineage F15/LAM4/KZN shows a high bootstrap-support (83%). Bioinformatics analysis revealed three lineage-specific mutations in virulence genes: T44C in mce3B, T1076C in cyp125, A221C in vapC25, and C140A in vapB34 (table 4).
Fig. 3.—

Phylogenetic trees of the Mycobacterium tuberculosis sublineges. (a) Phylogenetic tree of the LAM lineage. The tree is constructed for 337 isolates of the LAM lineage. Three sublineages are separated in different branches with high bootstrap support. (b) Phylogenetic tree of the Beijing-B0/W-148 sublineage. The tree is constructed for 87 isolates of the Beijing-B0/W-148 sublineage. Note two Belarus clusters and B0/N-90 sublineage. (c) Phylogenetic tree of the Ural lineage. The three is constructed for 83 isolates of Ural sublineage. All isolates are divided into two geographical clusters. The trees were rooted on H37Rv reference genome. Bootstrap values >50% are shown.
Table 4.
Lineage-Specific Mutations in Virulence Genes of the F15/LAM4/KZN and B0/N-90 Sublineages
| Gene | Group | Function of Product Protein | SNP | Effect of SNP |
|---|---|---|---|---|
| F15/LAM4/KZN | ||||
| mce3B | Cell wall proteins | Adhesion or invasion | T44C F-S | A (0/3) |
| cyp125 | Cholesterol catabolism | Degradation of cholesterol, participation in synthesis of virulence-associated lipids PDIM | T1076C I-T | A (0/3) |
| vapC25 | TA systems | Toxin, RNase, participation in developing of dormant state | A221C N-T | N (3/0) |
| vapB34 | Antitoxin, participation in developing of dormant state | C140A A-E | D (2/1) | |
| B0/N-90 | ||||
| mce3F | Cell wall proteins | Adhesion or invasion | A1229C D-A | D (2/1) |
| vapC46 | TA systems | Toxin, participation in pathogenesis and developing of dormant state | C113G A-G | D (2/1) |
| irtB | Metal transporter proteins | ABC transporter, import of iron | G523A A-T | N (3/0) |
Note.—Effect of SNP was defined using three algorithms from online-tool PredictSNP. Numbers shown in parentheses are algorithms predicting neutral effect of mutations versus algorithms predicting deleterious effect. Note that mutation in mce3B and cyp125 are likely to affect protein function. All provided mutations have Pearson coefficient >0.95. A, affected; D, doubtful; N, neutral.
The mutation in mce3B is located in the uncharacterized part of the protein, and in cyp125, the mutation is located in the cytochrome p450 domain. In vapC25, which encodes a toxin, the mutation is located in the ribonuclease PIN domain, and vapB34, which encodes the antitoxin, is not divided into functional parts. The unambiguous predictions of SNP’s effect on protein function for mutations in mce3B and cyp125 allow supposing that the virulence phenotype of F15/LAM4/KZN can be changed by these mutations. The mutations in TA system genes vapC25 and vapB34 hardly change protein function.
Comparative Analysis of the Beijing-B0/W-148 Sublineage
Sublineage Beijing-B0/W-148 belongs to the virulent Beijing-modern group. Beijing-B0/W-148 is prevalent in Russia and it demonstrates high virulence level (Shitikov et al. 2014). Its phylogenetic tree is shown in figure 3b. The tree has three distinct branches; two of them are formed only by Belarus isolates. The third branch is formed by five isolates: two from Russia (SP21 and 13-4152), two from Belarus (XTB13-176 and XTB13-200) and one from Sweden (BTB07-170). The expansion of this sublineage to different countries proposes a better adaptation to various environmental conditions. This sublineage was named B0/N-90 and it has three lineage-specific nsSNPs in virulence genes: A1229C in mce3F, G523A in irtB, and C113G in vapC46 (table 4). The mutation in mce3F can possibly affect protein function. Presumably, this SNP can alter protein function like the mutation in mce3B in the F15/LAM4/KZN sublineage. The mutation in vapC46 also may change protein function (promising PredictSNP results). The mutation in irtB is less likely to affect protein function. These three mutations were not detected in other lineages.
We have carried out a more detailed whole genome SNP analysis of this sublineage (fig. 4). Eleven core SNPs were identified, and a set of common SNPs for Russian and Belarus isolates were detected. High numbers of individual SNPs in isolates SP21 and B9741 (see the next section) are most probably caused by poor assembly quality. Detailed SNP information is provided in supplementary material S2, Supplementary Material online. On an average, isolates differ by ∼20 SNPs. When applying a mutation rate of 0.5 mutation per year (Walker et al. 2013), the sublineage emerged <50 years ago. The mutation rate used here was estimated from epidemiologic studies and is slightly higher than the substitution rate obtained from ancient DNA analysis (Eldholm et al. 2016).
Fig. 4.—
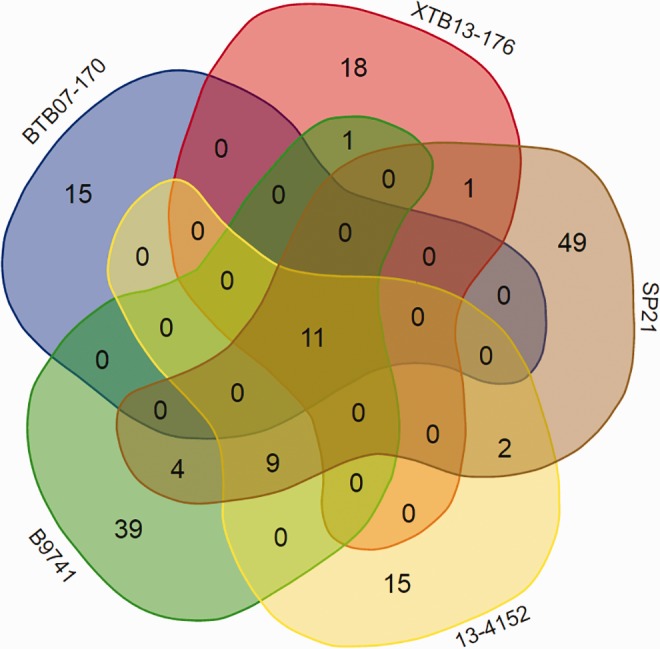
Distribution of SNPs between B0/N-90 isolates. Comparative diagram of SNPs in five isolates of the B0/N-90 sublineage. Both common and individual mutations are shown. The distribution of SNPs shows close evolutionary relationships between Russian isolates, whereas the Swedish isolate shows no common mutations except core ones.
Further search for B0/N-90 members was conducted in the GMTV database that stores 140 Beijing-B0/W-148 genomes (most of them isolated in Russia). Twenty-two new Russian isolates of B0/N-90 sublineage were detected, most of them isolated in Samara Oblast, Russia. Presumably, the outbreak of B0/N-90 started there at a hospital and then spread across Russia and other countries. Detailed information about all B0/N-90 isolates is given in supplementary material S2, Supplementary Material online.
The fact that this sublineage was formed inside the high-virulent Beijing-B0/W-148 group and has mutations possibly affecting protein function, brings on the assumption that B0/N-90 can show high virulence at least for some populations. The genome 13-4152 was isolated from a patient with diagnosed caseous pneumonia that often develops on the background of low immunity (Maslov, Shur, et al. 2015); therefore, it is possible that B0/N-90 poses a threat for immunocompromised patients.
Comparative Analysis of Strain B9741 Isolated from HIV-Positive Patient
Mycobacterium tuberculosis strain B9741 was isolated from a HIV-positive patient from Irkutsk, Russia. The strain is resistant to streptomycin, isoniazid, pyrazinamide and rifampicin. According to lineage-specific mutations (G757C in oxcA and A96G in vapC12) the strain has been classified to Beijing-B0/W-148 sublineage. Read mapping on W-148 genome revealed that genomes differ by ∼50 SNPs. These mutations include three lineage-specific mutations of the B0/N-90 sublineage which makes the B9741 strain a member of this sublineage.
PCR-Based Detection of Sublineage B0/N-90
We have developed a set of oligonucleotides for genes containing lineage-specific SNPs (supplementary material S2, Supplementary Material online). The primer sequences meet the standard requirements for universality and high specificity. The length of the DNA fragment is selected to be appropriate for various sequencing methods.
To validate the primers we have formed a collection of 14 Beijing-B0/W-148 isolates including five isolated from HIV-positive patients, five—from HIV-negative and four from patients with unknown HIV status. We also included 13-4152 isolate as a positive control (Maslov, Shur, et al. 2015; Maslov, Zaîchikova, et al. 2015) that contained all three lineage-specific SNPs (A1229C in mce3F, C113G in vapC46 and G523A in irtB). After amplification of these DNA fragments, these mutations were found only in isolate 13-4152.
Ural Lineage Phylogenetic Analysis
The Ural lineage is one of the MT sublineages that emerged in the 20th century (Mokrousov 2012). First this lineage was detected in Russia; nowadays it has spread across Europe. The phylogenetic tree of the Ural lineage is clearly divided into two geographical sublineages (fig. 3c). The first sublineage includes isolates from Sweden (23 isolates), and the second—isolates from other countries, mainly Moldova (42), Belarus (7) and Romania (5).
Lineage-specific nsSNPs of these two clusters are listed in supplementary material S2, Supplementary Material online. The second group was identified by a set of mutations in the mce-operons. These SNPs have been previously reported in a study focused on Russian Ural isolates (absent in this work), and were present in all set (Sinkov et al. 2016). Hence, Ural isolates from Russia belong to the second sublineage.
Distribution of nsSNPs in Mce Genes
Noticing that mutations in mce genes appear in every studied sublineage (B0/N-90, F15/LAM4/KZN, Ural) we decided to conduct more a detailed analysis of nsSNPs distribution in these genes. As previously mentioned, mce-proteins play crucial roles in bacterial virulence by participating in adhesion, invasion, survival inside macrophages and cholesterol transport. Mce genes have numerous mutations (table 5), from which 26 are lineage-specific. Besides the lineage-specific nsSNPs, mutations located in groups of two or more isolates were counted and their effect on protein function was measured by PredictSNP. Such narrowly spread mutations are more likely to appear in the last decades, i.e., in the era of widespread antibiotic use and sharp change in population’s immune status caused globalization, stresses and change of lifestyle. Hence, these functional mutations are more likely to be the result of bacterial adaptation to new conditions. Mce1 operon had the largest number of nsSNPs, while mce4 operon had the lowest. Interestingly, mce4 operon showed to have the biggest number of mutations affecting protein function (63%).
Table 5.
Nonsynonymous SNPs in mce1–4 operons
| Operon | Number of nsSNPs |
|||
|---|---|---|---|---|
| Total | Lineage-Specific | Specific to Small Groups |
||
| Total | Affected | |||
| mce1 | 48 | 11 (22%) | 37 | 10 (27%) |
| mce2 | 35 | 5 (14%) | 30 | 15 (50%) |
| mce3 | 32 | 8 (25%) | 24 | 12 (50%) |
| mce4 | 26 | 2 (8%) | 24 | 15 (63%) |
Note.—A total number of nsSNPs in mce genes was counted. All identified mutations were divided into two groups: specific to an established lineage and located in small groups of at least two isolates. Number of affected mutations was counted using PredictSNP. Note that mce4 operon has the biggest (63%) and mce1—the smallest (27%) percent of small group mutations possibly affecting protein function. Full version of the table with descriptions of SNPs and groups is listed in supplementary material S2, Supplementary Material online.
Currently, mce proteins are promising candidates for new vaccine development because of their antigenic activity (Ahmad et al. 2004; Obregón-Henao et al. 2011; Singh et al. 2016). Mutations in these genes can possibly affect protein structure and antigenic activity level as discussed in (Pasricha et al. 2011). New DNA-vaccines based on mce genes with SNPs specific to particular lineages may leverage the specificity of immunity.
Discussion
Today M. tuberculosis evolves in a multifactor way. The leading selection factors in MT evolution are the use of the antibiotic therapy, global immune status change caused by immunosuppression diseases expansion, environmental conditions, and microbiota dysbiosis (Carding et al. 2015). All of these factors lead to significant changes inside MT and the emergence of new lineages with higher drug resistance and virulence (Mokrousov 2013; Shitikov et al. 2014; Luo et al. 2015; Bespyatykh et al. 2016). For instance, the latest discovered Beijing sublineage has been detected in Afghanistan (Eldholm et al. 2016). Higher virulence poses a threat for patients, not only because it worsens the infection, but also because it increases a risk of drug resistance development. One of the most essential examples of a gene that influences both of these characteristics is the transcription factor whiB7. WhiB7 modulates the expression of the regulation that encodes virulence and drug resistance genes (Larsson et al. 2012). Other examples are type VII system secretion genes, two-component systems, katalase katG and TA systems (Forrellad et al. 2013; Prozorov et al. 2013).
Defining key factors that influence the selection of sublineages with increased virulence levels against particular population groups is one of the most urgent problems in the modern physiology. It is necessary to identify the interaction between virulence and drug resistance networks, and to detect mutations in regulatory and structural virulence genes. Although a considerable part of these mutations is lineage-specific, we propose that virulence change is caused not by mass nonsynonymous mutations, but rather by several crucial ones that influence the activity of the gene products. The same idea is expressed in (Hershberg et al. 2008). Thus, detection of new sublineages by nsSNPs affecting pathogen phenotype seems to be promising in regards to mitigating MT outbreaks. Using these SNPs for discrimination of known lineages is of particular interest.
Studying the relationship between mutations in virulence genes and F15/LAM4/KZN sublineage, which has high virulence, has revealed three lineage-specific nonsynonymous SNPs in virulence genes. Sublineage F15/LAM4/KZN is characterized by extremely high host tissue necrosis during the infection (Smith et al. 2014). One possibility that accounts for this phenotype is the mutation in cyp125. This gene participates in the catabolism of cholesterol—the main component of cell membrane. Higher invasion of F15/LAM4/KZN isolates may be caused by the mutation in mce3B virulence gene. The favorable environment, formed by high HIV burden since 1990s, resulted in a tuberculosis epidemic (Cohen et al. 2015). Perhaps the virulence change also contributed to the “success” of KZN. The change in virulence might be caused by discovered here mutations. Rapid transmission between immunodeficient patients poses a threat of acquiring compensatory mutations than can allow mycobacteria to be easily transmitted from one person to another, including patients with normal immune status.
The new sublineage B0/N-90, emerged <50 years ago, has been detected inside Beijing-B0/W-148 sublineage. This sublineage can also be identified by nsSNPs in virulence genes. Of the approximately 30 members of B0/N-90 that were found, most were isolated in Russia. We conclude that the new MT sublineage was formed inside the high virulent Beijing-B0/W-148 sublineage. We assume that mutations in irtB and mce3F can increase survivability in host macrophages and invasiveness of B0/N-90 isolates. These SNPs can be used for immediate detection of this sublineage in whole-genome sequencing, multilocus sequence typing or PCR-based diagnosis. The primer sequences mentioned in this article can be used for SNP detection both in real-time PCR and multilocus PCR with some modifications.
The present work may give an insight into the change in pathogenesis for some M. tuberculosis lineages. As the link between the virulence level and nsSNPs in virulence genes is subject to experimental verification, all SNPs analyzed here in silico need checking in vivo. Future work will focus on searching new functional subgroups inside other lineages and studying the association between virulence levels and drug resistance profile.
Another promising perspective is the development of DNA vaccines based on M. tuberculosis target genes. The currently used standard BCG vaccine is not an optimal one as it shows a variable efficiency (from 0% to 80%) (Zhang et al. 2016) depending on the immune status, age, and even ethnicity of the patient (Orme 2006; Valdez et al. 2014). Besides, BCG vaccination does not work in immunodeficient patients (Dietrich et al. 2003). The existence of lineages with various virulence levels and people with various immune statuses assumes the development of new genetically engineered vaccines, efficient in the particular patient’s health state and the pathogen’s lineages. There are perspectives for DNA vaccines based on genes involved in MT pathogenesis (Teimourpour et al. 2015). Mce3 proteins are of interest as they are located in the cell wall, are potentially involved in antigenic activity, and are absent in BCG genome (Ahmad et al. 2004; Obregón-Henao et al. 2011; Pasricha et al. 2011; Parida et al. 2015; Singh et al. 2016). The use of a mutant gene to genetically engineer a vaccine, can increase the selectivity for population groups and M. tuberculosis lineages of particular area.
Supplementary Material
Supplementary materials are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Prof Gail Cassell, Prof Gil Alterovitz, Prof Alan Christoffels and Prof Qian Gao for useful advices on the design of the study. This work is conducted as a part of TBResist consortium (http://projects.iq.harvard.edu/tbresist).
Literature Cited
- Ahmad S, El-Shazly S, Mustafa AS, Al-Attiyah R. 2004. Mammalian cell-entry proteins encoded by the mce3 operon of Mycobacterium tuberculosis are expressed during natural infection in humans. Scand J Immunol. 60(4):382–391. [DOI] [PubMed] [Google Scholar]
- Ashiru OT, Manormoney P, Sturm WA. 2010. Adhesion to and invasion of pulmonary epithelial cells by the F15/LAM4/KZN and Beijing strains of Mycobacterium tuberculosis. J Med Microbiol. 59(5):528–533. [DOI] [PubMed] [Google Scholar]
- Bach H, et al. 2008. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe 3(5):316–322. [DOI] [PubMed] [Google Scholar]
- Bashyam MD, Seyed EH. 2004. The extracytoplasmic function Sigma factors: role in bacterial pathogenesis. Infect Genet Evol. 4(4):301–308. [DOI] [PubMed] [Google Scholar]
- Behar SM, et al. 2011. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 4(3):279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendl J, et al. 2014. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol. 10(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bespyatykh J, et al. 2016. Proteome analysis of the Mycobacterium tuberculosis Beijing B0/W148 cluster. Sci Rep. 6:28985.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burian J, et al. 2013. The mycobacterial antibiotic resistance determinant WhiB7 acts as a transcriptional activator by binding the primary sigma factor SigA (RpoV). Nucleic Acids Res. 41(22):10062–10076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carding S, et al. 2015. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis. 26:26191.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernyaeva EN, et al. 2014. Genome-wide Mycobacterium tuberculosis variation (GMTV) database: a new tool for integrating sequence variations and epidemiology. BMC Genome 15(1):308.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobat A, Orlova M, Barrera LF, Schurr E. 2013. Host genomics and control of tuberculosis infection. Public Health Genome 16(1–2):44–49. [DOI] [PubMed] [Google Scholar]
- Cohen KA, et al. 2015. Evolution of extensively drug-resistant tuberculosis over four decades: whole genome sequencing and dating analysis of Mycobacterium tuberculosis isolates from KwaZulu-Natal. PLoS Med 12(9):1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll F, et al. 2014. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun. 5:4812.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Converse PJ, et al. 2009. Role of the dosR-dosS two-component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect Immun. 77(3):1230–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C, Ghosh TS, Mande SS. 2011. Computational analysis of the Esx-1 region of Mycobacterium tuberculosis: insights into the mechanism of type VII secretion system. PLoS One 6(11):e27980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich G, Viret JF, Hess J. 2003. Mycobacterium Bovis BCG-based vaccines against tuberculosis: novel developments. Vaccine 21(7–8):667–670. [DOI] [PubMed] [Google Scholar]
- Eldholm V, et al. 2016. Armed conflict and population displacement as drivers of the evolution and dispersal of Mycobacterium tuberculosis. PNAS. 113(48):13881–13886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasani RA, Michael AS. 2013. Molecular mechanisms of multiple toxin-antitoxin systems are coordinated to govern the persister phenotype. PNAS 110(27):2528–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerriegel S, Claudio UK, Stefan N. 2014. Phylogenetic polymorphisms in antibiotic resistance genes of the Mycobacterium tuberculosis complex. J Antimicrob Chemother. 69(5):1205–1210. [DOI] [PubMed] [Google Scholar]
- Filliol I, et al. 2006. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into Tuberculosis evolution, phylogenetic accuracy. J Bacteriol. 188(2):759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein S, Wyk N, Warren RM, Sampson SL. 2015. Phylogeny to function: PE/PPE protein evolution and impact on Mycobacterium tuberculosis pathogenicity. Mol Microbiol. 96(5):901–916. [DOI] [PubMed] [Google Scholar]
- Ford CB, et al. 2013. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 45(7):784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrellad MA, et al. 2013. Virulence factors of the Mycobacterium tuberculosis complex. Virulence 4(1):3–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S, et al. 2006. Variable host-pathogen compatibility in Mycobacterium tuberculosis. PNAS 103(8):2869–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S, Peter MS. 2007. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis. 7(5):328–337. [DOI] [PubMed] [Google Scholar]
- Gandhi NR, et al. 2013. Nosocomial transmission of extensively drug-resistant tuberculosis in a rural hospital in South Africa. J Infect Dis. 207(1):9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein AE, et al. 2006. Structure/function studies of Ser/Thr and Tyr protein phosphorylation in Mycobacterium tuberculosis. J Mol Microbiol Biotechnol. 9(3–4):167–181. [DOI] [PubMed] [Google Scholar]
- He H, et al. 2006. MprAB is a stress-responsive two-component system that directly regulates expression of Sigma factors SigB and SigE in mycobacterium. J Bacteriol. 188(6):2134.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, et al. 2008. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 6(12):2658–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AVS. 2001. The genomics and genetics of human infectious disease susceptibility. Annu Rev Genomics Hum Genet. 2:373–400. [DOI] [PubMed] [Google Scholar]
- Homolka S, et al. 2012. High resolution discrimination of clinical Mycobacterium tuberculosis complex strains based on single nucleotide polymorphisms. PLoS One 7(7):e39855.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagielski T, et al. 2014. Current methods in the molecular typing of Mycobacterium tuberculosis and other mycobacteria. Biomed Res. 2014:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Narayanan S. 2012. PknE, a serine/threonine kinase of Mycobacterium tuberculosis modulates multiple apoptotic paradigms. Infect Genet Evol. 12(4):737–747. [DOI] [PubMed] [Google Scholar]
- Kurtz S, et al. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5(2):R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzas F, Karakousis PC, Sacchettini JC, Ioerger TR. 2013. Multidrug-resistant tuberculosis in Panama is driven by clonal expansion of a multidrug-resistant Mycobacterium tuberculosis strain related to the KZN extensively drug-resistant M. tuberculosis strain from South Africa. Clin Microbiol. 51(10):3277–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson C, et al. 2012. Gene expression of Mycobacterium tuberculosis putative transcription factors whiB1-7 in redox environments. PLoS One 7(7):e37516.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P. 2007. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23(1):127–2128. [DOI] [PubMed] [Google Scholar]
- Luo T, et al. 2015. Southern East Asian origin and coexpansion of Mycobacterium tuberculosis Beijing family with Han Chinese. PNAS 112(26):8136–8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonneuve E, Lana JS, Mikkel GJ, Kenn G. 2011. Bacterial persistence by RNA endonucleases. PNAS 108(32):13206–13211. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Maslov DA, Shu KV, et al. 2015. Draft genome sequences of two pyrazinamide-resistant clinical isolates, Mycobacterium tuberculosis 13-4152 and 13-2459. Genome Announ. 3(4):3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov DA, Zachikova MV, et al. 2015. Resistance to pyrazinamide in Russian Mycobacterium tuberculosis isolates: PncA sequencing versus Bactec MGIT 960. Tuberculosis 95(5):608–612. [DOI] [PubMed] [Google Scholar]
- Mokrousov I. 2012. The quiet and controversial: Ural family of Mycobacterium tuberculosis. Infect Genet Evol. 12(4):619–629. [DOI] [PubMed] [Google Scholar]
- Mokrousov I. 2013. Insights into the origin, emergence, and current spread of a successful Russian clone of Mycobacterium tuberculosis. Clin Microbiol Rev. 26(2):342–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obregón-Henao A, et al. 2011. Vaccination of guinea pigs using Mce operon mutants of Mycobacterium tuberculosis. Vaccine 29(26):4302–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orme IM. 2006. Preclinical testing of new vaccines for tuberculosis: a comprehensive review. Vaccine 24(1):2–19. [DOI] [PubMed] [Google Scholar]
- Pandey AK, Sassetti CM. 2008. Mycobacterial persistence requires the utilization of host cholesterol. PNAS 105(11):4376–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parida SK, et al. 2015. Totally drug-resistant tuberculosis and adjunct therapies. J Intern Med. 277(4):388–405. [DOI] [PubMed] [Google Scholar]
- Parish T, et al. 2003. The senX3-regX3 two-component regulatory system of Mycobacterium tuberculosis is required for virulence. Microbiology 149(6):1423–1435. [DOI] [PubMed] [Google Scholar]
- Pasricha R, et al. 2011. Single nucleotide polymorphism in the genes of mce1 and mce4 operons of Mycobacterium tuberculosis: analysis of clinical isolates and standard reference strains. BMC Microbiol. 11(1):41.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prozorov AA, Fedorova IA, Bekker OB, Danilenko VN. 2014. The virulence factors of Mycobacterium tuberculosis: genetic control, new conceptions. Russ J Genet. 50(8):775–797. [PubMed] [Google Scholar]
- Prozorov AA, Danilenko VN. 2011. Mycobacteria of the tuberculosis complex: genomics, molecular epidemiology, and evolution trends. Biol Bull Rev. 1(6):483–495. [Google Scholar]
- Prozorov AA, Zaichikova MV, Danilenko VN. 2013. Systems of genes and proteins affecting mycobacteria virulence and their homologs participation in conjugation of Mycobacterium smegmatis. Russ J Genet. 49(1):41–125. [DOI] [PubMed] [Google Scholar]
- Quadri LEN. 2008. Iron uptake in mycobacteria In: Mycobacterial cell envelope. In Daffé M; Reyrat JM, editors. Herndon: ASM Press; 167–184. [Google Scholar]
- Raynaud C, et al. 2002. The functions of OmpATb, a pore-forming protein of Mycobacterium tuberculosis. Mol Microbiol. 46(1):191–201. [DOI] [PubMed] [Google Scholar]
- Reiling N. 2013. Clade-specific virulence patterns of Mycobacterium tuberculosis. mBio 4(4):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan J, Bloom BR, Rubin EJ. 2005. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. PNAS 102(23):8327–8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GM, Smith I. 2006. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J Bacteriol. 188(2):424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander P, et al. 2004. Lipoprotein processing is required for virulence of Mycobacterium tuberculosis. Mol Microbiol. 52(6):1543–1552. [DOI] [PubMed] [Google Scholar]
- Sarkar S, et al. 2016. Strains of Mycobacterium tuberculosis differ in affinity for human osteoblasts and alveolar cells in vitro. SpringerPlus 5(1):163.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitikov EA, et al. 2014. Unusual large-scale chromosomal rearrangements in Mycobacterium tuberculosis Beijing B0/W148 cluster isolates. PLoS One 9(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shur KV, et al. 2016. Draft genome sequence of Mycobacterium tuberculosis strain B9741 of Beijing B0/W lineage from HIV positive patient from Siberia. Genomics Data 10:61–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Katoch VM, Mohanty KK, Chauhan DS. 2016. Analysis of expression profile of Mce operon genes (mce1, mce2, mce3 operon) in different Mycobacterium tuberculosis isolates at different growth phases. Indian J Med Res. 143(4):487.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkov VV, Ogarkov OB, Mokro IV, Zhdanova SN. 2016. Evolutionary significance of non-synonymous substitution for Mycobacterium tuberculosis of Ural genotype. Mol Med. 14(4):44–50. [Google Scholar]
- Sirakova TD, Dubey VS, Cynamon MH, Kolattukudy PE. 2003. Attenuation of Mycobacterium tuberculosis by disruption of A MAS-like gene or a chalcone synthase-like gene, which causes deficiency in dimycocerosyl phthiocerol synthesis. J Bacteriol. 185(10):2999–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KLJ, et al. 2014. Reduced virulence of an extensively drug-resistant outbreak strain of Mycobacterium tuberculosis in a murine model. PLoS One 9(4):e94953.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GR, et al. 2005. Mycobacterial mutants with defective control of phagosomal acidification. PLoS Pathog. 1(3):0269–0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, et al. 2010. Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine raw 264.7 macrophages. PLoS One 5(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teimourpour R, et al. 2015. Construction of a DNA vaccine encoding Mtb32C and HBHA genes of Mycobacterium tuberculosis. Jundishapur J Microbiol. 8(8):e21556.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari P, et al. 2015. MazF ribonucleases promote Mycobacterium tuberculosis drug tolerance and virulence in guinea pigs. Nat Commun. 6:6059.. [DOI] [PubMed] [Google Scholar]
- Valdez Y, Brown EM, Finlay BB. 2014. Influence of the microbiota on vaccine effectiveness. Trends Immunol. 35(11):526–537. [DOI] [PubMed] [Google Scholar]
- Walker TM, et al. 2013. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 13(2):137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters SB, et al. 2006. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol Microbiol. 60(2):312–330. [DOI] [PubMed] [Google Scholar]
- Wards BJ, de Lisle GW, Collins DM. 2000. An esat6 knockout mutant of Mycobacterium bovis produced by homologous recombination will contribute to the development of a live tuberculosis vaccine. Tubercle Lung Dis. 80(4–5):185–189. [DOI] [PubMed] [Google Scholar]
- Zahrt TC, Deretic V. 2002. Reactive nitrogen and oxygen intermediates and bacterial defenses: unusual adaptations in Mycobacterium tuberculosis. Antioxid Redox Signal. 4(1):141–159. [DOI] [PubMed] [Google Scholar]
- Zaychikova MV, et al. 2015. Mycobacterium tuberculosis type II toxin-antitoxin systems: genetic polymorphisms and functional properties and the possibility of their use for genotyping. PLoS One 10(12):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, et al. 2016. Variable virulence and efficacy of BCG vaccine strains in mice and correlation with genome polymorphisms. Mol Ther. 24(2):398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Xie J. 2011. Mycobacterium tuberculosis proteases and implications for new antibiotics against tuberculosis. Crit Rev Eukaryot Gene Expr. 21(4):347–361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.