

Abstract
Phytophthora megakarya (Pmeg) and Phytophthora palmivora (Ppal) are closely related species causing cacao black pod rot. Although Ppal is a cosmopolitan pathogen, cacao is the only known host of economic importance for Pmeg. Pmeg is more virulent on cacao than Ppal. We sequenced and compared the Pmeg and Ppal genomes and identified virulence-related putative gene models (PGeneM) that may be responsible for their differences in host specificities and virulence. Pmeg and Ppal have estimated genome sizes of 126.88 and 151.23 Mb and PGeneM numbers of 42,036 and 44,327, respectively. The evolutionary histories of Pmeg and Ppal appear quite different. Postspeciation, Ppal underwent whole-genome duplication whereas Pmeg has undergone selective increases in PGeneM numbers, likely through accelerated transposable element-driven duplications. Many PGeneMs in both species failed to match transcripts and may represent pseudogenes or cryptic genetic reservoirs. Pmeg appears to have amplified specific gene families, some of which are virulence-related. Analysis of mycelium, zoospore, and in planta transcriptome expression profiles using neural network self-organizing map analysis generated 24 multivariate and nonlinear self-organizing map classes. Many members of the RxLR, necrosis-inducing phytophthora protein, and pectinase genes families were specifically induced in planta. Pmeg displays a diverse virulence-related gene complement similar in size to and potentially of greater diversity than Ppal but it remains likely that the specific functions of the genes determine each species’ unique characteristics as pathogens.
Keywords: cacao, black pod rot, Phytophthora, genome, transcriptome
Introduction
Cacao (Theobroma cacao L.) is grown around the world where favorable environments occur. It is the source of cocoa, the critical ingredient in chocolate. Black pod rot, caused by species of Phytophthora, is the most economically important disease on cacao globally. Losses to black pod rot were estimated at 700,000 metric tons in 2012 (Ploetz 2016) or approximately 16% of the annual production. The cacao crop is currently valued at 12.6 billion US dollars per year (https://www.icco.org, last accessed 16 February 2017). Phytophthora palmivora (Ppal), the most common species causing black pod rot, occurs in cacao growing countries around the globe. Ppal has more than one hundred recorded hosts (Mchau and Coffey 1994), showing its extreme adaptability. Ppal causes yield losses on cacao of 20–30% annually (Flood et al. 2004). Phytophthora megakarya (Pmeg) occurs only in the countries of West and Central Africa and represents a relatively new disease. Pmeg is the most virulent species causing black pod rot; it can cause 60–100% crop losses if not managed (Opoku et al. 2000). Pmeg was first identified taxonomically as a species in 1979 (Brasier and Griffin 1979), although it was likely causing losses in Nigeria in the early 1950s (Thorold 1959). By the mid-1980s, Pmeg was dominant in Cameroon, Equatorial Guinea, Gabon and Togo, (Guest 2007) and was confirmed in Ghana in 1985 (Dakwa 1987). Pmeg is not known to cause significant disease on any plant species other than Theobroma cacao, but can be isolated from roots of many tree species (Akrofi et al. 2015).
In recent years, Pmeg has displaced Ppal from cacao in Cameroon and Nigeria (Nyasse et al. 1999; Ndubuaku and Asogwa 2006; Djocgoue et al. 2007) where isolation of Ppal from cacao is no longer routine. Ppal tends to have a more rapid growth rate than Pmeg in culture and in planta, and can cause accelerated necrosis in wounded cacao tissues compared with Pmeg (Ali et al. 2016). Wounding is less relevant for Pmeg infection (Ali et al. 2016), possibly due to its ability to rapidly form appressoria in greater numbers than Ppal. Ppal typically enters unwounded tissues through stomata (Ali et al. 2016); something that Pmeg can do with similar efficiency. Pmeg can survive in dry soil longer than Ppal, a trait that may contribute to its carryover ability during dry cycles (Bailey et al. 2016). Beyond these possibilities, there is no explanation for the dominance of Pmeg over Ppal in Africa.
Phytophthora is a genus of filamentous oomycetes, within the kingdom Stramenopila (Tyler et al. 2006; Bollmann et al. 2016). There are over 120 known species (Kroon et al. 2012) and most are pathogens causing disease in a large range of plant hosts. Due to their economic and environmental impact, there is expanding interest in Phytophthora genetics and genomics, resulting in the recent releases of genome sequences of P. ramorum, P. sojae, P. infestans, P. capsici and P. litchii (Tyler et al. 2006; Haas et al. 2009; Lamour et al. 2012; Ye et al. 2016). The identification of gene families encoding classes of toxins, elicitors, and effectors shared among the Phytophthora species is critical to understanding the disease process. Comparative genomics of P. ramorum, P. sojae, and P. infestans have aided in understanding the role of the RxLR (synonym- Avr and Avh) and CRN (crinkling and necrosis inducing protein) super families of effectors in pathogenicity and host specificity. Other complex families of elicitors/protein toxins such as necrosis-inducing phytophthora proteins (NPPs) and elicitins, also important determinants in the Phytophthora-plant interaction, have been described (Kanneganti et al. 2006). Another common observation has been the involvement of transposable elements (TEs) in influencing Phytophthora genome sizes and compositions (Judelson 2012).
The genus Phytophthora has been divided into ten clades and both Ppal and Pmeg fall within clade 4 (Blair et al. 2008). Being related but with differential pathogenic potential on the same host makes the duo excellent candidates for comparative genomic studies to understand the expanding phytopathogenic ability of Phytophthora species. Therefore, we report here the draft genome sequences of Ppal and Pmeg and carry out a comparative study between the two genomes to identify and better understand possible virulence determinants, an understanding that may prove critical to their management and preventing the spread of Pmeg into new cacao production areas.
Materials and Methods
Selection of the Pmeg and Ppal Strains to Be Sequenced
Pmeg isolate ZTHO120, isolated from black pod infected cacao in Cameroon, and Ppal isolate SBR112.9, isolated from black pod infected cacao in Ivory Coast, were sequenced. Isolates were collected as described (Ali et al. 2016). The isolates were identified to species using sequencing of the ribosomal RNA gene internal transcribed spacer (JX315263, JX315268).
Isolation of Pmeg and Ppal Genomic DNA and RNA
See supplementary materials and methods, Supplementary Material online, for genomic DNA extraction, and mycelia and zoospore RNA extraction from Pmeg and Ppal isolates ZTHO120 and SBR112.9, respectively. For RNA sequencing from infected plant material and validation of the RNA-Seq expression profiles, Pmeg and Ppal isolates ZTHO120 and SBR112.9 used in the original genome sequencing and mycelia and zoospore RNA-Seq analysis were replaced by isolates ZTHO145 and Gh-ER1349, respectively. The ZTHO120 culture could not be maintained and SBR112.9 showed low virulence compared with other Ppal isolates. The zoospore-inoculated pod husk infection assays were carried out essentially as described (Ali et al. 2016) and the RNA extraction procedure is described in the supplementary materials and methods, Supplementary Material online.
Genome Sequencing and Assembly
Pmeg and Ppal genomic DNA were sequenced using Illumina paired-end short-read technology (library preparation and sequencing performed by Beijing Genome Institute, Shenzhen, China). The short reads (90 bp), comprising 5,233 and 8,053 Mb of data for Pmeg and Ppal, respectively, were assembled using “SOAPdenovo” (http://soap.genomics.org.cn/soapdenovo.html, last accessed 16 February 2017; version: 1.05). Key parameter K was set at 47 and 59 for Pmeg and Ppal, respectively which produced the optimal assembly result. An overview of the assembly algorithm (Li et al. 2010) is shown in the supplementary figure S1, Supplementary Material online. Assembled scaffolds based on SOAPdenovo were linked by SSPACE; version: 1.1 (Boetzer et al. 2011). Reads were mapped to the draft assembly and mapping statistics were obtained. Reads were aligned onto the assembled sequences and 500-bp nonoverlapping sliding windows along the assembled sequence were used to calculate GC content and average read depth. Repeat rate was calculated based on contig length and depth analysis. Each contig was categorized as unique, repeat, similar, or error. Error was based on contigs with less than 0.1-fold coverage; repeat was based on contigs with more than 1.8-fold coverage; similar was defined as having similarity higher than 0.95% between contigs of the same length with the average coverage between 0.1- and 1.8-fold; the remaining were classified as unique. Repeat contigs included short contigs with lengths <100 bp and coverage no less than 1.5-fold and long contigs with length >100 bp and coverage no less than 1.8-fold. The complete nucleotide sequence assemblies from both species and their annotations can be found online at the genome resource site of NCBI (BioProject ID: PRJNA318028 and PRJNA318026).
Ab Initio Putative Gene Model Prediction
The ab initio putative gene model (PGeneM) prediction was performed using two different approaches. Initially PGeneMs were predicted from the assembly results using AUGUSTUS version 2.6.1 (Stanke et al. 2004), trained with P. sojae gene models (Joint Genome Institute Phytophthora sojae v3.0 database). Secondly, PGeneMs were predicted using MAKER (Cantarel et al. 2008). The MAKER pipeline masked repeats with RepeatMasker (Cantarel et al. 2008), used SNAP (Korf 2004) and AUGUSTUS for de novo gene prediction, and used Phytophthora peptides and Pmeg/Ppal RNA-Seq transcriptomes for evidence-based gene prediction. SNAP was trained with Core Eukaryotic Genes Mapping Approach (CEGMA) genes identified from either the Pmeg or Ppal genome, respectively, whereas AUGUSTUS was trained with the previously identified genes of P. infestans, P. parasitica, P. capsici, P. cinnamomi, P. sojae, and P. ramorum. The predicted proteins were compared against NCBI nonredundant (NR) protein databases by BLASTp to identify biological functions (Altschul et al. 1997). The predictions in overlapping PGeneMs from the two different approaches were ranked according to the following criteria and the “best” ab initio PGeneM according to the stipulated criteria was selected for functional annotation. The criteria for the ranking of the PGeneMs were the following: 1) Manual annotation had priority over all other evidence; 2) Prediction with similarities to known proteins then had priority. A prediction was considered to be similar to a known protein if it had an E-value of at most 1e-10 (BLASTP against NR+ Phytophthora protein set); 3) If two overlapping PGeneM predictions had similarity with known proteins, the one with better a coverage score had priority; 4) In clusters without similarity with known proteins and without expressed sequence tag (EST) evidence, the priority was for the MAKER PGeneM prediction. Open reading frames were also annotated using Blast2GO (http://www.blast2go.com/b2ghome, last accessed 16 February 2017) (Conesa et al. 2005) and the KEGG-database of metabolic pathways (Moriya et al. 2007).
Validation of Duplicate PGeneMs
Various single-copy housekeeping (HK) PGeneMs of Pmeg were selected and used to search for orthologs in the Ppal genome, most often resulting in the identification of two copies of homeologous PGeneMs. To validate that multiple Ppal isolates commonly carry duplicate HK PGeneMs, two homeolog-specific polymerase chain reaction (PCR) primer sets were designed for the SUMO-conjugating enzyme (SCE) (see supplementary table S1, Supplementary Material online) and the homeologs were PCR-amplified using DNA from various isolates of Pmeg and Ppal.
Identification of Core Eukaryotic Genes
To assess transcriptome completeness and to compare with other published Phytophthora genomes, PGeneMs were analyzed with the CEGMA pipeline. CEGMA was developed to identify a subset of 248 highly conserved core eukaryotic genes (CEGs) in eukaryotic genomes (Parra et al. 2007). For comparison purposes, the P. sojae and P. ramorum genomes were downloaded from the Joint Genome Institute (Tyler et al. 2006) and the P. infestans genome was downloaded from the Broad Institute (Haas et al. 2009) and analyzed with CEGMA. Assemblies were run using default settings. Subsequently, the Benchmarking Universal Single-Copy Orthologs (BUSCO) strategy was also used to test the completeness of both assemblies and PGeneMs using the eukaryote profile (Simão et al. 2015).
Synteny Analysis
Synteny between Pmeg and Ppal contigs (10,000 bp or larger) and synteny within Pmeg and Ppal whole genomes were assessed using the whole-genome aligner MUMmer3.22 (Delcher et al. 2002; Kurtz et al. 2004). For the reciprocal synteny analysis of pairs of contigs, the NUCmer parameters were: breaklen = 200, maxgap = 90, mincluster = 65, minmatch = 20. For the synteny analysis across the entire genome, a higher stringency was selected with breaklen = 400, maxgap = 50, mincluster = 65, minmatch = 50.
Transcriptome Sequencing
RNA-Seq analysis from mycelia and zoospores was carried out by Beijing Genome Institute and RNA-Seq analysis of infected plant material was carried out by the National Center for Genome Resources (Santa Fe, NM). cDNA was generated using the RNA library preparation TruSeq protocol developed by Illumina Technologies (San Diego, CA). Using the kit, mRNA was first isolated from total RNA by performing a polyA selection step, followed by construction of single-end sequencing libraries with an insert size of 160 bp. Single-end sequencing was performed using the Illumina HiSeq2000 platform. Samples were multiplexed with unique six-mer barcodes generating filtered (for Illumina adapters/primers and PhiX contamination) 1 × 50 bp reads. The sequences acquired by RNA-Seq were verified by comparison to the Pmeg and Ppal genomes assembled in this study. RNA reads from RNA-Seq libraries ranging from 50 to 70 million reads in fastq format were aligned using memory-efficient short-read aligner Bowtie-2-2.1.0 (Langmead and Salzberg 2012) to the coding sequences (CDS) of the Pmeg and Ppal genomes. Tabulated raw counts of reads to each CDS were obtained from the bowtie alignment. Raw counts were normalized using the DESeq package in the R statistics suite (Anders and Huber 2010). For DESeq’s default normalization method, scaling factors are calculated for each lane as median of the ratio, for each gene, of its read count of its geometric mean across all lanes and apply to all read counts.
Validation of RNA-Seq Analysis Using RT-qPCR
After RNA-Seq, 13 and 21 PGeneMs of Pmeg and Ppal, respectively, were chosen for analysis by quantitative reverse transcription PCR (RT-qPCR) across mycelia, zoospores, and infected pod husks. Three replicate samples of mycelia, zoospores, and infected pod husks RNA were extracted as described above for both species. RT-qPCR analysis was conducted following Bailey et al. (2013). Primer sources and sequences for the Phytophthora PGeneMs are in the supplementary Excel file S1, sheet 1: RNA-Seq-RTqPCR comparison, Supplementary Material online. Phytophthora PGeneMs were selected based upon results of RNA-Seq analysis. The delta–delta Ct method was used to calculate fold changes among the mycelia, zoospores, or in planta samples (Livark and Schmittgen 2001).
Determining Secretomes
Pmeg and Ppal protein-coding sequences were scanned for possible signal peptides using SignalP, version 3.0 (Petersen et al. 2011). The amino acid sequences containing predicted signal peptides were scanned for transmembrane proteins using the TMHMM program (prediction of transmembrane helices in proteins) (Sonnhammer et al. 1998). Proteins with no more than one transmembrane domain were considered potential components of the secretome.
RxLR Motif Finding
The predicted RxLR effector protein set of P. sojae and P. ramorum was used to perform PSI-BLAST against the Pmeg and Ppal ab initio PGeneM. Significant hits (E-value <1e-5) were manually checked for the presence of an RxLR–dEER domain and N-terminal signal peptide. By using the program HMMER 2.3.2 (http://hmmer.org/download.html, last accessed 16 February 2017) (Eddy 1998), two hidden Markov models (HMMs) were built from the candidates mentioned above, one using the RxLR motif and ten amino acids on the left side and the other using the complete RxLR–dEER domains, with variable spacing arbitrarily placed in between. The predicted protein set, as well as all six-frame translations from both genomes were screened using the HMMs. Proteins with a significant HMM score (E-value <0.05) were considered as candidates. BLASTp searches within and between Pmeg and Ppal RxLR PGeneMs were conducted to identify relationships within and between the Pmeg and Ppal RxLR gene family members. Similar RxLR PGeneMs were identified based on at least 50% sequence identity with E-value less than 1e-10 between two or more PGeneMs.
Transcript Profile Analysis by Self-Organizing Map Analysis
Tabulated raw counts of CDS reads were obtained from the bowtie alignment. Read counts were normalized using the DEseq package (Anders 2010) and R x64 2.15.2 program (http://www.r-project.org/, last accessed date 16 February 2017). Normalized RNA-Seq count were log10 transformed and neural network self-organizing map (SOM) analysis (Kohonen 1982, 1990) was employed (using MATLAB Neural Network Toolbox) to provide an unsupervised multivariate and nonlinear classification of the transcriptome into 24 different classes. The number of classes was chosen based on the number of treatments (mycelia, zoospores, and infected tissue), number of samples, and allowing for differences in gene expression levels. The classification used a multivariate input vector describing the transcription response of each PGeneM.
In order to further examine the relationships among the 24 classes, results from 20 of the most highly expressed PGeneMs from each of the 24 SOM classes were used as input for Principal Coordinates Analysis (PCoA) (via GenAlEx 6.5) based on the covariance matrix with data standardization of log10-transformed RNA-Seq count data. The PCA results were then used to generate a 3D scatter plot via an Excel macro add-in.
Result and Discussion
Genome Sequencing and Assembly
Whole-genome shotgun sequencing generated genome assemblies for Pmeg and Ppal with total contig lengths of 101.18 and 107.42 Mb, respectively (table 1). The Illumina read coverage of the assembly was examined at 90 base intervals (distribution shown in the supplementary figs. S2A and S3A, Supplementary Material online), yielding peak coverages of approximately 33× for Pmeg and 44× for Ppal (table 1). The total length of repeat fragments in the Pmeg and Ppal genomes was 42.4 and 36.8 Mb, respectively (supplementary table S2, Supplementary Material online). Considering the additional dimension of depth of Illumina read coverage, we identified assembly regions that represented separately assembled haplotypes (half the expected coverage depth), diploid consensus regions (at the expected coverage depth) and tetraploid consensus regions (twice the expected coverage depth). These regions appear as secondary peaks before and after the primary diploid consensus peak (see supplementary figs. S2B and S3B, Supplementary Material online). The proportions of the assembled genomes that fall into these categories are shown in the supplementary table S3, Supplementary Material online. Subtracting half of the haploid contribution, doubling the tetraploid contribution, and adding the repeat fragment contribution result in more accurate estimates of genome sizes for Pmeg (126.88 Mb) and Ppal (151.23 Mb). The discrepancy between the contig assembly size and the true genome size is common for oomycetes due to poor assembly of repeat rich regions (Gijzen 2009; Baxter et al. 2010). The estimated genome sizes of Pmeg and Ppal are larger than those of P. sojae (95 Mb) and P. ramorum (65 Mb) (Tyler et al. 2006) which are from clades 7 and 8, respectively. They are also larger than another clade 4 species, P. litchii, which has a 58-Mb genome (Ye et al. 2016). However, they are considerably smaller than P. infestans (240 Mb) (Haas et al. 2009) from clade 1 (Blair et al. 2008). The average GC% was similar for both Pmeg and Ppal (48.9%) as well as for P. litchii (Ye et al. 2016), but less than three other Phytophthora genomes (51–54.4%) (Tyler et al. 2006; Haas et al. 2009). The sequence and annotations are available from NCBI (BioProject ID: PRJNA318028 and PRJNA318026).
Table 1.
Genome Assembly and Annotation Statistics
| P. megakarya | P. palmivora | |
|---|---|---|
| Genome | ||
| Estimates genome sizes (Mb)a | 126.88 | 151.23 |
| Total Contig length | 101,182,312 | 107,423,419 |
| Contig numbers | 27,143 | 28,632 |
| CEGMA Completeness (%) | 94.35 | 96.37 |
| GC content (%) | 48.92 | 48.91 |
| N50 Contig length | 6,902 | 6,456 |
| Scaffold number | 24,070 | 24,815 |
| K-mer analysis | ||
| Read data (Mb) | 5,233 | 8,053 |
| Ave. read length | 90 | 90 |
| K-mer length | 15 | 15 |
| Coverage depth | 33.41 | 44.63 |
| K-mer number | 4,388,028,112 | 5,057,947,212 |
| K-mer depth | 28.01 | 28.03 |
| Contig representation (%) | 64.58 | 59.53 |
| Putative gene model | ||
| Gene number | 42,036 | 44,327 |
| Total gene length | 42,723,254 | 45,995,141 |
| Average gene length | 969.78 | 1,038.15 |
| Average gene densityb | 0.272 | 0.254 |
| Number of expressed genesc | 14,915 | 25,617 |
| Genes with GO annotationd | 15,431 | 21,276 |
| Genes within KEGG pathway | 14,789 | 15,717 |
| Core orthologous genese | 14,624 | 14,624 |
| Genes with unknown functions/hypothetical protein | 26,940 | 22,056 |
See supplementary table S2, Supplementary Material online.
CDS bases/total genome bases.
Only gene models with ≥10 reads, either in mycelia, zoospore, or in planta are reported.
Gene models with E < 10−4 for BLASTn against Uniport Gene Ontology database.
Core orthologs were estimated based on bidirectional best BLASTp hits between the inferred Pmeg and Ppal proteomes. To be considered as an ortholog, BLASTp matches should span at least 50% of the sequence with E-value less than 1e-10.
The initial approach of ab initio gene prediction using AUGUSTUS generated 25,493 and 25,852 PGeneMs in the Pmeg and Ppal genomes, respectively. Using a similar prediction pipeline, Ye et al. (2016) recently reported 13,155 PGeneMs from the 38-Mb assembled genome of P. litchi. The second approach of ab initio gene prediction using MAKER including SNAP and AUGUSTUS as part of the pipeline generated 33,614 and 37,283 PGeneMs in the Pmeg and Ppal genomes, respectively. The average annotation edit distance (AED) was 0.27 and 0.23 for Pmeg and Ppal, respectively (AED of 0 indicates perfect agreement between the model and the union of the supporting evidence; AED of 1 indicates no agreement [Yandell and Ence 2012]) and only three PGeneMs had an AED of 1. Using the MAKER pipeline on a published P. sojae genome (v1.0), 21,447 PGeneMs were obtained (data not shown), an increase of 2,420 over the previously published gene numbers for the species (v1.0; Tyler et al. 2006) but less than the most recent assembly and annotation (v3.0; 26,584 PGeneMs) based on gap closing, and EST and RNA-Seq evidence (http://gp-next2.jgi.doe.gov/Physo3/Physo3.info.html, last accessed 16 February 2017). Manually combining the two ab initio gene prediction approaches generated 42,036 PGeneMs in the Pmeg genome and 44,327 PGeneMs in the Ppal genome (table 1; see supplementary Excel file S2, sheet 1 for Pmeg PGeneMs and sheet 2 for Ppal PGeneMs, Supplementary Material online). Of these, 14,624 PGeneMs were identified as core orthologs shared between Pmeg and Ppal (table 1). Phytophthora infestans, P. sojae and P. ramorum originally had 18,179, 19,027 and 15,743 predicted PGeneMs, respectively (Tyler et al. 2006; Haas et al. 2009), though those gene number estimates did not benefit from RNA-Seq data. As evidenced by the comparison to the latest version of the P. sojae genome (v3.0) the 33,614 and 37,283 PGeneMs predicted by MAKER for the Pmeg and Ppal genomes, respectively, are likely to be conservative estimates, whereas the higher numbers obtained by merging the two methods (42,036 in Pmeg 44,327 in Ppal) may be an upper limit. The gene predictors used (AUGUSTUS and MAKER) work by statistically profiling protein-coding, intergenic and boundary regions using various classifiers. Gene callers can be compromised by factors such as genomic islands of differing GC content, pseudogenes and genes with programmed or artificial frameshifts, leading to variability between PGeneM predictions (Pati et al. 2010). To check the efficacy of the gene predictors, we looked for CEGs within the Pmeg and Ppal genomes. The Pmeg and Ppal genomes harbored 94.3% and 96.3%, respectively, of the 248 CEGs (Parra et al. 2007). This is similar to the other published Phytophthora genomes (fig. 1B). BUSCO, a more current strategy for the quantitative assessment of genome completeness, was also used. Of the genes anticipated to be present as single copies in an eukaryote, the benchmarking strategy indicated that the Pmeg PGeneM set contained 92.1% of examined loci (87% complete genes and 5.1% fragmented genes) and the Ppal PGeneM set contained 91% of examined loci (77% complete genes and 14% fragmented genes) out of the 429 queried (supplementary table S4, Supplementary Material online).
Fig. 1.—

Comparison of the gene complements of P. megakarya and P. palmivora. (A) The ratio between gene model numbers of selected P. megakarya and P. palmivora gene families. (HK housekeeping gene; # gene families with a single-copy gene model in P. megakarya; * gene families related to plant-infection). (B) Presence of CEGs within different Phytophthora spp. based on the CEGMA output report (Parra et al. 2007). (C) Evolutionary relationship between eight randomly selected gene models where there is only a single copy in P. megakarya but there are two homeologous copies in P. palmivora. Amino acid sequence similarities were inferred using guide trees generated by WebPRANK (http://www.ebi.ac.uk/goldman-srv/webprank/, last accessed 16 February 2017). Branch length represents the number of amino acid substitutions per site. (S.dic, Saprolegnia diclina; P.meg, P. megakarya; P.pal, P. palmivora; P.par, P. parasitica; P.vit, Plasmopara viticola; P.soj, P. sojae; T.ree, Trichoderma reesei; P.inf, P. infestans; R.sol, Ralstonia solanacearum; P.hal, Plasmopara halstedii). (D) PCR amplification of SCE with two P. palmivora homeolog-specific PCR primer sets across 12 P. palmivora and 2 P. megakarya isolates (Ali et al. 2016).
Transcript Analysis of the PGeneMs
To validate the expression of genes for both species, RNA-Seq was performed on RNA from mycelium and zoospores, and from infected pod husks at 72 h postinfection. The RNA-Seq analysis identified 11,733, 13,179, and 12,468 Pmeg PGeneMs (with ≥10 normalized reads) for the mycelia, zoospore and in planta samples, respectively. Similarly RNA-Seq analysis for Ppal has identified 21,162, 23,305, and 22,047 PGeneMs (with ≥10 normalized read numbers) for the mycelia, zoospore and in planta samples, respectively. Combining these data for Pmeg, transcripts from 14,915 gene models could be detected in RNA-Seq data from mycelium, zoospores, and infected pod husks, leaving 27,121 without transcripts. In Ppal, transcripts were detected for 25,617 PGeneMs whereas 18,710 did not show transcripts. In a digital gene expression study involving the RNA from multiple life stages, 14,969 out of the 18,093 (83%) mappable P. sojae PGeneMs matched at least one tag (Ye et al. 2011). In an RNA-Seq analysis of three in vitro life stages of P. capsici and 15,111 (13,731 with ≥10 reads) out of 19,805 PGeneMs were considered transcribed (Chen et al. 2013, supplementary material S). Only 9,109 out of 18,179 P. infestans PGeneMs showed hits from RNA sequences from P. infestans-infected tomato plants (Zuluaga et al. 2016).
Why would these two Phytophthora species, especially Pmeg, carry so many nonexpressed PGeneMs? Many of the nontranscribed PGeneMs show high sequence similarity to functional PGeneMs, and so presumably could encode functional proteins if expressed. For example, among the 184 NPP PGeneMs of Pmeg and Ppal, there is high sequence similarity between expressed and nonexpressed PGeneMs (see supplementary fig. S4, Supplementary Material online). Although the majority of the nonexpressed PGeneMs have no putative function, many do, including virulence-associated PGeneMs like the NPPs and RxLR effectors.
In P. sojae there are examples of RxLR avirulence genes that have become silenced in order to avoid detection by soybean resistance genes (Tyler and Gijzen 2014), but they encode fully functional proteins. It is plausible that for nontranscribed genes in Phytophthora genomes, unsilencing could occur and be selected for when the genes confer an advantage. For example, this process might have contributed to the adaptation of Pmeg to cacao as a new encounter host (Bailey et al. 2016).
Whole-Genome Duplication in Ppal
A comparison between the proteomes of Pmeg and Ppal was performed using a bidirectional BLASTp search with ≥50% coverage and an E-value of 1.00e-10 as the cut-off for two proteins to be considered orthologs. There were 27,037 PGeneMs (64.3%) from Pmeg with this level of similarity to Ppal PGeneMs and 34,762 PGeneMs (78.4%) from Ppal with this level of similarity to Pmeg PGeneMs. Of these orthologous PGeneMs only 14,624 were unique, and thus were considered as core orthologous genes. Similarly, BLASTp analysis within each genome with an E-value cutoff of 1.00e-10 found 35,671 PGeneMs (84.9%) from Pmeg had similarity to at least one other PGeneM within the Pmeg genome, and 41,401 PGeneMs (93.4%) from Ppal had similarity to at least one other PGeneM within the Ppal genome (see supplementary Excel file S2, Supplementary Material online).
To examine the possibility of genome duplications in either species, given the large numbers of PGeneMs, gene copy number was examined for a set of putative HK PGeneMs based on the list of mammalian HK genes described by Eisenberg and Levanon (2013). The ratio between PGeneM numbers in Pmeg and Ppal was around 0.5 for most families of HK PGeneMs (fig. 1A and see supplementary Excel file S1, sheet 2: PGeneM nos, Supplementary Material online). For many genes there were two copies in Ppal compared with one copy in Pmeg. The average number of copies per CEG in the case of Ppal was 2.36. Furthermore almost 70% of CEGs had more than one copy in Ppal, whereas in four other Phytophthora species including Pmeg, only 20–34% had more than one copy (fig. 1B). This observation suggests that Ppal has gone through a whole-genome duplication (WGD) event. The BUSCO analysis of complete, full length genes also indicated that 49% of the expected single-copy Ppal genes were duplicated, whereas only 26% of the expected single-copy Pmeg genes were duplicated (supplementary table S4, Supplementary Material online). The presence of a tetraploid consensus region within the read cover depth distribution (supplementary fig. S3B, Supplementary Material online) further supports this conclusion. The presence of a haploid consensus region within the read cover depth distribution of Pmeg (supplementary fig. S2B, Supplementary Material online) indicates the presence of haplotypes, produced by a high level of heterozygosity, as observed in the animal pathogen oomycete Saprolegnia parasitica (Jiang et al. 2013).
Analysis of the evolutionary relationship between eight randomly selected PGeneMs with a gene ratio of 0.5 revealed a pattern in which there was a single copy in Pmeg and two homeologous copies in Ppal (fig. 1C) consistent with postspeciation WGD in Ppal. The close relationships between the two Ppal HK PGeneMs (fig. 1C), compared with closely related Phytophthora species including Pmeg, support the hypothesis that Ppal, having gone through WGD, is functionally an autotetraploid. PCR amplification of one such PGeneM, SCE, from multiple Ppal isolates from cacao (Ali et al. 2016) using two homeolog-specific PCR primer sets suggested that the duplication is widespread if not ubiquitous within the species (fig. 1D). Slight variations in the PCR product sizes suggest some sequence variation among the isolates.
In the case of Ppal, the number of PGeneMs is 44,327 compared with the 13,155–26,584 reported in other Phytophthora species. Though WGD in Ppal has doubled its core PGeneM number, there was sequence divergence between duplicate copies and for some PGeneMs closely related copies could not been identified in the current version of the sequence assembly. Although the apparent sequence divergence and missing homeologs could simply result from problems with the sequence assembly or annotation, in some cases the observed gene loss/modification/creation might represent birth-and-death evolution (Nei and Rooney 2005) and neofunctionalization processes (Albertin and Marullo 2012). Duplicated genes have relaxed constraints on function, and can diverge to produce new phenotypes, which might confer a selective advantage over a long time period (Osborn et al. 2003).
Possible genomic processes contributing to the large number of predicted PGeneMs and the variation from the estimated core gene number include WGD, segmental duplication (SD), and TE driven gene duplication (TDD). SD of small gene blocks is common in Phytophthora, but has been attributed to ancient WGD events in the Phytophthora ancestor (Martens and Van de Peer 2010). Van Hooff et al. (2014) disputed this claim and instead suggested TEs as the source of these gene duplications. Large numbers of TE-related sequences are found in Phytophthora genomes (Tyler et al. 2006; Haas et al. 2009).
Amplification of Virulence-Related Genes and Genes of Unknown Function in Pmeg
As Pmeg does not show evidence of WGD, it is surprising that its genome size and predicted PGeneM numbers approach that of Ppal. Ppal and Pmeg also have similar PGeneM densities (table 1). Unlike general and HK gene families, many gene families related to plant-infection processes have a ratio of Pmeg PGeneM numbers to Ppal PGeneM numbers above 0.5 or even above 1.0 (fig. 1A). Phylogenetic analysis of the amino acid sequences of the NPP protein family demonstrated species-specific increases in PGeneM lineages subsequent to species separation (see supplementary fig. S4, Supplementary Material online). By comparison to Ppal which likely accumulated many of its expanded gene complement through a single event of WGD, the accumulation of PGeneM for virulence-related genes in Pmeg may have occurred in association with its development as a pathogen. Thus, Pmeg may have achieved increased PGeneM numbers through other genetic mechanisms. Enhanced gene duplication in Pmeg appears to be associated with disproportionate accumulation of untranscribed PGeneMs compared with Ppal. The percentage of untranscribed PGeneMs for gene families NPP, protease inhibitor, elicitin, CRN and RxLR were 71.5%, 48.3%, 20.0%, 66.4% and 71.5%, respectively, for Pmeg; whereas for Ppal, the percentage of untranscribed PGeneMs for the same families were 36.0%, 17.5%, 1.8%, 55.4% and 58.1%, respectively. A comprehensive tree including all the NPP proteins from both species indicates that most of the duplicated genes in Pmeg are not transcribed (supplementary fig. S4, Supplementary Material online).
The largest gains in species-specific PGeneM copy number in Pmeg over Ppal were in genes with unknown function. Pmeg has 26,940 PGeneMs (7,715 transcribed) with unknown function compared with 22,056 PGeneMs (11,025 transcribed) in Ppal (see supplementary Excel file S2, Supplementary Material online). Among the Pmeg PGeneMs with unknown functions, 15,216 (56.4%) PGeneMs have orthologs in Ppal (≥50% coverage and E ≤ 1.00e-10) and 19,063 (70.6%) PGeneMs have homologs in other Phytophthora species (E ≤ 1.00e-10). In Ppal, 15,429 (69.9%) PGeneMs have orthologs in Pmeg and 15,916 (72.1%) PGeneMs have homologs in other Phytophthora species. Thus, the majority of the PGeneMs of unknown function in both species show high sequence similarity with called genes in other species.
The early observations that Ppal has 9–12 comparatively small chromosomes whereas Pmeg has 5–6 extra-large chromosomes (Sansome et al. 1975; Brasier and Griffin 1979), fits well with the hypothesis that WGD occurred in Ppal whereas SD and/or TDD have occurred at a higher rate in Pmeg.
Transcription of the Expanded Genomes of Pmeg and Ppal
Transcripts from 14,915 Pmeg PGeneMs and 25,617 Ppal PGeneMs could be detected. If all PGeneMs with at least one read in the raw data were considered transcribed (see supplementary Excel file S2, Supplementary Material online) it would increase the number of potentially expressed PGeneMs to 17,531 and 28,534 in Pmeg and Ppal, respectively. Considering that both members of duplicated gene pairs in Ppal would be identified as transcribed by the sequence matching software, the 25,617 transcribed genes in Ppal could correspond to 12,808 gene pairs, comparable to the 14,915 transcribed PGeneMs in Pmeg. As the RNA-Seq experiment estimated transcript expression levels via 90-bp read counts, reads aligning similarly to PGeneM pairs were sorted randomly. This is a common problem with next-generation RNA-Seq methods (Ilut et al. 2012) and a problem in Ppal due to its WGD. Although the RNA-Seq can verify at least one member of a closely related PGeneM pair is expressed, it cannot easily verify that both copies are expressed.
Partial or WGD has the general effect of increasing gene expression levels on a per cell basis in proportion to the gene dosage conferred by ploidy level, as was shown for most genes in a euploid series (monoploid, diploid, triploid, and tetraploid) of maize (Guo et al. 1996). It is difficult to ascertain this in the case of Ppal because of the difficulty of separating homeolog expression levels using RNA-Seq analysis.
Transposons in the Pmeg and Ppal Genomes
Phytophthora genomes carry many TE-related sequences (Tyler et al. 2006; Haas et al. 2009). Both the Pmeg and Ppal genomes contain rich and diverse populations of TEs (supplementary Excel file S1, sheet 3 & 4: Pmeg/Ppal TE, Supplementary Material online). TE-related PGeneMs represent 15.0% (6,288; 174 expressed) and 13.6% (6,025; 281 expressed) of the Pmeg and Ppal PGeneMs, respectively. There are 550 Helitron helicase-related PGeneMs in Pmeg compared with 246 in Ppal, and 273 in P. infestans (Haas et al. 2009). Similarly there are 35 Ty3–gypsy retrotransposons identified in Pmeg compare with just 9 in Ppal. Many gene duplications might have resulted from transposon activity (Van Hooff et al. 2014).
Class I retrotransposons, such as Gypsy elements, create duplicate genes in new genomic positions through the reverse transcription of transcripts from expressed genes. Class II DNA-transposons, such as helitrons, are mobile DNA elements that utilize a transposase and single- or double-strand DNA breaks to replicate and transpose gene segments (Richard et al. 2008). In both cases, the duplicated gene copy does not usually carry its promoter and may die out (Mighell et al. 2000), although the associated gene sequences, “spare parts,” often remain in the genome. Functional retroposed genes are normally chimeric, either retroposed coding regions with a new regulatory sequence or retroposed coding regions encoding new protein fragments that are recruited from the targeted site resulting in new functions (Long et al. 2003). This possibility offers potential advantages to organisms carrying large loads of silenced genes or pseudogenes. We hypothesize that Phytophthora species, especially Pmeg, carries many “spare parts” (silent genes or pseudogenes) within their genomes which have potential, through gene conversion, rearrangement and activation under selective pressure, to support adaptability to future challenges.
Kasuga et al. (2012) reported that when P. ramorum infected the dead end host tanoak (Notholithocarpus densiflorus), silencing of many genes, including transposons, was suspended. In addition, extensive genome rearrangements and aneuploidy were induced in the pathogen (Kasuga et al. 2016). These results were considered support for the “epi-transposon hypothesis” which proposes that stress disrupts epigenetic silencing of TE which, when activated, stimulate genome diversification. Expression of effectors in the ascomycete Leptosphaeria maculans was also under epistatic control and their expression was associated with change to a pathogenic life style (Soyer et al. 2014).
Differences in Gene Family Repertoires of Pmeg and Ppal
Reciprocal BLASTp searches identified 4,267 PGeneMs in Pmeg and 2,740 in Ppal that do not have a homolog in the other species’ genome at a threshold of ≥50% sequence coverage (E ≤1e-10). Considering the WGD of Ppal, Pmeg had a much faster rate of PGeneM gain, resulting mostly in apparent silent or pseudogenes. Of the species-specific PGeneMs, only 689 of the 4,267 PGeneMs were transcriptionally active in Pmeg, whereas only 981 of the 2,740 PGeneMs were transcriptionally active in Ppal (see supplementary Excel file S1, sheet 5 & 6: Pmeg/Ppal specific PGeneM, Supplementary Material online). Among these transcribed species-specific PGeneMs, the largest category was PGeneMs of unknown function (542 in Pmeg; 653 in Ppal) followed by RxLR PGeneMs (37 in Pmeg; 51 in Ppal). Other infection-related differences include six CRNs, in Pmeg and five elicitins in Ppal (see supplementary Excel file S1, sheet 5 & 6: Pmeg/Ppal specific PGeneM, Supplementary Material online). These PGeneMs may be interesting targets for future studies to understand their role in virulence.
PGeneM Density in Different Gene Families
In general, Phytophthora genomes are organized into blocks having conserved PGeneM order, high PGeneM density and low repeat content, separated by regions in which PGeneM order is not conserved, PGeneM density is low and repeat content is high (Haas et al. 2009). The conserved blocks represent almost 90% of the core orthologous PGeneMs for three reported Phytophthora species (Haas et al. 2009). The average length of Pmeg scaffolds harboring HK PGeneMs ranged between 14,800 and 23,759 bp in length, with a gene density of 2.17–2.63 kb/gene and an average GC content of 50.07–51.21%. Scaffolds harboring virulence-related PGeneMs ranged between 6,917 and 14,216 bp in length with a gene density of 2.04–2.63 kb/gene and an average GC content of 44.62–48.79% (table 2). In Ppal there was no distinction between the average scaffold size harboring HK PGeneMs (6,466–9,198 bp) and virulence-related PGeneMs (5,686–10,032 bp). However, the average GC content of scaffolds harboring HK PGeneMs (50.41–51.39%) was also higher than the scaffolds harboring virulence-related PGeneMs (46.24–50.28%) (table 2). Conversely, the average PGeneM density of Ppal was slightly higher in the scaffolds harboring HK PGeneMs (2.0–2.17 kb/gene) compared with the scaffolds harboring virulence-related PGeneMs (2.17–2.68 kb/gene) (table 2). In fungi and plants, conserved genetic regions are GC-rich compared with dispersed repetitive regions (Meyers et al. 2001; Muñoz et al. 2015). The Pmeg and Ppal HK PGeneMs were associated with GC-rich scaffolds whereas virulence-related PGeneMs were associated with GC-poor scaffolds. The codon preferences of oomycete genes are biased toward higher GC content codons, and this is reflected in the tRNA gene composition (Tripathy and Tyler 2006). The codon distribution of conserved genes reflects this bias (Prat et al. 2009; De La Torre et al. 2015). In rapidly evolving genes, the evolutionary forces driving diversification outpace the forces driving codon optimization. As a result, the codon composition of rapidly evolving genes drifts away from the host organism’s codon bias.
Table 2.
Average Length, Gene Density, and GC Content of Scaffolds Harboring Putative Gene Models from Different Gene Families of Phytophthora megakarya and Phytophthora palmivora
| Gene Family |
P. megakarya |
P. palmivora |
|||||
|---|---|---|---|---|---|---|---|
| Average Scaffold Length | Gene Density (kb per gene)a | Average GC Content (%) | Average Scaffold Length | Gene Density (kb per gene) | Average GC Content (%) | ||
| Housekeeping | |||||||
| Amino acyl tRNA ligases | 23,759 | 2.17 | 51.21 | 6,719 | 2.13 | 51.39 | |
| 40s ribosomal proteins | 14,803 | 2.17 | 50.99 | 9,198 | 2.00 | 50.71 | |
| Translation initiation factors | 18,358 | 2.63 | 50.54 | 8,466 | 2.17 | 50.41 | |
| Elongation factors | 21,475 | 2.38 | 50.07 | 6,466 | 2.04 | 51.29 | |
| General | |||||||
| Alcohol dehydrogenases | 11,820 | 2.38 | 49.05 | 8,174 | 2.08 | 49.75 | |
| MFS transporters | 15,585 | 2.56 | 48.80 | 7,284 | 2.44 | 49.50 | |
| Virulence-related | |||||||
| Pectinases | 13,247 | 2.63 | 48.87 | 5,686 | 2.17 | 49.71 | |
| Proteases | 13,219 | 2.38 | 48.84 | 8,359 | 2.33 | 49.32 | |
| Elicitins | 14,216 | 2.13 | 48.79 | 7,777 | 2.22 | 50.28 | |
| CRNs | 8,281 | 2.04 | 46.84 | 8,770 | 2.50 | 47.23 | |
| NPPs | 6,917 | 2.22 | 45.58 | 9,458 | 2.63 | 47.49 | |
| RxLRs | 8,917 | 2.49 | 44.62 | 10,032 | 2.68 | 46.34 | |
Density of all the PGeneMs in the scaffolds harboring one or more PGeneMs from different gene families. kb/gene is scaffold length (kb) divided by the number of genes per scaffold.
Synteny and Genome Expansion
Synteny between Pmeg and Ppal was analyzed with MUMmer using the NUCmer setting that compares the contigs from both genomes at the nucleotide level. Ppal contigs were queried using Pmeg contigs as the reference sequence. Contigs (≥10 kb) with high sequence identity resulted in a straight line graph without any parallel shift (fig. 2A). In addition, there were 392 points of secondary alignment (fig. 2A), a likely consequence of WGD in Ppal. Synteny analysis within the Ppal genome showed 74.54 Mb of secondary hits suggesting 69.38% of the genome has a duplicate homologous region (supplementary fig. S5B, Supplementary Material online). Only 8.41 Mb or 8.31% of the Pmeg genome showed secondary hits (supplementary fig. S5A, Supplementary Material online).
Fig. 2.—

Synteny- and repeat-driven genome expansion between the P. megakarya and P. palmivora genomes. (A) MUMmer alignment dot plot of P. megakarya and P. palmivora contigs (≥10 kb) (NUCmer parameters: breaklen = 200, maxgap = 90, mincluster = 65, minmatch = 20). Red dots represent positive strand alignments whereas blue dots represent negative strand alignments. The left panel is an enlargement of the bottom-left corner of the right panel. (B) Conserved gene order and genome expansion across two homologous contigs of the P. megakarya and P. palmivora genomes. Phytophthora megakarya contig 180 and P. palmivora contig 1 were aligned using Lasergene’s MegAlign Pro and gene model locations (indicated by block arrows) were determined manually. Red bands represent homologous regions (≥65% nucleotide sequence identity in either orientation). (C) LALIGN pairwise sequence alignment (Huang and Miller 1991) of the two contigs showing repeat-driven expansion in three different regions of the contigs, in addition to a 2.3-kb insertion in the P. megakarya contig. (D) Sequence similarities among the PGeneMs within the two contigs and their expression profiles as determined by RNA-Seq analysis. Amino acid identities between closely related PGeneMs (>70% identity) were inferred using guide trees generated by WebPRANK (http://www.ebi.ac.uk/goldman-srv/webprank/, last accessed 16 February 2017). Branch lengths in the tree represent the number of nucleic acid substitutions per site. RNA-Seq read counts for zoospore and in planta libraries were normalized by read counts in the mycelium library and values were LOG-transformed to linearize the data. The heat map was generated using CIMminer (http://discover.nci.nih.gov/cimminer, last accessed 16 February 2017). Underlying % Identity Matrix data are shown in the supplementary Excel file S1, sheet 7: %IM Pmeg-Ppal contig, Supplementary Material online.
Figure 2A shows the alignment of a set of contigs ≥10 kb from Ppal representing 11.35 Mb against homologous Pmeg contigs ≥10 kb representing 16.4 Mb. The aligned regions display a 44% genome expansion in Pmeg. A comparison involving Ppal contig 1 (30.7 kb) and Pmeg contig 180 (36.1 kb) demonstrates possible tandem repeat-driven genome expansion in both the species, and an increase of five PGeneMs (fig. 2B) in Pmeg along with a net increase of 5.4 kb compared with Ppal. The contigs were neither highly conserved nor highly diverged as the similarity index (Nucleotide Match/[Match + Mismatch + Gaps]) between Ppal contig 1 and Pmeg contig 180 was 0.46. There was co-linearity between gene orthologs within these contigs; eight PGeneMs in the Ppal contig had unique matching syntenic Pmeg PGeneMs with a nucleotide sequence identity that ranged from 80.5% to 89.5%. There was also high nucleotide sequence similarity (>70%) among the 11 PGeneMs within the two contigs (fig. 2D; see supplementary Excel file S1, sheet 7: %IM Pmeg-Ppal contig, Supplementary Material online) resulting from multiple tandem duplications (fig. 2C) that disrupt the synteny between the contigs. Tandem repeats of paralogs within gene families were common in both the species (see supplementary Excel file S2, Supplementary Material online). The transcript levels determined by RNA-Seq analysis indicated conserved expression levels between species for some pairs in figure 2B (e.g., M17 and P12) whereas other orthologous pairs showed different expression levels (e.g., M9 and P5) (fig. 2D). Similar results were obtained for a second pair of contigs (supplementary fig. S6, Supplementary Material online). The similarity index between Ppal contig 42 and Pmeg contig 299 was 0.52. There were several identified PGeneMs, especially in the Pmeg contig 299, where the PGeneMs were not identified by the gene predictors in the opposite species, despite gene “footprints” being detected manually (see black arrows in the supplementary fig. S6A, Supplementary Material online); these undetected footprints may indicate gene sequences in the process of divergence and loss. In P. infestans, massive genome expansion has resulted from a proliferation of repetitive elements that is largely confined to the nonconserved intergenic regions (Haas et al. 2009). As a result, P. infestans has a similar number of PGeneMs compared with P. sojae and P. ramorum despite having a genome size expanded by almost 3-fold. Co-linearity between orthologs was also preserved between P. sojae and P. ramorum, with interruptions by gene sparse regions containing transposons and PGeneMs associated with plant-infection (Tyler et al. 2006).
Relationships among Expression Profiles
To validate PGeneM expression levels as measured by RNA-Seq, RT-qPCR was conducted across replicated samples of mycelium, zoospores, and infected pod husks for both species. Among the 11 putative HK PGeneMs selected based on the RNA-Seq read counts from mycelium, zoospores and infected pod husks samples (supplementary Excel file S1, sheet 8: HK genes tested, Supplementary Material online), a transmembrane protein (PmTP/PpTP) showed uniform expression levels in different life stage-specific RNA samples for both the species as shown by RT-qPCR analysis (results not shown) and was used to normalize the RT-qPCR data. For the RT-qPCR analysis of the Ppal PGeneMs, homeolog-specific primers were designed to avoid the cumulative expression conferred by duplicate PGeneMs. Eleven out of the 13 and 17 out of 21 PGeneMs selected on the basis of RNA-Seq results showed correlation coefficients greater than 0.80 between the RNA-Seq and RT-qPCR results for Pmeg and Ppal, respectively (fig. 3 and supplementary Excel file S1, sheet 1: RNA-Seq-RTqPCR comparison, Supplementary Material online). In general, PGeneMs with limited treatment response variation or low RNA-Seq read alignments across treatments gave lower correlations between RNA-Seq and RT-qPCR results. Five Ppal homeologous pairs were included in the RNA-Seq/RT-qPCR analysis (fig. 3; supplementary Excel file S1, sheet 1: RNA-Seq-RTqPCR comparison, Supplementary Material online). As expected, RNA-Seq could not distinguish the transcript levels of the individual homeologs, and near identical profiles were assigned by the software. On the other hand, the RT-qPCR analysis identified strong differences in the expression levels between the individual pairs of homeologs. In particular the four Hsp homeologs showed 20- to 75-fold differences in expression levels between pairs (fig. 3; supplementary Excel file S1, sheet 1: RNA-Seq-RTqPCR comparison, Supplementary Material online). Differential expression of homeologous gene pairs is common in polyploid plants (Liu and Adams 2007), the result of relaxed evolutionary constraints on duplicated genes. Determining the full extent to which this occurs in Ppal will require further study.
Fig. 3.—

Heatmap comparison of RNA-Seq and RT-qPCR data. Fourteen P. megakarya genes and 20 two P. palmivora genes were identified by RNA-Seq analysis as being differentially expressed in mycelium, zoospores, and in planta. Relative mRNA expression levels were quantified relative to that of the housekeeping PGeneM PmTP/PpTP by the 2−ΔΔCt method (Livark and Schmittgen 2001). Both the global normalized value of RNA-Seq and the relative mRNA expression levels were LOG10-transformed to linearize the data, then the heat map was created using CIMminer (http://discover.nci.nih.gov/cimminer). Gene names with similar superscript letters are homeologs. In planta RNA-Seq and all RT-qPCR results are based on three biological replicates. Underlying data are shown in the supplementary Excel file S1, sheet 1: RNA-Seq-RTqPCR comparison, Supplementary Material online.
To compare gene expression profiles between the two species, we used neural network software to produce SOMs (Kohonen 1982, 1990) from the RNA-Seq data. This enabled us to identify common patterns of gene expression within and between the two species without matching genes one-to-one between the two species. SOM is an unsupervised classification technique that reduces the dimension of data through the use of self-organizing neural networks, not imposing a priori class assumptions, but allowing the data to determine how PGeneMs should be classified. The user inputs the number of classes to be generated which in this case was chosen giving consideration for the number of treatments, number of samples, and allowing for differences in gene expression levels. SOMs reduce dimensionality by producing a feature map that objectively plots the similarities of the data, grouping similar data together. SOMs learn to classify input vectors according to how they are grouped in the input space, and then learn to recognize neighboring sections of the input space. Thus, SOMs learn both the distribution and topology of the input vectors they are trained on.
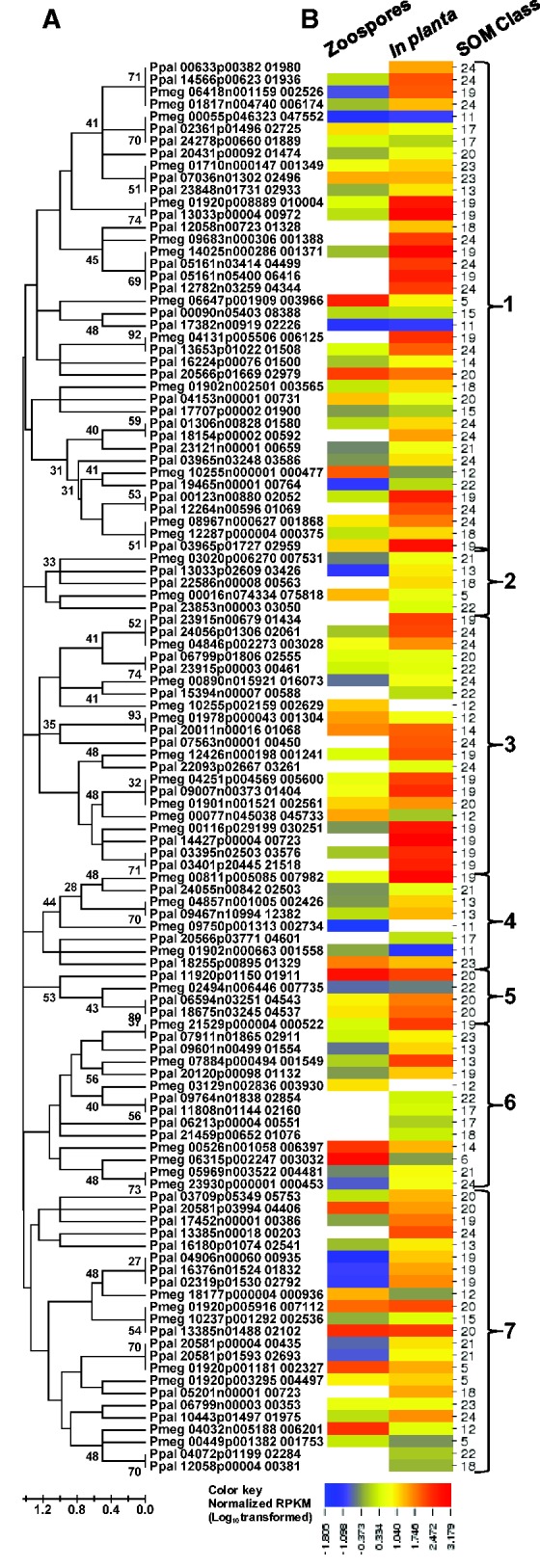
Pmeg and Ppal PGeneMs were classified into 24 multivariate and nonlinear SOM classes (fig. 4A). Among the 24 SOM classes generated, classes 6, 11, 12, 14, 15, 16, 17, 18, 21, and 22 showed constitutive expression across all the RNA-Seq libraries with expression level being the primary divider. Class 1 contained the most highly expressed PGeneMs with more than 30% of these being ribosomal proteins. Classes 7, 13, 19, and 23 contained PGeneMs that were induced in mycelium and zoospores but repressed in planta. Classes 1, 20, and 24 were induced in zoospores only. Classes 8 and 9 were induced in zoospores and in planta, whereas classes 2, 3, 4, 5, and 10 were induced in planta only (fig. 4A). The last two groups of SOM classes (i.e., classes 2, 3, 4, 5, 8, 9, and 10) which contained 2,092 and 3,229 PGeneMs from Pmeg and Ppal, respectively, are of interest in terms of plant pathogenicity.
Fig. 4.—

SOM classification of P. megakarya and P. palmivora gene expression profiles based on the RNA-Seq count data. (A) P. megakarya and P. palmivora gene models were classified into 24 different multivariate, nonlinear SOM classes based on their expression patterns (Kohonen 1982, 1990). (B) PCoA of the log10 transformed RNA-Seq count data of the 20 most highly expressed gene models from the 24 SOM classes, plotted on a 3D scatter plot.
Principal coordinate analysis was carried out using log10-transformed RNA-Seq count data of the 20 most highly expressed PGeneMs from each of the 24 SOM classes. A 3D scatter plot comparison of principal coordinates 1, 2, and 3 is presented (fig. 4B). These coordinates account for 53.8%, 24.6%, and 13.34% of the data variation, respectively. As expected, PGeneMs from each of the SOM classes clustered separately, according to transcript level and tissue specificity.
The distribution of PGeneMs among these SOM classes was examined for HK and Phytophthora virulence-related gene families. RxLR, NPP, and pectinase PGeneMs were primarily induced in planta (fig. 5). Some, virulence-related gene families, namely elicitins and CRNs, did not show a preferential pattern of expression. As expected, HK PGeneMs like those encoding elongation factors, amino-acyl tRNA ligases, and 40S ribosomal proteins (40S RP) were grouped among the constitutively expressed SOM classes (fig. 5).
Fig. 5.—

Distributions of gene models from selected gene families among the 24 SOM classes. SOM classes are the same as in figure 4. Expression levels were based on the average read counts of each RNA-Seq library. Gene families included are RxLR, necrosis-inducing proteins (NPP), pectinases, elicitins, crinklers (CRN), elongation factors (EF), amino-acyl tRNA ligases, and 40S ribosomal proteins (40S RP). The percentage of genes from each gene family within a SOM class was used to generate the heat map using CIMminer (http://discover.nci.nih.gov/cimminer).
Potential Phytophthora Virulence-Related Genes
Ppal has a wide host range while Pmeg mainly causes disease on cacao where it is more virulent than Ppal (Bowers et al. 2001; Ali et al. 2016). An interesting question is whether genes involved in host interactions have diverged between the two species since their evolutionary separation. The WGD in Ppal may have relaxed functional constraints on genes, allowing divergence in sequence, function, and/or regulation. Ppal and Pmeg secreted proteins are candidates for mediators of cacao interactions (Kamoun 2006). There were 3,757 (1,779 transcribed) and 3,865 (2,633 transcribed) predicted proteins within the secretomes of Pmeg and Ppal, respectively (see supplementary Excel file S1, sheet 9 & 10: Pmeg/Ppal secretome, Supplementary Material online). The ratios between the predicted secretome gene numbers from the two species (0.97; 0.67 transcribed) are notably above the 0.5 ratio observed for HK genes due to WGD in Ppal.
Plant pathogenic Phytophthora species are considered hemibiotrophs, initially deriving nutrition from living tissue (biotrophic stage), but switching to a necrotrophic lifestyle once infection is established (necrotrophic stage). Pmeg and Ppal are expected to produce gene products enabling them to evade/suppress the plant’s defense responses during their biotrophic phase and produce gene products capable of killing plant tissue during their necrotrophic phase. Phytophthora species secrete two broad categories of effector proteins that alter host physiology and facilitate colonization: Apoplastic proteins accumulate in the plant intercellular space and cytoplasmic proteins are translocated into the plant cell (Whisson et al. 2007; Dou, Kale, Wang, Jiang, et al. 2008). Apoplastic proteins include enzymes such as proteases, lipases, and glycosyl hydrolases that attack plant tissues; enzyme inhibitors to protect against host defense enzymes; lipid transfer proteins (elicitins); and necrotizing toxins such as the NPPs and PcF-like small cysteine-rich proteins. Among the cytoplasmic proteins, the most notable are the RxLR and CRN effectors. Both apoplastic and cytoplasmic effectors can interact with plant defense receptors (Bozkurt et al. 2012).
Table 3 summarizes PGeneM numbers for a variety of secreted hydrolytic enzymes, effectors, and other proteins encoded by the Pmeg and Ppal genomes, compared with P. sojae and P. ramorum. Within each gene family in each species, there were large differences between the transcribed PGeneM numbers versus the total PGeneM numbers. This difference is higher in Pmeg supporting the hypothesis that TE-driven gene multiplication in Pmeg may have created many nontranscribed gene copies. Despite lacking WGD, Pmeg compared with Ppal has nearly the same number or in some cases a larger number of PGeneMs for gene families like RxLRs, elicitins, protease inhibitors, CRNs, NPPs, PcF-like small cysteine-rich proteins, glucanase inhibitors, and proteases; in all but a few cases Pmeg has fewer transcribed PGeneMs than Ppal, but more than a 0.5 ratio (table 3). Although the gene prediction software used in this study differed from earlier Phytophthora studies, Pmeg and Ppal have many larger gene families compared with P. sojae and P. ramorum: Proteases, glycosyl hydrolases, ABC super-family, protease inhibitors, NPPs, CRNs, and RxLRs (table 3). There were 1,181 and 991 RxLR PGeneMs for Pmeg and Ppal, respectively, which is dramatically larger than the estimated 350–395 RxLR PGeneMs in P. sojae and P. ramorum (table 3).
Table 3.
Potential Infection-Related Putative Gene Models in the Phytophthora megakarya and Phytophthora palmivora Genomes
| Gene Family |
P. megakarya |
P. palmivora |
Core Orthologa |
P. sojae | P. ramorum | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | TSb | TS-SPc | Total | TSb | TS-SPc | Total | TSb |
||||
| Pmeg | Ppal | ||||||||||
| Proteases, all | 500 | 306 | 76 | 644 | 532 | 106 | 233 | 226 | 224 | 282 | 311 |
| Serine proteases | 177 | 105 | 28 | 196 | 167 | 37 | 78 | 76 | 76 | 119 | 127 |
| Metalloproteases | 62 | 42 | 7 | 91 | 75 | 12 | 32 | 71 | 71 | 71 | 86 |
| Cysteine proteases | 78 | 40 | 11 | 73 | 62 | 21 | 28 | 27 | 27 | 67 | 74 |
| Aspartic protease | 49 | 5 | 1 | 37 | 7 | 3 | 3 | 3 | 3 | 13 | 14 |
| Glycosyl Hydrolases | 322 | 261 | 105 | 508 | 458 | 128 | 195 | 192 | 193 | 190 | 173 |
| Chitinases | 7 | 7 | 4 | 2 | 2 | 2 | 1 | 1 | 1 | 5 | 2 |
| Cutinases | 9 | 5 | 5 | 7 | 6 | 1 | 1 | 1 | 1 | 16 | 4 |
| Glucanase inhibitor | 16 | 9 | 8 | 5 | 5 | 4 | 4 | 4 | 4 | NK | NK |
| Pectinases, all | 67 | 43 | 31 | 100 | 72 | 52 | 36 | 33 | 35 | NK | NK |
| Pectin (methyl)-esterase | 17 | 9 | 8 | 21 | 14 | 14 | 7 | 7 | 7 | 19 | 15 |
| Polygalacturonase | 22 | 15 | 12 | 34 | 31 | 20 | 13 | 12 | 13 | NK | NK |
| Pectate lyase | 21 | 15 | 12 | 27 | 19 | 19 | 12 | 12 | 12 | 43 | 41 |
| Lipases | 53 | 38 | 16 | 105 | 93 | 311 | 30 | 28 | 30 | 171 | 154 |
| ABC super-family | 462 | 297 | 27 | 913 | 440 | 63 | 161 | 157 | 158 | 134 | 135 |
| PDR | 62 | 45 | 7 | 113 | 89 | 8 | 35 | 34 | 35 | 45 | 46 |
| MRP | 55 | 26 | 2 | 85 | 60 | 9 | 24 | 24 | 24 | 23 | 22 |
| Phytotoxic peptides | |||||||||||
| NPP | 109 | 31 | 22 | 75 | 48 | 36 | 27 | 15 | 24 | 29 | 40 |
| PcF | 6 | 0 | 0 | 3 | 2 | 2 | 0 | 0 | 0 | 19 | 4 |
| Protease inhibitor, all | 60 | 31 | 17 | 57 | 47 | 34 | 22 | 20 | 21 | 22 | 19 |
| Kazal | 30 | 14 | 6 | 23 | 19 | 13 | 7 | 6 | 6 | 15 | 12 |
| PAMPs | |||||||||||
| Elicitins | 50 | 40 | 33 | 54 | 53 | 45 | 26 | 23 | 26 | 57 | 48 |
| Transglutaminase | 14 | 9 | 7 | 43 | 28 | 16 | 10 | 10 | 10 | NK | NK |
| Effectors | |||||||||||
| CRN | 152 | 51 | 4 | 137 | 61 | 4 | 30 | 19 | 22 | 40 | 8 |
| RxLR | 1,181 | 336 | 336 | 991 | 415 | 414 | 106 | 43 | 56 | 395 | 350 |
Note.—NK, not known.
Core orthologous genes shared by Ppal and Pmeg were estimated based on bidirectional BLASTp hits. To be considered as an ortholog, BLASTp should produce an alignment of at least 50% of the sequence E-value less than 1e-10.
Transcribed sequence. Genes with >10 reads, either in mycelia, zoospores, or in planta are considered transcribed.
Transcribed genes with predicted signal peptide sequence (secretomes).
Pectinases
During infection, pathogens produce a range of cell wall-degrading enzymes including glycosyl hydrolases and pectinases. Pectinases degrade pectin which is a component of the plant primary cell wall and middle lamella. Pectinases are divided into three types by mode of action: Polygalacturonases, pectin methyl esterases, and pectate lyases (Yadav et al. 2009). There were 100 PGeneMs for predicted pectinases in Ppal of which 72 were expressed. Pmeg has 67 pectinase PGeneMs of which 43 were expressed (table 3). Among the expressed pectinases, 31 and 52 proteins from Pmeg and Ppal, respectively, have predicted secretion signal peptides. Among the 167 pectinase genes, 36 were identified as core orthologs between Ppal and Pmeg. Of these, 33 were transcribed in Pmeg and 35 were transcribed in Ppal. This indicates extensive evolutionary divergence between the two species. All of the 115 expressed pectinases could be grouped into seven major phyletic groups, each including PGeneMs from both species (fig. 6A). As expected, most expressed pectinases were highly induced in planta. Forty Ppal pectinases were induced in planta compared with 27 in Pmeg, (fig. 6B) presumably reflecting the WGD. Ppal also exhibited a larger number of transcribed PGeneMs for other plant cell wall-degrading enzymes (table 3); this may influence its much broader host range.
Fig. 6.—
Evolutionary relationships and transcription profiles of 115 transcribed pectinase gene models from P. megakarya and P. palmivora. (A) Amino acid sequences were aligned using MUSCLE (Edgar 2004) and evolutionary relationships were inferred using the Neighbor-Joining method (Saitou and Nei 1987) with bootstrap (100 replicates). Branch lengths represent the number of amino acid differences per sequence. Branches are labeled with bootstrap values. There were a total of 30 informative positions in the final data set. Evolutionary analyses were conducted in MEGA5 (Tamura et al. 2011). (B) For the relative transcription profiles, RNA-Seq read counts for zoospore and in planta libraries were normalized by read counts in the mycelium library and values were LOG10-transformed. The heat map was generated using CIMminer (http://discover.nci.nih.gov/cimminer). Self-organizing classes were as in figure 4. Large bold numbers indicate phyletic groups. White blocks indicate no detectable transcription.
Necrosis-Inducing Phytophthora Proteins
NPPs within Phytophthora are highly diverse (Tyler et al. 2006) and exist in higher numbers compared with other plant pathogens (Fellbrich et al. 2002; Qutob et al. 2002; Tyler et al. 2006). The NPPs tend to be more highly expressed in planta. Several studies have demonstrated NPPs are induced in planta during the shift from the biotrophy to necrotrophy in association with plant cell death (Fellbrich et al. 2002; Qutob et al. 2002). NPPs have expanded in Pmeg, but only 31 out of 100 PGeneMs were transcribed, whereas 48 out of 75 were transcribed in Ppal. Among the expressed NPPs, 22 and 36 proteins from Pmeg and Ppal, respectively, exhibited signal peptides for extracellular secretion (table 3). Most of the transcribed genes in both species were highly induced in planta (supplementary fig. S7, Supplementary Material online). Though NPPs are quite diverse in these two species, there was no clade specific to either of the species (supplementary fig. S7, Supplementary Material online).
CRN Effectors
CRN effectors are a major class of well-studied cytoplasmic effectors showing extensive expansion in all sequenced Phytophthora species (Torto et al. 2003). Pmeg displays an enormously increased number of CRN genes (152) whereas Ppal has 137, but only 30 of these are shared as core-orthologs. Both species have larger families than observed in any other Phytophthora species, so far. The 51 and 61 functional (i.e., transcribed) CRNs from Pmeg and Ppal, respectively, show high sequence diversification and mostly constitutive expression (supplementary fig. S8, Supplementary Material online). Similar constitutive expression of CRN genes in mycelia and in planta was reported for P. infestans (Haas et al. 2009). CRNs from P. sojae suppress cell death induced by pathogen-associated molecular patterns (PAMPs) or other elicitors (Song et al. 2013). The majority of P. capsici CRN effectors were localized to plant nuclei, indicating that they target and perturb host nuclear processes when exerting effector activity (Stam et al. 2013). Most of the Pmeg and Ppal CRN effectors were not identified as having predicted signal peptides for extracellular secretion; however, this is a common feature of CRNs in other oomycete species due to unusually high variation in the signal peptide sequences that interferes with effective prediction.
Elicitins
Elicitins are a diverse family of secreted proteins produced by Phytophthora and Phytopythium species. Many induce hypersensitive cell death in plants (Yu 1995). Although elicitins are recognized by certain plants as PAMPs, their intrinsic function is to bind lipids and sequester sterols, presumably fulfilling a biological function in Phytophthora and Pythium species which cannot synthesize sterols (Ponchet et al. 1999). The numbers of total and expressed elicitins are similar in Pmeg and Ppal (table 3). Among the expressed elicitins, 33 and 45 proteins from Pmeg and Ppal, respectively, have signal peptides for secretion. Elicitins represent a diverse gene family both phylogenetically and based on expression patterns (supplementary fig. S9, Supplementary Material online). Elicitins showed different expression profiles in Pmeg and Ppal, with 6 Pmeg and 13 Ppal elicitins being induced in planta (28%) whereas 12 Pmeg and 6 Ppal elicitins were induced in zoospores. The majority of the remaining elicitins are expressed constitutively (fig. 5 and supplementary fig. S9B, Supplementary Material online).
RxLR Effectors
The largest and most diverse family of virulence-related genes identified in the Pmeg and Ppal genomes is the superfamily of RxLR effectors, also known as Avh genes. They possess signal peptides and N-terminal amino acid motifs (RxLR and dEER) required for targeting into host cells together with diverse, rapidly evolving carboxy-terminal effector domains (Tyler et al. 2006; Jiang et al. 2008). These C-terminal domains exhibit virulence activities such as suppression of host cell death (Bos et al. 2006; Dou, Kale, Wang, Chen, et al. 2008; Wang et al. 2011). Using HMM searches, 1,181 and 991 RxLR PGeneMs were detected, of which, 336 and 415 matched transcripts in Pmeg and Ppal, respectively (see supplementary Excel file S1, sheet 11 & 12: Pmeg/Ppal RxLRs, Supplementary Material online). These are the largest numbers of putative RxLRs detected in any Phytophthora species so far. In the case of the RxLR PGeneMs, the consequences of WGD on gene family diversity in Ppal are obvious. Ppal has 751 (332 expressed) RxLR PGeneMs that show high sequence similarity to one or more of its other RxLRs (% identity ≥ 70; E ≤ 1.00e-10). Pmeg has only 452 (122 expressed) RxLR PGeneMs that show high sequence similarity to one or more of its other RxLR PGeneMs (% identity ≥ 70; E ≤ 1.00e-10). If these are accounted for along with the orthologs shared by both species, Pmeg carries 620 (174 expressed) unique RxLR PGeneMs compared with 162 (58 expressed) unique RxLR PGeneMs in Ppal. The variation in RxLR amino acid sequences prevented direct determination of evolutionary relationships using the Neighbor-Joining method (Saitou and Nei 1987). Instead, we generated a percent identity matrix of the combined 751 Pmeg- and Ppal-expressed RxLRs (see supplementary Excel file S1, sheet 13: %IM Pmeg & Ppal RxLRs, Supplementary Material online) using the sequence alignment program Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/, last accessed 16 February 2017). Ppal exhibited 248 expressed RxLRs having ≥70% sequence identity with at least one paralog. In contrast Pmeg RxLRs were highly diverse; only 96 expressed RxLRs had ≥70% sequence identity with at least one paralog. There was only one pair of expressed RxLRs sharing ≥70% sequence identity between Pmeg and Ppal. Out of the 751 transcribed RxLR PGeneMs, 245 and 228 from Pmeg and Ppal, respectively, fell within the SOM classes exhibiting elevated expression in both zoospores and in planta or in planta alone (fig. 5). SOM classes 4, 5, 9, and 10 represent the highly transcribed PGeneMs and 70 Pmeg RxLRs and 59 Ppal RxLRs were found in these SOM classes, making them targets for future studies concerning virulence. The 129 RxLRs were grouped into 25 phyletic groups (supplementary fig. S10, Supplementary Material online), though these groups were not statistically significant. Most phyletic groups exhibited a similar distribution of PGeneMs from both species.
In contrast to the genes encoding the core proteome, RxLR genes typically occupy a gene sparse and repeat-rich genomic environment (Tyler et al. 2006; Haas et al. 2009). The mobile elements contributing to the dynamic nature of these repetitive regions may enable recombination events resulting in the higher rates of gene gain and loss observed for effectors (Haas et al. 2009). In addition to the RxLR effectors, there were an additional 324 (82 transcribed) and 241 (105 transcribed) RxLR protein-like PGeneMs in Pmeg and Pal, respectively (see supplementary Excel file S2, Supplementary Material online). These PGeneMs exhibited sequence similarities with published RxLR effectors, but they were not detected by HMM searches. These PGeneMs may correspond to pseudogenes that have lost the RxLR–dEER domain or N-terminal signal peptide or they may be incomplete due to the partial genome assembly.
Conclusions
Pmeg and Ppal present a unique example of postspeciation divergent evolutionary trajectories of two pathogens capable of parasitizing the same host, Ppal having an extremely broad host range and Pmeg having a narrow host range in Africa. The pathogenic potential of Ppal appears to have been augmented via WGD or tetraploidy. In contrast, extensive gene duplication especially among virulence-related gene families, possibly mediated by TEs, has expanded the pathogenic potential of Pmeg. Many virulence-associated genes in both species were highly induced in planta. The Pmeg–cacao interaction represents a recent encounter of less than 200 years (Zhang and Motilal 2016). Thus, the adaptive events resulting in the virulence of Pmeg on cacao likely occurred prior to this encounter. A greater understanding of the origins of Pmeg and its genetic diversity, together with characterization of its native host interactions, will be important if these adaptive events are to be understood.
It is unclear how long the Ppal–cacao interaction has existed. Some estimates have placed the center of origin of Ppal in Southeast Asia (Mchau and Coffey 1994). If so, the interaction of Ppal with cacao is also relatively recent. Ppal causes significant disease on a wide range of host plants, and individual isolates vary in their virulence on cacao (Ali et al. 2016). Given that Ppal has numerous hosts, the selective pressure on Ppal to adapt to cacao and to compete effectively with Pmeg may be low. On the other hand, Ppal with its WGD seems to have the mechanisms to adapt and it will be interesting to determine whether these mechanisms have contributed to its wide host range.
Considering what we currently know about their genomes, we propose a working hypothesis to guide future dissection of the Pmeg and Ppal interactions with cacao. Ppal, through doubling of its gene complement through WGD and subsequent gene diversification, has consequently expanded its genetic capacity for nutrient acquisition and breakdown of complex structures, for example cell walls. This capacity makes Ppal vigorous and fast growing, and capable of attacking many hosts, even without extended coevolution. Ppal is thus a generalist, not specifically adapted to cacao and perhaps not to most hosts. As a result, although Ppal can attack cacao directly, it gains additional benefit from wounds where its many hydrolytic enzymes have direct access to host nutrients and structures. Without wounds, these same enzymes may generate elicitors of plant defense slowing down the infection process and reducing infection success. The many virulence-associated gene products produced by Ppal are likely less diverse due to the limitations of genome doubling. On the other hand, having two copies of all critical virulence-associated gene products also creates a potential advantage as the extra copies are presumably free to evolve, enabling adaptation to a wider diversity of host interactions.
In Pmeg, on the other hand, adaptation has been enabled through extensive amplification of specific genes sets including many virulence determinants. It is unclear whether this adaptation may have occurred over a prolonged time through interactions with an unknown host or over a short time period during some unique event, for example, the availability of a new host, cacao. An intriguing possibility is suggested by the genomic-destabilizing response of P. ramorum to infection of the novel host tanoak. Perhaps, epigenetic derepression of transposon activity and effector expression triggered by the stresses of the new encounter with cacao resulted in extensive gene amplifications and activations, which through selection resulted in a new highly adapted pathogen.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was funded by the USDA/ARS and The Pennsylvania State University’s College of Agricultural Sciences, The Huck Institutes of the Life Sciences, Penn State Endowed Program in Molecular Biology of Cacao. Special thanks are given to the Hershey Company which supported the sequencing of the Phytophthora megakarya genome through a gift to the Penn State Endowed Program in Molecular Biology of Cacao. This work was supported by a grant from the Hershey Company to MJG, The Pennsylvania State University's College of Agricultural Sciences, The Huck Institutes of the Life Sciences, the Penn State Endowed Program in Molecular Biology of Cacao and by the USDA National Institute of Food and Agriculture Hatch project 1003147. References to a company and/or product by the USDA are only for the purposes of information and do not imply approval or recommendation of the product to the exclusion of others that may also be suitable. USDA is an equal opportunity provider and employer.
Literature Cited
- Akrofi AY, Amoako-Atta I, Assuah M, Asare EK. 2015. Black pod disease on cacao (Theobroma cacao, L) in Ghana: spread of Phytophthora megakarya and role of economic plants in the disease epidemiology. Crop Prot. 72:66–75. [Google Scholar]
- Albertin W, Marullo P. 2012. Polyploidy in fungi: evolution after whole-genome duplication. Proc R Soc Lond B Biol Sci. 279:2497–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali SS, et al. 2016. PCR-based identification of cacao black pod causal agents and identification of biological factors possibly contributing to Phytophthora megakarya's field dominance in West Africa. Plant Pathol. 65:1095–1108. [Google Scholar]
- Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S. 2010. Analysing RNA-Seq data with the "DESeq" package. Mol Biol. 43:1–17. [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey BA, Ali SS, Akrofi AY, Meinhardt LW. 2016. Phytophthora megakarya, a causal agent of black pod rot in Africa.In: Bailey BA, Meinhardt LW, editors. Cacao diseases. Springer International Publishing, New York, USA. p. 267–303. [Google Scholar]
- Bailey BA, et al. 2013. Dynamic changes in pod and fungal physiology associated with the shift from biotrophy to necrotrophy during the infection of Theobroma cacao by Moniliophthora roreri. Physiol Mol Plant Pathol. 81:84–96. [Google Scholar]
- Baxter L, et al. 2010. Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science 330:1549–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair JE, Coffey MD, Park S-Y, Geiser DM, Kang S. 2008. A multi-locus phylogeny for Phytophthora utilizing markers derived from complete genome sequences. Fungal Genet Biol. 45:266–277. [DOI] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27:578–579. [DOI] [PubMed] [Google Scholar]
- Bollmann S, Fang Y, Press C, Tyler BM, Grünwald NJ. 2016. Diverse evolutionary trajectories for small RNA biogenesis genes in the oomycete genus Phytophthora. Front Plant Sci. 7:284.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JI, et al. 2006. The C‐terminal half of Phytophthora infestans RXLR effector AVR3a is sufficient to trigger R3a‐mediated hypersensitivity and suppress INF1‐induced cell death in Nicotiana benthamiana. Plant J. 48:165–176. [DOI] [PubMed] [Google Scholar]
- Bowers JH, Bailey BA, Hebbar PK, Sanogo S, Lumsden RD. 2001. The impact of plant diseases on world chocolate production. Plant Health Prog. doi:10.1094/PHP-2001-0709-01-RV (online). [Google Scholar]
- Bozkurt TO, Schornack S, Banfield MJ, Kamoun S. 2012. Oomycetes, effectors, and all that jazz. Curr Opin Plant Biol. 15:483–492. [DOI] [PubMed] [Google Scholar]
- Brasier C, Griffin M. 1979. Taxonomy of Phytophthora palmivora on cocoa. Trans Br Mycol Soc. 72:111–143. [Google Scholar]
- Cantarel BL, et al. 2008. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18:188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X-R, Xing Y-P, Li Y-P, Tong Y-H, Xu J-Y. 2013. RNA-Seq reveals infection-related gene expression changes in Phytophthora capsici. PLoS One. 8:e74588.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, et al. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. [DOI] [PubMed] [Google Scholar]
- Dakwa J. 1987. A serious outbreak of black pod disease in a marginal area of Ghana. In: Proceedings of the 10th International Cocoa Research Conference, Santo Domingo, Dominican Republic. COPAL, Lagos, Nigeria. pp. 447–451.
- De La Torre AR, Lin Y-C, Van de Peer Y, Ingvarsson PK. 2015. Genome-wide analysis reveals diverged patterns of codon bias, gene expression, and rates of sequence evolution in Picea gene families. Genome Biol Evol. 7:1002–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Phillippy A, Carlton J, Salzberg SL. 2002. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 30:2478–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djocgoue P, et al. 2007. Heritability of phenols in the resistance of Theobroma cacao against Phytophthora megakarya, the causal agent of black pod disease. J Phytopathol. 155:519–525. [Google Scholar]
- Dou D, Kale SD, Wang X, Chen Y. et al. 2008. Conserved C-terminal motifs required for avirulence and suppression of cell death by Phytophthora sojae effector Avr1b. Plant Cell 20:1118–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou D, Kale SD, Wang X, Jiang RHY, et al. 2008. RXLR-mediated entry of Phytophthora sojae effector Avr1b into soybean cells does not require pathogen-encoded machinery. Plant Cell 20:1930–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. 1998. Profile hidden Markov models. Bioinformatics 14:755–763. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, Levanon EY. 2013. Human housekeeping genes, revisited. Trends Genet. 29:569–574. [DOI] [PubMed] [Google Scholar]
- Fellbrich G, et al. 2002. NPP1, a Phytophthora‐associated trigger of plant defense in parsley and Arabidopsis. Plant J. 32:375–390. [DOI] [PubMed] [Google Scholar]
- Flood J, et al. 2004. Cocoa under attack In: Flood J, Murphy R, editors. Cocoa futures. London: CABI BioSciences/The Commodities Press; p. 164. [Google Scholar]
- Gijzen M. 2009. Runaway repeats force expansion of the Phytophthora infestans genome. Genome Biol. 10:241.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest D. 2007. Black pod: diverse pathogens with a global impact on cocoa yield. Phytopathology 97:1650–1653. [DOI] [PubMed] [Google Scholar]
- Guo M, Davis D, Birchler JA. 1996. Dosage effects on gene expression in a maize ploidy series. Genetics 142:1349–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, et al. 2009. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461:393–398. [DOI] [PubMed] [Google Scholar]
- Huang X, Miller W. 1991. A time-efficient, linear-space local similarity algorithm. Adv Appl . 12:337–357. [Google Scholar]
- Ilut DC, et al. 2012. A comparative transcriptomic study of an allotetraploid and its diploid progenitors illustrates the unique advantages and challenges of RNA-seq in plant species. Am J Bot. 99:383–396. [DOI] [PubMed] [Google Scholar]
- Jiang RH, et al. 2013. Distinctive expansion of potential virulence genes in the genome of the oomycete fish pathogen Saprolegnia parasitica. PLoS Genet. 9:e1003272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang RH, Tripathy S, Govers F, Tyler BM. 2008. RXLR effector reservoir in two Phytophthora species is dominated by a single rapidly evolving superfamily with more than 700 members. Proc Natl Acad Sci U S A. 105:4874–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judelson HS. 2012. Dynamics and innovations within oomycete genomes: insights into biology, pathology, and evolution. Eukaryot Cell. 11:1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamoun S. 2006. A catalogue of the effector secretome of plant pathogenic oomycetes. Phytopathology 44:41.. [DOI] [PubMed] [Google Scholar]
- Kanneganti T-D, Huitema E, Cakir C, Kamoun S. 2006. Synergistic interactions of the plant cell death pathways induced by Phytophthora infestans Nep1-like protein PiNPP1.1 and INF1 elicitin. Mol Plant Microbe Int. 19:854–863. [DOI] [PubMed] [Google Scholar]
- Kasuga T, et al. 2016. Host-induced aneuploidy and phenotypic diversification in the sudden oak death pathogen Phytophthora ramorum. BMC Genomics 17:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuga T, et al. 2012. Phenotypic diversification is associated with host-induced transposon derepression in the sudden oak death pathogen Phytophthora ramorum. PLoS One 7:e34728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohonen T. 1982. Self-organized formation of topologically correct feature maps. Biol Cybern. 43:59–69. [Google Scholar]
- Kohonen T. 1990. The self-organizing map. Proc IEEE. 78:1464–1480. [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon LP, Brouwer H, de Cock AW, Govers F. 2012. The genus Phytophthora anno 2012. Phytopathology 102:348–364. [DOI] [PubMed] [Google Scholar]
- Kurtz S, et al. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5:R12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamour KH, et al. 2012. Genome sequencing and mapping reveal loss of heterozygosity as a mechanism for rapid adaptation in the vegetable pathogen Phytophthora capsici. Mol Plant Microbe Int. 25:1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, et al. 2010. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20:265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Adams KL. 2007. Expression partitioning between genes duplicated by polyploidy under abiotic stress and during organ development. Curr Biol. 17:1669–1674. [DOI] [PubMed] [Google Scholar]
- Livark K, Schmittgen T. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C (T)) method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- Long M, Betrán E, Thornton K, Wang W. 2003. The origin of new genes: glimpses from the young and old. Nat Rev Genet. 4:865–875. [DOI] [PubMed] [Google Scholar]
- Martens C, Van de Peer Y. 2010. The hidden duplication past of the plant pathogen Phytophthora and its consequences for infection. BMC Genomics 11:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mchau GR, Coffey MD. 1994. Isozyme diversity in Phytophthora palmivora: evidence for a southeast Asian centre of origin. Mycol Res. 98:1035–1043. [Google Scholar]
- Meyers BC, Tingey SV, Morgante M. 2001. Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res. 11:1660–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mighell A, Smith N, Robinson P, Markham A. 2000. Vertebrate pseudogenes. FEBS Lett. 468:109–114. [DOI] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35:182–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz JF, et al. 2015. The dynamic genome and transcriptome of the human fungal pathogen Blastomyces and close relative Emmonsia. PLoS Genet. 11:e1005493.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndubuaku T, Asogwa E. 2006. Strategies for the control of pests and diseases for sustainable cocoa production in Nigeria. Afr Sci. 7:209–216. [Google Scholar]
- Nei M, Rooney AP. 2005. Concerted and birth-and-death evolution of multigene families. Annu Rev Genet. 39:121.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyasse S, et al. 1999. Diversity of Phytophthora megakarya in Central and West Africa revealed by isozyme and RAPD markers. Mycol Res. 103:1225–1234. [Google Scholar]
- Opoku I, Appiah A, Akrofi A, Owusu G. 2000. Phytophthora megakarya: a potential threat to the cocoa industry in Ghana. Ghana J Agric Sci. 33:237–248. [Google Scholar]
- Osborn TC, et al. 2003. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 19:141–147. [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I. 2007. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23:1061–1067. [DOI] [PubMed] [Google Scholar]
- Pati A, et al. 2010. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods. 7:455–457. [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 8:785–786. [DOI] [PubMed] [Google Scholar]
- Ploetz R. 2016. The impact of diseases on cacao production: a global overview.In: Bailey BA, Meinhardt LW, editors. Cacao diseases. Springer International Publishing, New York, USA. p. 33–59. [Google Scholar]
- Ponchet M, et al. 1999. Are elicitins cryptograms in plant-Oomycete communications? Cell Mol Life Sci. 56:1020–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat Y, Fromer M, Linial N, Linial M. 2009. Codon usage is associated with the evolutionary age of genes in metazoan genomes. BMC Evol Biol. 9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qutob D, Kamoun S, Gijzen M. 2002. Expression of a Phytophthora sojae necrosis‐inducing protein occurs during transition from biotrophy to necrotrophy. Plant J. 32:361–373. [DOI] [PubMed] [Google Scholar]
- Richard G-F, Kerrest A, Dujon B. 2008. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol Mol Biol Rev. 72:686–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 4:406–425. [DOI] [PubMed] [Google Scholar]
- Sansome E, Brasier C, Griffin M. 1975. Chromosome size differences in Phytophthora palmivora, a pathogen of cocoa. Nature 255:704–705. [DOI] [PubMed] [Google Scholar]
- Simão FA, et al. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31: 3210–3212. [DOI] [PubMed] [Google Scholar]
- Song S, et al. 2013. The bHLH subgroup IIId factors negatively regulate jasmonate-mediated plant defense and development. PLoS Genet. 9:e1003653.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer EL, Von Heijne G, Krogh A. 1998. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol. 6:175–182. [PubMed] [Google Scholar]
- Soyer JL, et al. 2014. Epigenetic control of effector gene expression in the plant pathogenic fungus Leptosphaeria maculans. PLoS Genet. 10:e1004227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam R, et al. 2013. Identification and characterisation CRN effectors in Phytophthora capsici shows modularity and functional diversity. PLoS One 8:e59517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Steinkamp R, Waack S, Morgenstern B. 2004. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32:309–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28:2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorold C. 1959. Methods of controlling black pod disease (caused by Phytophthora palmivora) of Theobroma cacao in Nigeria. Ann Appl Biol. 47:708–715. [Google Scholar]
- Torto TA, et al. 2003. EST mining and functional expression assays identify extracellular effector proteins from the plant pathogen Phytophthora. Genome Res. 13:1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathy S, Tyler BM. 2006. The repertoire of transfer RNA genes is tuned to codon usage bias in the genomes of Phytophthora sojae and Phytophthora ramorum. Mol Plant Microbe Int. 19:1322–1328. 2 [DOI] [PubMed] [Google Scholar]
- Tyler BM, Gijzen M. 2014. The Phytophthora sojae genome sequence: foundation for a revolution In: Dean RA, Lichens-Park A, Kole C, editors. Genomics of plant-associated fungi and oomycetes: dicot pathogens. Springer, New York, USA. p. 133–157. [Google Scholar]
- Tyler BM, et al. 2006. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science 313:1261–1266. [DOI] [PubMed] [Google Scholar]
- Van Hooff JJ, Snel B, Seidl MF. 2014. Small homologous blocks in Phytophthora genomes do not point to an ancient whole-genome duplication. Genome Biol Evol. 6:1079–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, et al. 2011. Transcriptional programming and functional interactions within the Phytophthora sojae RXLR effector repertoire. Plant Cell 23:2064–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whisson SC, et al. 2007. A translocation signal for delivery of oomycete effector proteins into host plant cells. Nature 450:115–118. [DOI] [PubMed] [Google Scholar]
- Yadav P, Singh V, Yadav S, Yadav K, Yadav D. 2009. In silico analysis of pectin lyase and pectinase sequences. Biochemistry (Mosc) 74:1049–1055. [DOI] [PubMed] [Google Scholar]
- Yandell M, Ence D. 2012. A beginner's guide to eukaryotic genome annotation. Nat Rev Genet. 13:329–342. [DOI] [PubMed] [Google Scholar]
- Ye W, et al. 2011. Digital gene expression profiling of the Phytophthora sojae transcriptome. Mol Plant Microbe Int. 24:1530–1539. [DOI] [PubMed] [Google Scholar]
- Ye W, et al. 2016. Sequencing of the litchi downy blight pathogen reveals it is a Phytophthora species with downy mildew-like characteristics. Mol Plant Microbe Int. 29:573–583. [DOI] [PubMed] [Google Scholar]
- Yu LM. 1995. Elicitins from Phytophthora and basic resistance in tobacco. Proc Natl Acad Sci U S A. 92:4088–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Motilal L. 2016. Origin, dispersal, and current global distribution of cacao genetic diversity.In: Bailey BA, Meinhardt LW, editors. Cacao diseases. Springer International Publishing, New York, USA. p. 3–31. [Google Scholar]
- Zuluaga AP, et al. 2016. Transcriptional dynamics of Phytophthora infestans during sequential stages of hemibiotrophic infection of tomato. Mol Plant Pathol. 17:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.