

Abstract
Transposable elements (TEs) are selfish, autonomously replicating DNA sequences that constitute a major component of eukaryotic genomes and contribute to genome evolution through their movement and amplification. Many fungal genomes, including the anther-smut fungi in the basidiomycete genus Microbotryum, have genome defense mechanisms, such as repeat-induced point mutation (RIP), which hypermutate repetitive DNA and limit TE activity. Little is known about how hypermutation affects the tempo of TE activity and their sequence evolution. Here we report the identification of a massive burst-like expansion of Gypsy-like retrotransposons in a strain of Microbotryum. This TE expansion evidently occurred in the face of RIP-like hypermutation activity. By examining the fitness of individual TE insertion variants, we found that RIP-like mutations impair TE fitness and limit proliferation. Our results provide evidence for a punctuated pattern of TE expansion in a fungal genome, similar to that observed in animals and plants. While targeted hypermutation is often thought of as an effective protection against mobile element activity, our findings suggest that active TEs can persist and undergo selection while they proliferate in genomes that have RIP-like defenses.
Keywords: transposable elements, genome defense, repeat-induced point mutation
Introduction
Transposable elements (TEs) are selfish DNA sequences capable of self-replication within their host genomes (Doolittle and Sapienza 1980; Orgel and Crick 1980). TEs have been remarkably successful: they are found in nearly every organism studied to date (Wicker et al. 2007) and compose a large fraction of many eukaryotic genomes (Sanmiguel and Bennetzen 1998; Daboussi and Capy 2003; Bennetzen 2005; Hua-Van et al. 2005). TE activity drives the evolution of genome architecture and organismal phenotype by shuffling and duplicating genes (Lai et al. 2005; Morgante et al. 2005) and their regulatory sequences (Kunarso et al. 2010; Lynch et al. 2011; Schmidt et al. 2012; Brawand et al. 2014). In addition, TE content is a major contributor to the wide variation in genome size observed among plants, animals, and fungi (Sanmiguel and Bennetzen 1998; Kidwell 2002; Bennetzen 2005; Neumann et al. 2006; Piegu et al. 2006; Tenaillon et al. 2011; Raffaele and Kamoun 2012).
In many plant and animal genomes, TEs have proliferated in short-lived, burst-like waves of expansion that punctuate periods of population stasis (Neumann et al. 2006; Piegu et al. 2006; Pritham and Feschotte 2007; Hawkins et al. 2008; Schmidt et al. 2012; Brawand et al. 2014). Expansions of TE families may be facilitated by the disruption of genome regulatory mechanisms, such as RNA silencing and DNA methylation, which function as genome defenses by suppressing TE activity (Selker et al. 1987; Miura et al. 2001; Galagan and Selker 2004; Buchon and Vaury 2005; Cerutti and Casas-Mollano 2006; Brennecke et al. 2007; Chung et al. 2008). Despite the important role of TEs in genome evolution across kingdoms of life, the pace and tempo of TE activity in fungal genomes have not been characterized.
Many fungal genomes have a defense mechanism known as repeat-induced point mutation (RIP), which hypermutates duplicated nuclear DNA (Selker 2002; Clutterbuck 2011; Horns et al. 2012). For example, the RIP process of the ascomycete Neurospora crassa introduces C-to-T mutations at CpA dinucleotides mostly within linked sequences that are longer than ∼400 bp (or ∼1000 bp in the case of unlinked sequences; Watters et al. 1999) and sharing greater than ∼80% nucleotide identity (Cambareri et al. 1991). RIP mutations can regulate TE activity by causing inactivating substitutions in TE genes (Kinsey et al. 1994; Cambareri et al. 1998; Margolin et al. 1998) and inducing silencing DNA methylation (Singer et al. 1995; Lewis et al. 2009). RIP mutations also reduce sequence similarity between initially identical TE insertions, thereby reducing the rate of ectopic recombination.
Repetitive elements in basidiomycete fungi of the genus Microbotryum display hypermutation patterns that resemble a RIP-like defense targeting the trinucleotide TpCpG (Horns et al. 2012). Nevertheless, these genomes contain a large fraction of TEs (≥15%) from diverse families, including LTR (Copia-like and Gypsy-like) and non-LTR retrotransposons, and Helitron elements (Hood 2005). A RIP-like genome defense may be expected to impact TE activity, population structure, and evolution. While observations of such effects have been lacking, co-occurrence of TEs and a RIP-like hypermutation process in Microbotryum offers an opportunity to provide new insights.
Here, we investigated the evolutionary dynamics of Gypsy-like retrotransposons in Microbotryum, a genus known as the anther-smut fungi. This common name derives from the pathogenic life cycle of the fungi, which includes the systemic colonization of plants in the carnation family (Caryophyllaceae) and replacement of the host's pollen in the flowers with fungal spores. Insect pollinators then spread the spores from diseased to healthy plants. By sequencing Gypsy-like TE populations from two strains of Microbotryum and analyzing their sequence variation in a phylogenetic framework, we found that Gypsy-like TEs have undergone a recent, strain-specific expansion in a lineage of Microbotryum. We found evidence that RIP-like hypermutations impact the fitness of TEs, limiting their proliferation and imposing a selective sieve that favors TE insertions that evade the RIP defense.
Materials and Methods
We initially assessed TE content in four field isolates of Microbotryum from host plants in the genus Dianthus, which we identify here by the abbreviation “MvD” followed by their geographic origin (table 1): MvD-Vinadio, MvD-Sestriere, MvD-Pesio, and MvD-Cesana. Previously subsumed in the epithets Microbotryum violaceum or Microbotryum dianthorum, the anther-smut fungi on Dianthus hosts are not yet resolved taxonomically (Le Gac et al. 2007; Denchev et al. 2009; Kemler et al. 2012). Identification of the samples used in this study and inference of the relationships among them was performed by comparing DNA sequences of the internal transcribed spacer (ITS) regions 1 and 2 of the ribosomal RNA complex (as in Freeman et al. 2002) and the intron of the γ-tubulin gene (as in Le Gac et al. 2007). Based upon γ-tubulin sequences, the named Microbotryum species closest to the current samples were M. carthusianorum for strains MvD-Vinadio and MvD-Sestriere, M. superbum for MvD-Pesio, and M. shykoffianum for MvD-Cesana (Denchev et al. 2009). The sister relationships among the four samples inferred using ribosomal ITS regions and γ-tubulin were the same (fig. 1).
Table 1.
Origin of Microbotryum Samples, Showing the Host-of-Origin and Geographic Location
| Strain ID | Host Species | Location | Lat., Long. |
|---|---|---|---|
| MvD-Pesio | Dianthus pavonius | Valle Pesio | 44.196, 7.680 |
| MvD-Cesana | Dianthus sylvestris | Cesana Torinese | 44.937, 6.805 |
| MvD-Sestriere | Dianthus carthusianorum | Colle Sestriere | 44.976, 6.884 |
| MvD-Vinadio | Dianthus pavonius | Sant'Anna di Vinadio | 44.260, 7.123 |
Fig. 1.—

Evolutionary relationships and genomic TE content of Microbotryum strains. Bars display the fraction of whole genome sequencing reads aligning to repetitive element families. Error bars indicate 95% confidence intervals based on binomial sampling. Neighbor-joining phylogeny of Microbotryum strains was constructed based on γ-tubulin DNA sequences.
To estimate the TE content of each genome, we sequenced the genomes using the GS-FLX platform (454 Life Sciences) with 250 bp reads covering 20–71% of each genome at ≥1X coverage. We compared all reads to the Repbase database of repetitive sequences (Jurka 2000) using tBLASTx. We estimated the genomic abundance of each TE family represented in Repbase by calculating the fraction of reads having significant similarity to that family (E-value < 10−2). Because mobile elements from the Pucciniomycotina are poorly represented in the Repbase database, the divergence between query sequences and target sequences in the genomes that we studied might be high. We therefore chose to use the translated BLAST search because it has high sensitivity to detect divergent sequences. This genomic shotgun sequencing approach combined with translated BLAST analysis has given predictions of relative TE content in different isolated chromosomes (Hood et al. 2004; Fontanillas et al. 2015) that were later verified by analyses of high-coverage annotated whole-genome sequencing (Perlin et al. 2015).
Next, we sampled Gyspy-like TEs from strains MvD-Vinadio and MvD-Sestriere using a chromosome-structured sampling approach that allowed distinguishing TE insertions with identical sequences on different chromosomes (Hood et al. 2005). We isolated DNA from individual chromosomes by separating the karyotypes of haploid meiotic products using pulsed-field gel electrophoresis. Electrophoresis was conducted on a CHEF-DRII pulsed-field system (Bio-Rad) with run conditions of 14 °C, 96 h, 2.7 V/cm, 0.8% agarose, 200 s initial and 1100 s final switch times. To separate larger chromosomes, separate electrophoresis runs were conducted with conditions of 2.1 V/cm, 2% agarose, 1100 s initial and 1800 s final switch times. We extracted chromosome-specific DNA by removing an agarose gel plug from each karyotype band using a glass Pasteur pipette. The plugs were then pulverized and twice frozen and thawed in 1X Tris-EDTA buffer. Most karyotype bands were well-resolved and therefore likely contained DNA from a single chromosome. One agarose gel plug from MvD-Vinadio came from a less well-resolved region and may have contained DNA from more than one chromosome.
We amplified by PCR, cloned, and sequenced Gypsy-like TE fragments from each chromosome. PCR primers were designed to amplify 938bp of the consensus of an alignment of shotgun sequence reads from Microbotryum strains tiled against a full-length Gypsy-like TE from Laccaria bicolor strain S238N-H82 (gi 170112043): forward primer MvGf 5′ AAAGTCCCCACGGCGTTAT and reverse primer MvGr 5′ GTTGAGAAACTGGGGTTAGCTGT. These primer sequences contain no TpCpG hypermutation target sites and flank a fragment of the pol gene that spans about half of the conserved reverse transcriptase domain and half of the protease domain. PCR was performed in a total volume of 10 μL containing 1 μL of chromosome-specific DNA solution, 0.25 μM forward and reverse primer, 1X PCR buffer, 1.5 mM MgCl2, 0.05 mM dNTP, and 0.1 units Taq polymerase (New England Biolabs). The thermal profile consisted of initial denaturation at 95°C for 3 min; 30 cycles of amplification with each comprising 95°C for 30 s, touchdown from 64°C to 59°C with a 1°C decrease after every six cycles for 30 s, and 68°C for 1 min; and final extension at 68°C for 7 min. We cloned the resulting products using a TOPO TA cloning kit (Invitrogen), then re-amplified forty clones from each chromosome using the PCR protocol described earlier, except with the initial denaturation step increased to 10 min. We purified the PCR products using ExoSAP-IT (Affymetrix). Clones were sequenced using the BigDye Terminator kit on an ABI Prism 377XL sequencer (Applied Biosystems) at the University of Massachusetts Amherst DNA sequencing facility. We excluded short sequence reads (< 700 bp) from our analysis.
We obtained 329 sequences from MvD-Vinadio and 282 from MvD-Sestriere. Because identical cloned sequences within a chromosome sample might represent amplification products from the same locus, we conservatively included in our analysis only non-redundant sequences from each chromosome, which represent distinct TE insertions. After excluding redundant sequences, 274 sequences from MvD-Vinadio and 225 from MvD-Sestriere remained, with an average of 27 unique insertions per chromosome.
To analyze sequence variation within these TE populations, we aligned the sequences using MUSCLE (Edgar 2004) and constructed maximum-likelihood phylogenies using FastTree (Price et al. 2010). Because the mutational effects of TpCpG hypermutation could obscure phylogenetic relationships among the sequences, all potential hypermutation target sites, including unmutated (TpCpG and CpGpA) and mutated (TpTpG and CpApA) sites on both strands, were excluded from the phylogenetic analysis. Phylogenies were rooted using a Gypsy-like TE sequence from Microbotryum violaceum from host plants in the genus Silene as an outgroup. We reconstructed ancestral sequence states by maximum-likelihood. We inferred the relative fitness of the sampled sequences based on the branching patterns of the phylogenetic trees (Neher et al. 2014). Analyses were performed using custom scripts written in Java, Python, and R, which are available upon request.
Results
Elevated Abundance of Gypsy-Like TEs in MvD-Vinadio
Using whole-genome sequencing on the 454 GS-FLX platform to assess the TE content of each Microbotryum strain, we found that a significantly larger fraction of reads from MvD-Vinadio align with Gypsy-like TEs (2.0%) compared with any of the other closely related Microbotryum strains (0.9–1.1%) (fig. 1; P = 7 × 10−24, Pearson’s χ2 test, two-sided). In contrast, all four strains carry comparable amounts of other TE families that were present in the Repbase repeat database, including Copia-like, CcNgaro3-like, non-LTR, and Helitron-like TEs (minimum χ2 comparison P = 0.035). This suggests that an expansion of Gypsy-like TEs occurred in the lineage leading to MvD-Vinadio after it diverged from the MvD-Sestriere lineage, resulting in a two-fold increase in genomic copy number.
Recent Burst-Like Proliferation of Gypsy-Like TEs in MvD-Vinadio
Targeted sequencing of Gypsy-like TE insertions in MvD-Vinadio and its close relative MvD-Sestriere provided confirmation that Gypsy-like TEs have recently proliferated in MvD-Vinadio and enabled characterization of the tempo of TE activity. We sampled Gypsy-like TE insertions from the intragenomic populations of MvD-Vinadio and MvD-Sestriere, and sequenced a ∼1 kb fragment spanning the conserved reverse transcriptase and protease domains of the pol gene (Materials and Methods; Hood et al. 2005).
Intragenomic phylogenetic analysis of Gypsy-like TEs, 274 from MvD-Vinadio and 225 from MvD-Sestriere, uncovered evidence of a burst-like expansion of these TEs in MvD-Vinadio. In the genome of MvD-Vinadio ∼70% of the TEs belong to a single clade of highly similar elements having mean pairwise sequence identity of 99.0% (fig. 2A), while a group of such highly similar TEs is not present in MvD-Sestriere (fig. 2B). When we reconstructed a phylogenetic tree using aligned sequences from MvD-Vinadio and MvD-Sestriere together, we found that these TEs from MvD-Vinadio form a distinct strain-specific clade (fig. 2C). We note that numerous sequences from MvD-Vinadio cluster with those from MvD-Sestriere elsewhere in the tree, indicating that our approach was able to detect similar TE sequence diversity in both genomes. Furthermore, older insertions shared by descent between MvD-Vinadio and MvD-Sestriere are present at similar frequency, indicating that the MvD-Vinadio-specific clade of TEs is not explained by biased sampling.
Fig. 2.—

Phylogenies of Gypsy-like retrotransposon sequences from Microbotryum strains. Phylogenies of Gypsy-like TE sequences from (A) MvD-Vinadio, (B) MvD-Sestriere, and (C) both genomes were reconstructed by maximum-likelihood. In (A) and (B), color indicates chromosome of origin. In (C), color indicates genome of origin. Green shading in (C) indicates MvD-Vinadio-specific clade. Outgroup is a Gypsy-like TE sequence from Microbotryum lychnidis-dioicae from the host plant Silene latifolia (MvSl). Branch support values are indicated in supplemental figure 1 (Supplementary Material online).
Analysis of sequence diversity among the TE insertions in each genome supports this interpretation of a burst-like expansion of Gypsy-like TEs in MvD-Vinadio (fig. 3). We compared each sequence to the common ancestor of extant sequences in each genome inferred by maximum likelihood. For TEs from MvD-Vinadio, the distribution of sequence distances to their common ancestor has a sharp peak at ∼98.5% identity. This distribution is significantly different from the more balanced distribution seen for TEs from MvD-Sestriere (fig. 3A; P = 8.8 × 10−4; Kolmogorov–Smirnov test, two-sample). This finding suggests that most Gypsy-like TE insertions in MvD-Vinadio were born during a short temporal window, implying a burst-like expansion event. This conclusion is further supported by the distributions of pairwise sequence identity among extant sequences within each genome (fig. 3B). TEs from MvD-Vinadio exhibit higher pairwise sequence identity compared to TEs from MvD-Sestriere (P = 1.5 × 10−38; Mann-Whitney U test, two-sided), including a sharp peak at 99% identity composed of pairs of sequences belonging to the recent expansion. New TE insertions in MvD-Vinadio were found on all chromosomes at similar frequency (supplemental fig. 2, Supplementary Material online).
Fig. 3.—
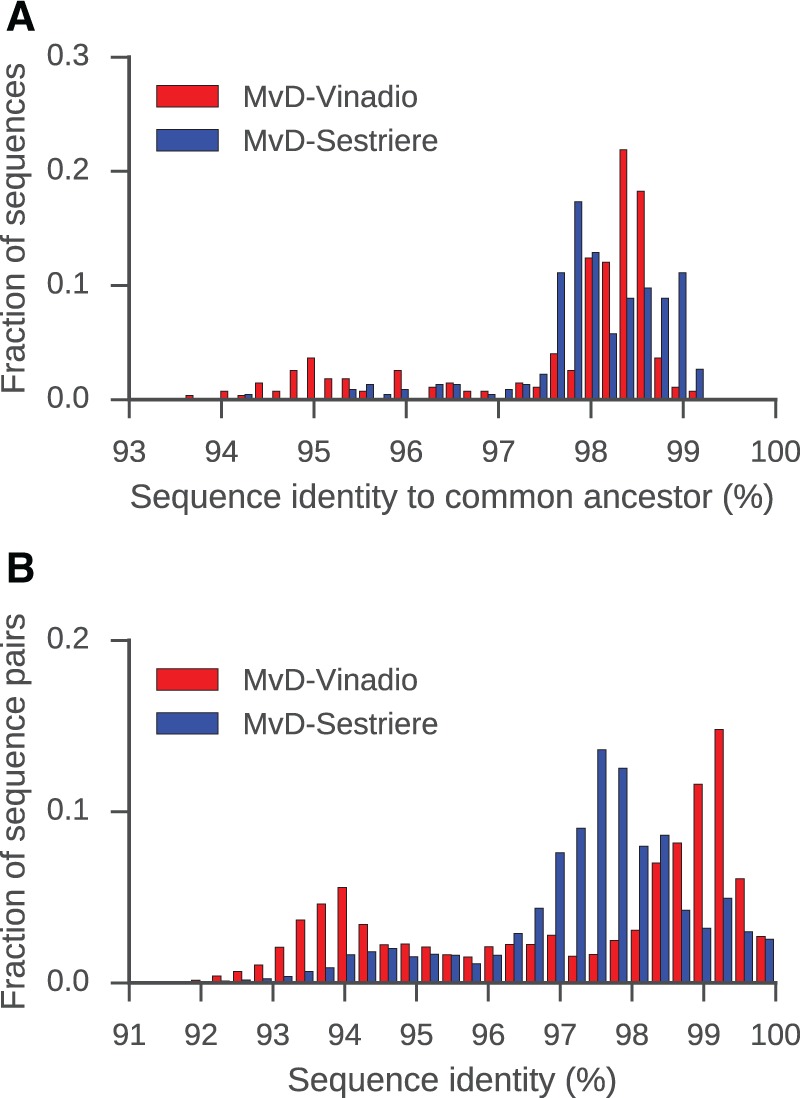
Sequence diversity of Gypsy-like retrotransposons from MvD-Vinadio and MvD-Sestriere. (A) Distributions of sequence identity to the common ancestor of all extant sequences in each genome. (B) Distributions of pairwise sequence identity among extant sequences in each genome.
Fitness Effects of RIP-Like Hypermutation on Gypsy-Like TEs
Next, we examined how the RIP-like genome defense influences Gypsy-like TE sequence evolution. Comparing the two focal strains, 68% of TEs from MvD-Vinadio and 55% of TEs from MvD-Sestriere display evidence of RIP-like, sequence context-dependent hypermutation (i.e., TpCpG to TpTpG transitions, or transitions in the reverse complement of this site) when compared to the common ancestor of extant elements. Each TE copy contains 1.7 ± 2.1 mutated TpCpG target sites (mean ± s.d.) among the 35 target sites present in the ancestral sequence. Mutation rates in TpCpG target sites were elevated compared to overall mutation rates of trinucleotide contexts (data not shown; see Horns et al. 2012).
By assessing mutation patterns of Gypsy-like TEs involved in the recent expansion in MvD-Vinadio, we found evidence that RIP-like mutations suppress TE activity. Strikingly, we found that TEs belonging to the recently expanded clade (N = 191 sequences) have fewer RIP-like mutations than other TEs (fig. 4A; P = 1.8 × 10−13; Mann-Whitney U test, two-sided). The median number of RIP-like mutations is 1 among TEs within the expanded clade, while the median is 4 among TEs outside the expanded clade. The DNA sequence of the ancestral TE that initiated the expansion in MvD-Vinadio, reconstructed by maximum likelihood, contained no mutations of TpCpG site in comparison with the common ancestor of extant elements. These findings suggest that evasion of RIP-like hypermutations was associated with TE proliferation. That some of the sequences within the expanded clade contain TpCpG site mutations suggests that the putative RIP-like genome defense was active during or after the expansion.
Fig. 4.—

Analysis of RIP-like mutations in Gypsy-like retrotransposons. (A) Distributions of the number of RIP-like mutations in sequences either belonging or not belonging to the strain-specific expansion in MvD-Vinadio. (B) Maximum-likelihood phylogeny of Gypsy-like TEs in MvD-Vinadio colored by inferred relative fitness based on local branching index. (C) Number of RIP-like mutations in extant sequences from MvD-Vinadio plotted against relative fitness rank (rank closer to 0 is higher fitness). (D) Number of RIP-like mutations in extant sequences in each quartile of relative fitness (quartile 1 is highest fitness). RIP-like mutations were determined by comparison with the ancestor of all extant sequences inferred by maximum-likelihood .
To examine how the RIP-like genome defense affects the fitness of Gypsy-like TEs in MvD-Vinadio, we inferred the relative fitness of each extant TE sequence based on the branching patterns of the phylogenetic tree (fig. 4B; Neher et al. 2014). This inference method is based on the notion that an individual with high fitness is expected to be found at the root of a rapidly branching clade. Similarly, extant individuals which are close descendants of these internal nodes are expected to have high fitness. Many Gypsy-like TEs in the expanded clade have high inferred fitness, reflecting the recent rapid proliferation of that clade. We found that the number of RIP-like mutations in extant TE sequences correlates significantly with their inferred fitness (fig. 4C; Spearman rank correlation coefficient 0.69; P = 2.0 × 10−39). When analyzed in quartiles, sequences in the top (first) quartile of ranked fitness contain TEs in the expanded clade and have significantly fewer RIP-like mutations compared to every other quartile (fig. 4D; P < 8.3 × 10−5; Mann-Whitney U test, two-sided). Conversely, TE sequences with more RIP mutations tend to have lower fitness. These findings suggest that Gypsy-like TEs which have not suffered RIP-like mutations proliferate more than TEs which have suffered such mutations. Notably, the second and third quartiles of inferred fitness also contained some TEs belonging to the expanded clade, and these had significantly more RIP-like mutations than the first quartile. Thus, even within the recent expansion, TEs that suffered more RIP-like mutations tended to proliferate less, suggesting that the RIP-like genome defense was actively limiting TE activity during the expansion, rather than only mutating sequences afterward.
Discussion
Despite the ubiquity of TEs and their important role in shaping genome evolution, their population and evolutionary dynamics remain poorly understood. By reconstructing the evolutionary history of Gypsy-like TEs within closely related genomes of Microbotryum, we have found evidence that a lineage-specific burst of Gypsy-like TE proliferation has dramatically remodeled the genome composition of the strain MvD-Vinadio, doubling the genomic representation of this TE family. In addition, our results show how a RIP-like hypermutation process shapes the evolution of Gypsy-like TEs and suggest that hypermutation imposes a selective sieve on TE insertions.
Punctuated patterns of retrotransposon proliferation have been described in plant (Neumann et al. 2006; Piegu et al. 2006; Hawkins et al. 2008) and mammalian genomes (Pritham and Feschotte 2007; Lynch et al. 2011; Schmidt et al. 2012). In this work, we have presented evidence for a massive burst-like expansion of retrotransposons in a fungal genome. While sequence differences due to PCR errors may be present in our analysis, this source of error would not be expected to affect the genomes differently nor to create the patterns consistent with proliferation that we observed. The narrow peak width in the distributions of sequence diversity among Gypsy-like TEs in MvD-Vinadio suggests that their proliferation occurred during a temporally compressed period. The proliferated TEs became dominant within the intragenomic population, indicating that this brief episode of proliferation has strongly driven the evolution of Gypsy-like TEs in this genome. While the causes underlying this episode of transposition activity remain undetermined, it is notable that the genus of Microbotryum has a history of hybridization (Devier et al. 2010). Hybridization (Bingham et al. 1982; Liu and Wendel 2000; Ungerer et al. 2006) or environmental stress (McClintock 1984) might trigger TE proliferation by compromising regulatory mechanisms such as DNA methylation or RIP.
Patterns of RIP-like hypermutation with a trinucleotide target preference of TpCpG are conserved among fungi of the Pucciniomycotina subphylum of the Basidiomycota (Horns et al. 2012). While caution is necessary in the absence of direct evidence for a RIP pathway, such as may be provided by transformation experiments (Toh et al. 2016), our data provide further evidence that a RIP-like hypermutation process targeting TpCpG has affected TE evolution in Microbotryum. Remarkably, the observed expansion of Gypsy-like TEs appears to have occurred in the face of concurrent activity of a RIP-like hypermutation process. Our results demonstrate that RIP-like mutations impact TE fitness by limiting the proliferation of individual TE insertions. This suggests that evading a RIP-like defense may be key for the success of retrotransposons in genome colonization. Selection may favor TE variants that are able to evade hypermutation, including variants that have shed RIP target sites. Proliferation of TE variants that evade RIP-like mutations exposes the host genome to elevated mutational load and risk of ectopic recombination. These costs in turn exert selective pressure on the host genome to curtail evasion and suppress TE proliferation, highlighting the potential of TE activity to drive an intragenomic arms race.
In other fungal species, RIP is thought of as a remarkably effective defense mechanism against selfish DNA, including TEs. Much of the evidence for this notion comes from studies of the ascomycete fungus Neurospora crassa and its close relatives, which have a well-defined RIP process that targets dinucleotide sequence motifs. Unlike Microbotryum, the genome of N. crassa harbors no intact mobile elements and contains numerous fossils of mobile elements that have evidently suffered RIP mutations (Galagan et al. 2003). Our results suggest that genomes having a RIP-like hypermutation process may be more permissive to the maintenance of TE activity than previously thought. We propose that the less frequently occurring trinucleotide target site sequence of the RIP-like hypermutation process in Microbotryum may render this defense less effective at suppressing TE activity than the dinucleotide-targeting RIP process found in Neurospora. Mechanisms that influence the pace and tempo of TE activity and permit coexistence of TEs and genome defenses deserve further investigation.
Supplementary Material
Acknowledgments
This work was supported by awards from the National Science Foundation (grant numbers NSF-DEB 1115765 and NSF-DEB 0747222 to M.E.H.).
Supplementary Material
Supplementary material are available at Genome Biology and Evolution online.
Literature Cited
- Bennetzen JL. 2005. Transposable elements, gene creation and genome rearrangement in flowering plants. Curr Opin Genet Dev. 15:621–627. [DOI] [PubMed] [Google Scholar]
- Bingham PM, Kidwell MG, Rubin GM. 1982. The molecular basis of P-M hybrid dysgenesis: the role of the P element, a P-strain-specific transposon family. Cell 29:995–1004. [DOI] [PubMed] [Google Scholar]
- Brawand D, et al. 2014. The genomic substrate for adaptive radiation in African cichlid fish. Nature 513:375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, et al. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in drosophila. Cell 128:1089–1103. [DOI] [PubMed] [Google Scholar]
- Buchon N, Vaury C. 2005. RNAi: a defensive RNA-silencing against viruses and transposable elements. Heredity 96:195–202. [DOI] [PubMed] [Google Scholar]
- Cambareri EB, Aisner R, Carbon J. 1998. Structure of the chromosome VII centromere region in Neurospora crassa: degenerate transposons and simple repeats. Mol Cell Biol. 18:5465–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambareri EB, Singer MJ, Selker EU. 1991. Recurrence of repeat-induced point mutation (Rip) in Neurospora Crassa. Genetics 127:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti H, Casas-Mollano J. 2006. On the origin and functions of RNA-mediated silencing: from protists to man. Curr Genet. 50:81–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W-J, Okamura K, Martin R, Lai EC. 2008. Endogenous RNA interference provides a somatic defense against Drosophila transposons. Curr Biol. 18:795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clutterbuck JA. 2011. Genomic evidence of repeat-induced point mutation (RIP) in filamentous ascomycetes. Fungal Genet Biol. 48:306–326. [DOI] [PubMed] [Google Scholar]
- Daboussi M-J, Capy P. 2003. Transposable elements in filamentous fungi. Annu Rev Microbiol. 57:275–299. [DOI] [PubMed] [Google Scholar]
- Denchev CM, Giraud T, Hood ME. 2009. Three new species of anthericolous smut fungi on Caryophyllaceae. Mycologia Balcanica 6:79–84. [Google Scholar]
- Devier B, Aguileta G, Hood ME, Giraud T. 2010. Using phylogenies of pheromone receptor genes in the Microbotryum violaceum species complex to investigate possible speciation by hybridization. Mycologia 102:689–696. [DOI] [PubMed] [Google Scholar]
- Doolittle WF, Sapienza C. 1980. Selfish genes, the phenotype paradigm and genome evolution. Nature 284:601–603. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanillas E, et al. 2015. Degeneration of the nonrecombining regions in the mating-type chromosomes of the anther-smut fungi. Mol Biol Evol. 32:928–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman AB, Duong KK, Shi T-L, Hughes CF, Perlin MH. 2002. Isolates of microbotryum violaceum from North American host species are phylogenetically distinct from their European host-derived counterparts. Mol Phylogenet Evol. 23:158–170. [DOI] [PubMed] [Google Scholar]
- Galagan JE, et al. 2003. The genome sequence of the filamentous fungus Neurospora crassa. Nature 422:859–868. [DOI] [PubMed] [Google Scholar]
- Galagan JE, Selker EU. 2004. RIP: the evolutionary cost of genome defense. Trends Genet. 20:417–423. [DOI] [PubMed] [Google Scholar]
- Hawkins J, Hu G, Rapp RA, Grafenberg JL, Wendel JF. 2008. Phylogenetic determination of the pace of transposable element proliferation in plants: copia and LINE-like elements in Gossypium. Genome 51:11–18. [DOI] [PubMed] [Google Scholar]
- Hood ME. 2005. Repetitive DNA in the automictic fungus Microbotryum violaceum. Genetica 124:1–10. [DOI] [PubMed] [Google Scholar]
- Hood ME, Antonovics J, Koskella B. 2004. Shared forces of sex chromosome evolution in haploid-mating and diploid-mating organisms: Microbotryum violaceum and other model organisms. Genetics 168:141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood ME, Katawczik M, Giraud T. 2005. Repeat-induced point mutation and the population structure of transposable elements in Microbotryum violaceum. Genetics 170:1081–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horns F, Petit E, Yockteng R, Hood ME. 2012. Patterns of repeat-induced point mutation in transposable elements of basidiomycete fungi. Genome Biol Evol. 4:240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua-Van A, Le Rouzic A, Maisonhaute C, Capy P. 2005. Abundance, distribution and dynamics of retrotransposable elements and transposons: similarities and differences. Cytogenet Genome Res. 110:426–440. [DOI] [PubMed] [Google Scholar]
- Jurka J. 2000. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 16:418–420. [DOI] [PubMed] [Google Scholar]
- Kemler M, et al. 2012. Contrasting phylogenetic patterns of anther smuts (Pucciniomycotina: Microbotryum. Org Divers Evol. 13:111–126. [Google Scholar]
- Kidwell MG. 2002. Transposable elements and the evolution of genome size in eukaryotes. Genetica 115:49–63. [DOI] [PubMed] [Google Scholar]
- Kinsey JA, Garrett-Engele PW, Cambareri EB, Selker EU. 1994. The Neurospora transposon Tad is sensitive to repeat-induced point mutation (RIP). Genetics 138:657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunarso G, et al. 2010. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet. 42:631–634. [DOI] [PubMed] [Google Scholar]
- Lai J, Li Y, Messing J, Dooner HK. 2005. Gene Movement by Helitron transposons contributes to the haplotype variability of maize. Proc Natl Acad Sci U S A. 102:9068–9073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gac M, Hood ME, Fournier E, Giraud T. 2007. Phylogenetic evidence of host-specific cryptic species in the anther smut fungus. Evolution 61:15–26. [DOI] [PubMed] [Google Scholar]
- Lewis ZA, et al. 2009. Relics of repeat-induced point mutation direct heterochromatin formation in Neurospora crassa. Genome Res. 19:427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Wendel JF. 2000. Retrotransposon activation followed by rapid repression in introgressed rice plants. Genome 43:874–880. [PubMed] [Google Scholar]
- Lynch VJ, Leclerc RD, May G, Wagner GP. 2011. Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat Genet. 43:1154–1159. [DOI] [PubMed] [Google Scholar]
- Margolin BS, et al. 1998. A methylated Neurospora 5S rRNA pseudogene contains a transposable element inactivated by repeat-induced point mutation. Genetics 149:1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. 1984. The significance of responses of the genome to challenge. Science 226:792–801. [DOI] [PubMed] [Google Scholar]
- Miura A, et al. 2001. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 411:212–214. [DOI] [PubMed] [Google Scholar]
- Morgante M, et al. 2005. Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nat Genet. 37:997–1002. [DOI] [PubMed] [Google Scholar]
- Neher RA, Russell CA, Shraiman BI. 2014. Predicting evolution from the shape of genealogical trees. eLife Sci. 3:e03568.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann P, Koblizkova A, Navratilova A, Macas J. 2006. Significant expansion of Vicia pannonica genome size mediated by amplification of a single type of giant retroelement. Genetics 173:1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel LE, Crick FH. 1980. Selfish DNA: the ultimate parasite. Nature 284:604–607. [DOI] [PubMed] [Google Scholar]
- Perlin MH, et al. 2015. Sex and parasites: genomic and transcriptomic analysis of Microbotryum lychnidis-dioicae, the biotrophic and plant-castrating anther smut fungus. BMC Genomics 16:461.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piegu B, et al. 2006. Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza Australiensis, a wild relative of rice. Genome Res. 16:1262–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. 2010. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritham EJ, Feschotte C. 2007. Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus: PNAS 104(6):1895–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaele S, Kamoun S. 2012. Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev. Microbiol. 10:417–430. [DOI] [PubMed] [Google Scholar]
- Sanmiguel P, Bennetzen JL. 1998. Evidence that a recent increase in maize genome size was caused by the massive amplification of intergene retrotransposons. Ann Bot 82:37–44. [Google Scholar]
- Schmidt D, et al. 2012. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 148:335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selker EU. 2002. Repeat-induced gene silencing in fungi. Adv Genet. 46:439–450. [DOI] [PubMed] [Google Scholar]
- Selker EU, Cambareri EB, Jensen BC, Haack KR. 1987. Rearrangement of duplicated DNA in specialized cells of Neurospora. Cell 51:741–752. [DOI] [PubMed] [Google Scholar]
- Singer MJ, Marcotte BA, Selker EU. 1995. DNA methylation associated with repeat-induced point mutation in Neurospora crassa. Mol Cell Biol. 15:5586–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon MI, Hufford MB, Gaut BS, Ross-Ibarra J. 2011. Genome size and transposable element content as determined by high-throughput sequencing in maize and Zea Luxurians. Genome Biol Evol. 3:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh SS, Treves DS, Barati MT, Perlin MH. 2016. Reliable transformation system for Microbotryum lychnidis. Arch Microbiol. 198:813–825 [DOI] [PubMed] [Google Scholar]
- Ungerer MC, Strakosh SC, Zhen Y. 2006. Genome expansion in three hybrid sunflower species is associated with retrotransposon proliferation. Curr Biol. 16:R872–R873. [DOI] [PubMed] [Google Scholar]
- Watters MK, Randall TA, Margolin BS, Selker EU, Stadler DR. 1999. Action of repeat-induced point mutation on both strands of a duplex and on tandem duplications of various sizes in Neurospora. Genetics 153:705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, et al. 2007. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 8:973–982. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.