

Abstract
The fungal kingdom displays an extraordinary diversity of lifestyles, developmental processes, and ecological niches. The MAPK (mitogen-activated protein kinase) cascade consists of interlinked MAPKKK, MAPKK, and MAPK, and collectively such cascades play pivotal roles in cellular regulation in fungi. However, the mechanism by which evolutionarily conserved MAPK cascades regulate diverse output responses in fungi remains unknown. Here we identified the full complement of MAPK cascade components from 231 fungal species encompassing 9 fungal phyla. Using the largest data set to date, we found that MAPK family members could have two ancestors, while MAPKK and MAPKKK family members could have only one ancestor. The current MAPK, MAPKK, and MAPKKK subfamilies resulted from duplications and subsequent subfunctionalization during the emergence of the fungal kingdom. However, the gene structure diversification and gene expansion and loss have resulted in significant diversity in fungal MAPK cascades, correlating with the evolution of fungal species and lifestyles. In particular, a distinct evolutionary trajectory of MAPK cascades was identified in single-celled fungi in the Saccharomycetes. All MAPK, MAPKK, and MAPKKK subfamilies expanded in the Saccharomycetes; genes encoding MAPK cascade components have a similar exon–intron structure in this class that differs from those in other fungi.
Keywords: fungi, MAPK cascades, gene family evolution, gene duplication, gene loss, evolutionary diversity
Introduction
The fungal kingdom, with an estimated 1.5 million species, displays extraordinary evolutionary diversity. This is reflected in different lifestyles, developmental processes, and ecological niches that make them central to every terrestrial ecosystem on the planet (Hawksworth 2001). Similarly to all other bioorganisms, fungi depend on the ability of their cells to sense and respond rapidly to changes in the surrounding environment; they exploit various signaling pathways, such as MAPK (mitogen-activated protein kinase) cascades, to coordinate cellular activities in response to environmental cues (Hamel et al. 2012).
An MAPK cascade generally consists of three interlinked components (the protein kinases MAPKKKs, MAPKKs, and MAPKs) that are activated sequentially. The activated MAPKs phosphorylate downstream substrates, affecting the biochemical properties of cells and leading to proper output responses. MAPK cascades have been functionally characterized in nonpathogenic fungi including Saccharomyces cerevisiae, Neurospora crassa, and Aspergillus nidulans (Park et al. 2008; Bayram et al. 2012; Furukawa and Hohmann 2013), and in pathogenic fungi such as the plant pathogenic fungi Magnaporthe oryzae, Ustilago maydis, and Fusarium graminearum (Xu 2000; Basse and Steinberg 2004; Jia and Tang 2015), the human pathogenic fungi Candida albicans, Cryptococcus neoformans, and Aspergillus fumigatus (Clarke et al. 2001; Fernandes et al. 2005), and the insect pathogenic fungi Metarhizium acridum and Beauveria bassiana (Zhang et al. 2009; Jin et al. 2014). These functional analyses showed that MAPK cascades regulate an extraordinary array of responses that are specific to each species. In contrast, MAPK cascades have long been considered to be evolutionarily conserved across the fungal kingdom (Rispail et al. 2009; Hamel et al. 2012). Therefore, the mechanism by which such a conserved signaling cascade can regulate so many output responses remains a fascinating question.
Evolutionary changes in the regulatory region, timing, or location of the gene expression of MAPK cascade components could be present among fungi. In addition, diversity also exists in signaling pathways interacting with MAPK cascades, such as calcium–calcineurin signaling, cAMP–PKA signaling pathway, G protein-coupled receptor signaling pathway, which could also facilitate MAPK-mediated regulation of fungal responses to diverse environmental conditions (Rispail et al. 2009). However, comparative genomics of MAPK cascade core components (designated as MAPK cascade components below)—that is, MAPKKK, MAPKK, and MAPK—in a limited number of saprophytic fungi, plant-associated fungi, and human pathogenic fungi also identified gene expansion in some fungi, which could be responsible for the adaptation of fungi to their respective ecological niches (Rispail et al. 2009; Hamel et al. 2012). Currently, a large number of genomes are available from taxonomically different fungi, which could facilitate a better understanding of the evolution of MAPK cascades in the fungal kingdom and their relationship with the diversity of fungal lifestyles, development processes, and adaptation to ecological niches. In this study, we identified the full complement of MAPK cascade components from 231 fungal species encompassing 9 fungal phyla. Together with MAPK cascade components from plants and animals, we investigated the evolutionary history of MAPK cascades in the fungal kingdom. We also found that the gene structure diversification and gain and loss of MAPK cascade components correlated with the evolution of fungal species and development of different lifestyles.
Materials and Methods
Identification of MAPK Cascade Components
An HMM (hidden Markov model) search was conducted to identify the full complement of MAPK cascade components from a fungal genome. The protein database of a fungal species was downloaded from National Center for Biotechnology or JGI (Joint Genome Institute). Nineteen MAPK, nine MAPKK, and eight MAPKKK proteins that have been functionally characterized in S. cerevisiae, U. maydis, M. oryzae, C r . neoformans, or C a . albicans (supplementary table S1, Supplementary Material online) were used as queries for training to determine the HMM parameters for searching MAPKs, MAPKKs, and MAPKKKs, respectively. We used MUSCLE (default parameters) to align the query proteins, and the command hmmbuild and hmmsearch in HMMER 3.1b1 (http://hmmer.org/; last accessed January 18, 2015) were used to implement the search experiments with default parameters (E-value = 10). Three selection criteria were exploited to remove false positives from the HMM search. The first criterion was the presence of at least one of the protein kinase domains of MAPK cascade components determined in the PFAM, SMART, and CDD databases. In the PFAM database, all of the MAPK cascade components have the protein kinase domain, PF00069; in the SMART database, the domain is smart00220. In the CDD database, four MAPK subfamilies have the domains cd07849, cd07857, cd07856, or cd07830. The three MAPKK subfamilies have cd06620, cd06621, and cd06622; and the three MAPKKK subfamilies have cd06626, cd06628, and cd06629. The second criterion was the presence of a conserved motif (D.K.N.*D.G.*PE.*D) in the MAPK cascade components as previously described (Hanks and Quinn 1991). After the above two rounds of removal, repeated sequences were manually deleted, and only one sequence was retained for further analysis.
The MAPK cascade components from plants and animals were retrieved with BLASTP using MAPK cascade components of S. cerevisiae as queries.
Phylogenetic Analysis and Classification of MAPK Cascade Components
Based on the annotation in the PFAM database, the protein kinase domain PF00069 of the MAPK cascade components was extracted using a local Perl script. The protein kinase domain sequences in each family were aligned using MUSCLE 3.7 (Edgar 2004) with default parameters. The alignments were then manually refined and end-trimmed to eliminate poor alignments and divergent regions. Unambiguously aligned positions were used to construct maximum-likelihood (ML) trees with MEGA version 6.0 (gaps treatment: partial deletion; model of evolution: WAG model; 100 bootstrap replications) (Tamura et al. 2013). The data sets for ML analysis were also used for Bayesian inference analysis with default parameters (model of evolution: WAG model; Mcmc ngen = 1000000; Samplefre = 1000) (Altekar et al. 2004).
An MAPK cascade component was classified based on phylogenetic and domain structure analysis. To classify a protein into a subfamily, it needed to meet two criteria. One was that, in the phylogenetic analysis, the protein needed to be in the clade that contained functionally characterized members of such subfamily (supplementary table S1, Supplementary Material online). The second was that, in the domain structure analysis, the protein should have the characteristic domain of such subfamily. Domain structure was analyzed by the online tools provided at the PFAM, CCD, and SMART databases.
Selection Pressure Assay
The selection pressure is presented as the Ka/Ks ratio. Protein sequences were aligned with MUSCLE v3.7 (Edgar 2004), which guided the alignment of their coding sequences with PAL2NAL (Suyama et al. 2006). Based on these two alignments, the nonsynonymous substitution rate (Ka), the synonymous substitution rate (Ks), and the Ka/Ks ratio were calculated pairwise using the MYN algorithm with the KaKs_Calculator v1.2 (Zhang et al. 2006). Compared with the mean Ks of proteins within a subfamily, pairs with unusually high Ks (range 10–99) or low Ks (near 0) were removed.
Results
Identification and Phylogenetic Analysis of MAPK Cascade Components
Using HMM search, we retrieved putative MAPK cascade components from 231 fungal species encompassing 9 fungal phyla: Ascomycota, Basidiomycota, Glomeromycota, Zygomycota, Blastocladiomycota, Chytridiomycota, Neocallimastigomycota, Microsporidia, and Cryptomycota (supplementary table S2, Supplementary Material online). HMM search identified many kinases other than MAPKs, MAPKKs, and MAPKKKs in all fungi (fig. 1), and the false positives were removed by examining the presence of characteristic domains and motifs of MAPK cascade components. In the end, 1,059 MAPK, 660 MAPKK, and 658 MAPKKK proteins were identified (fig. 1), which included all MAPKKKs, MAPKKs, and MAPKs identified in previous studies (Rispail et al. 2009; Hamel et al. 2012), suggesting that our method for retrieving MAPK cascade components from fungal genomes was effective.
Fig. 1.—
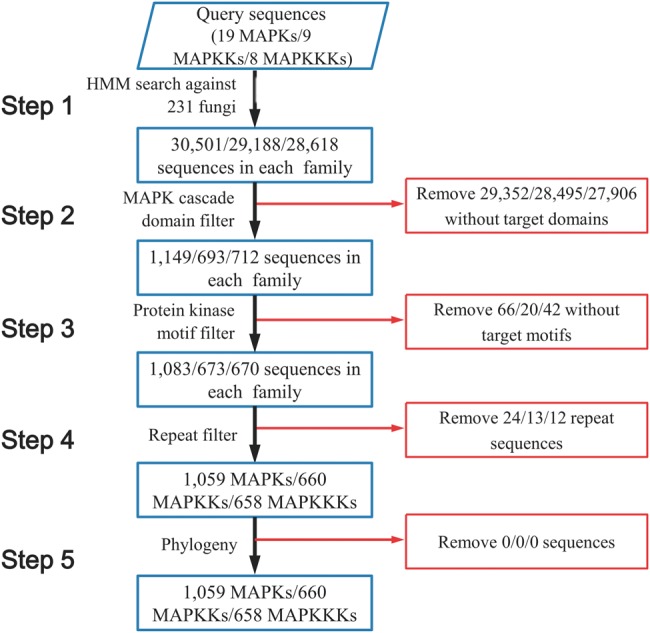
Flow chart of the identification of MAPK cascade components from 231 fungal species.
The kinase domain PF00069 from all of the MAPK cascade components were extracted and aligned using MUSCLE for the phylogenetic analyses. For the phylogenetic analysis of MAPK family members, 101 poorly aligned sequences were excluded (supplementary table S3, Supplementary Material online), and the remaining 958 sequences (supplementary data set 1, Supplementary Material online) were subjected for constructing phylogenetic trees using two methods: ML and Bayesian Inference (fig. 2A ). The resulting two trees consistently had four major clades with high statistical support, which contained the functionally characterized members from the Fus3/Kss1, Hog1, Slt2/Smk1, and Ime2-MAPK subfamilies, respectively (fig. 2A ). In each clade, all proteins had the characteristic domain of the functionally characterized subfamily members (supplementary table S4, Supplementary Material online). Therefore, the fungal MAPK family members from the 231 fungal species can be divided into 4 subfamilies: the Fus3/Kss1, Hog1, Slt2/Smk1, and Ime2-MAPK subfamilies. In the Slt2/Smk1-MAPK subfamily clade, there were two subclades with a high bootstrap value (100%) in the Bayesian inference tree, but the value was only 56% in the ML tree. One of the subclades had 31 proteins, including the only functionally sporulation-specific characterized Smk1-MAPK protein from S. cerevisiae (Gustin et al. 1998); the other subclade contained functionally characterized Slt2-MAPK proteins. The evolutionary origin of the fungal MAPK family was then phylogenetically analyzed using the 958 fungal MAPK and 31 MAPK proteins from plants or animals (supplementary data set 2, Supplementary Material online). The resulting trees consistently had two major clades with high statistical support (supplementary fig. S1 A and B). One contained fungal Ime2-MAPK proteins and their homologs from plants and animals. The other had fungal Fus3/Kss1, Hog1, and Slt2/Smk1-MAPK proteins and their plant and animal homologs; but this clade showed differences in topology between ML and Bayesian trees. Smk1 and Slt2 subclades were separated in the ML tree while the two groups formed a single Slt2/Smk1 subclade in the Bayesian inference tree.
Fig. 2.—

Phylogenetic analysis of MAPK family members (A), MAPKK family members (B), and MAPKKK family members (C). The trees were constructed with the protein sequence of the kinase domain PF00069 of MAPK cascade components. Numbers at nodes represent Bayesian posterior probabilities (left) and bootstrap values of ML (right), respectively.
The phylogenetic analysis of the MAPKK family was conducted using 617 fungal MAPKK proteins (supplementary data set 3, Supplementary Material online); the resulting ML and Bayesian inference trees consistently had 3 major clades with high statistical support. The three clades contained the functionally characterized members from the Mkk1/Mkk2, Ste7, and Pbs2-MAPKK subfamilies, respectively (fig. 2B ); all proteins in each clade contained the characteristic domain of the functionally characterized subfamily members (supplementary table S4, Supplementary Material online). Thus, the MAPKK family members from the 231 fungal species can be divided into the Mkk1/Mkk2, Ste7, and Pbs2-MAPKK subfamilies. The ML and Bayesian trees constructed using the 617 fungal MAPKK and 48 MAPKK proteins from plants or animals (supplementary data set 4, Supplementary Material online) also consistently contained 3 major clades with plant and animal MAPKKs as outgroups (supplementary fig. S1 C, Supplementary Material online).
To perform the phylogenetic analysis of the MAPKKK family, 599 fungal MAPKKK proteins were used for constructing phylogenetic trees (supplementary data set 5, Supplementary Material online). The ML and Bayesian inference trees consistently had three major clades with high statistical support, which contained functionally characterized members from the Bck1, Ste11, and Ssk2-MAPKKK subfamily, respectively (fig. 2C ). In addition, all proteins in each clade had the same domain characteristic of a subfamily (supplementary table S4, Supplementary Material online). Therefore, the Bck1, Ste11, and Ssk2-MAPKKK subfamilies constitute the fungal MAPKKK family. The ML and Bayesian trees constructed with the 599 fungal MAPKKK and 19 MAPKKK proteins from plants or animals also consistently contained 3 major clades with plant and animal MAPKKKs being clustered in outgroups (supplementary data set 6 and fig. S1D, Supplementary Material online).
The 218 proteins (101 MAPK, 57 MAPKK, and 60 MAPKKK proteins) (supplementary table S3, Supplementary Material online) that were excluded in the above phylogenetic analyses appeared to randomly distribute in fungal lineages. Phylogenetic and domain structure analyses were also conducted to classify these 218 proteins. For phylogenetic analyses, they were included one by one in the data set of above ML trees for a new ML analysis, and each of these proteins were grouped, with statistical support values being over 62, into one of the above phylogenetically determined 4 MAPK subfamilies, 3 MAPKK subfamilies, and 3 MAPKKK subfamilies. Meanwhile, domain structure analyses showed that each protein contained the characteristic domain of its subfamily determined by above phylogenetic and domain analyses. In the end, the 218 proteins were successfully classified. Hence, all of the putative MAPK, MAPKK, and MAPKKK proteins identified by the HMM search and the following selections were classified by the phylogenetic and domain analyses. Therefore, the repertoire of MAPK cascade components of the 231 fungal species is determined and summarized in supplementary table S4, Supplementary Material online.
Gain and Loss of MAPK Cascade Components
To illustrate the expansion and contraction of the MAPKKK, MAPKK, and MAPK family, the number of MAPK cascade components in each of the 231 fungal species was mapped to a fungal phylogenetic tree that was modified from the fungal life tree at JGI by replacing the parts consisting of Basidiomycota, Ascomycota, and Zygomycota with published phylogenetic trees (James et al. 2006) (fig. 3). Most of the fungi (72%) had all four MAPK, three MAPKK, and three MAPKKK subfamilies (fig. 3A ). A complete loss of the MAPK cascades was found in 18 fungal species, including 17 microsporidia and the Neocallimastigomycota fungus Piromyces sp. (fig. 3A and D ). In the remaining 213 species, no apparent relationship was identified between the loss of MAPK cascade components and fungal phylogeny.
Fig. 3.—

Distribution of MAPK cascades and their components in the fungal kingdom. (A) Pie diagram showing the number of fungal species with a complete (green), incomplete (brown), and null (gray) complement of MAPK cascade components. The percentage shows the proportion of the total of 231 fungal species. Complete complement: Presence of all four MAPK, three MAPKK, and three MAPKKK subfamilies; incomplete complement: Lack of at least one of the four MAPK, three MAPKK, and three MAPKKK subfamilies; null complement: No MAPK cascade components. (B) Bar diagram showing the presence of the MAPK cascade components in the 231 fungal species. The number of fungal species with only one member within each of the MAPK, MAPKK, and MAPKKK subfamilies is shown in blue, multiple in red, and none in gray. The upper left pie shows the number of fungal species with a complete set (green), incomplete set (brown), and none (gray) of the four MAPK subfamilies, and the upper middle and right pie shows the presence of three MAPKK and three MAPKKK subfamilies, respectively. (C) Bar diagram showing the number of fungal species with complete (blue), incomplete (brown), and no (gray) Fus3/Kss1-MAPK, Hog1-MAPK, and Slt2/Smk1-MAPK cascades. Pie diagram showing the percentage of the 231 fungal species with all 3 complete cascades (green), and that lack at least one cascade (brown), and without cascade (gray). (D) The distribution of the ten subfamilies of MAPK cascades (middle) and three MAPK cascades (right) in the nine fungal phyla. The left tree was the modified fungal life tree described in the Result section. For the distribution of MAPK cascade components (middle), the portions of the colored pie diagram show the percentage of the total number of species (purple) in a taxon with multiple members (orange), one member (blue), and no members (gray) within a subfamily. For the three MAPK cascades (right), the portions of the colored pie diagram show the percentage of the total number of species in a taxon with a complete MAPK cascade (blue), incomplete MAPK cascade (brown), and no MAPK cascade (gray).
The average number of members per species in the Ime2-MAPK, Hog1-MAPK, Ste7-MAPKK, Mkk-MAPKK, Pbs2-MAPKK, Ssk2-MAPKKK, Ste11-MAPKKK, and Bck1-MAPKKK subfamilies ranged from 1 to 1.1; 85–91% of the 213 fungal species had one member within each of the 8 subfamilies, with several cases of lineage-specific gene duplication (fig. 3D ). However, the Fus3/Kss1-MAPK and Slt2/Smk1-MAPK subfamilies had 1.5 and 1.3 members per species, respectively, indicating that they had undergone significant expansion. The expansion of the Fus3/Kss1-MAPK and Slt2/Smk1-MAPK subfamilies was clearly lineage specific (fig. 3D ). Excluding 2 species in the class Wallemiomycetes, the Fus3/Kss1-MAPK subfamily expanded in the remaining 9 classes of the Basidiomycota; 18 of the 48 examined Basidiomycota species (42%) had more than 3 members in the Fus3/Kss1-MAPK subfamily. In the Saccharomycetes of Ascomycota, 33 of the 36 species contained multiple members of the Fus3/Kss1-MAPK subfamily (fig. 3D ). The Slt2/Smk1-MAPK subfamily had multiple members in 29% of the 231 species, most of which belonged to Basidiomycota, Saccharomycetes of the Ascomycota, and Mucoromycotian of the Zygomycota (fig. 3D ).
All four MAPK, three MAPKK, and three MAPKKK subfamilies simultaneously expanded in the Agaricomycetes of the Basidiomycota and the Saccharomycetes of the Ascomycota (fig. 3D ). Excluding the Ste11-MAPKKK subfamily, the other nine subfamilies experienced expansion in the Mucoromycotina of the Zygomycota. Among the 213 species (excluding 17 Microsporidia and 1 Neocallimastigomycota), the average number of MAPK cascade components per species was 11.2, but the Agaricomycetes, Saccharomycetes, and Mucoromycotina fungi had 12.8, 13, and 16.9 components per species, respectively.
An MAPK cascade with a complete set of interlinked MAPKKKs, MAPKKs, and MAPKs is defined as a complete cascade; otherwise it is incomplete. A complete Fus3-MAPK, Hog1-MAPK, and Slt2-MAPK cascade was found in 192, 197, and 190 species, respectively (fig. 3C ). These three complete cascades were identified simultaneously in 171 (74%) species, most (96.5%) of which were dikarya fungi. Incomplete MAPK cascades were found more often in early diverging fungi (fig. 3D ).
Selective Pressure Acting on MAPK Cascade Components
The nonsynonymous to synonymous rate ratio Ka/Ks value is an indication of the change in selective pressures. Ka/Ks values <1, 1, and >1 indicate purifying selection, neutral evolution, and positive selection of the gene involved, respectively. We calculated the Ka/Ks values of the four MAPK, three MAPKK, and three MAPKKK subfamilies across the fungal kingdom, and all of them were <1 (fig. 4). We also calculated the Ka/Ks values for the Ascomycota, Basidiomycota, and classes with more than six species; all of the Ka/Ks values were smaller than 0.13. These data suggest that all of the MAPK, MAPKK, and MAPKKK subfamilies were under purifying selection.
Fig. 4.—

Pairwise estimation of the Ka/Ks values of the MAPK cascade components in the fungal kingdom. (A) Pairwise estimates of Ka/Ks values within members of each of the 4 MAPK, 3 MAPKK, and 3 MAPKKK subfamilies from the 231 fungal species. (B) The mean Ka/Ks values of the four MAPK, three MAPKK, and thee MAPKKK subfamilies across the fungal kingdom, two dikarya fungal phyla, and six major classes.
Diversification of the Sequence in the Activation Loop of MAPK Family Members
MAPK proteins are activated by dual phosphorylation of the conserved threonine and tyrosine residues in TXY motifs that are located in activation loops in the protein kinase domain (Widmann et al. 1999). We analyzed the diversity of the TXY motifs in the 4 MAPK subfamilies from the 231 species. For the Hog1-MAPK subfamily, 99.2% of the members had two TXY motifs, TGY and TRY, which were separated by two conserved amino acids (V and S) (fig. 5), suggesting that this subfamily is remarkably conserved in the TXY motifs. Similarly, great conservation was also found in the TXY motifs of the Fus3/Kss1-MAPK subfamily members. Among 314 members of the Fus3/Kss1-MAPK subfamily, 309 had the TEY motif (fig. 5).
Fig. 5.—

Diversification of the amino acid sequences of TXY motifs in the activation loops among members from each of the four MAPK subfamilies. Middle table: The number of members from an MAPK subfamily had a specific TXY motif named at the top of the table. Right panel: Logos showing the sequence conservation of the TXY motifs in the four MAPK subfamilies. The inserted logo shows the sequence conservation of the TXY motifs in Slt2/Smk1-MAPK subfamily members from the Saccharomycetes fungi. Left panel: Phylogenetic tree of the four MAPK subfamilies (simplified from fig. 2A). The digits in parentheses are the number of members in an MAPK subfamily.
Compared with the Hog1 and Fus3/Kss1-MAPK subfamily, a greater diversity in the TXY motifs was found in the Slt2/Smk1-MAPK subfamily, in which 86.5% of the 277 members had TEY (fig. 5). Among the 37 members without a TEY motif, 33 had TNY in the kinase domains and they were all from the Saccharomycetes yeast fungi. Thirty-one of the 33 TNY-containing members formed the Smk1-MAPK subclade of the Slt2/Smk1-MAPK clade in the phylogenetic tree of the MAPK family (fig. 2A ).
The greatest diversity in TXY motifs was found in the Ime2-MAPK subfamily. All 226 Ime2-MAPK subfamily members had TXY motifs in their protein kinase domain; 46.5% of the members had TTY and 23.6% had TEY (fig. 5).
Exon–Intron Structure of MAPK Cascade Component-Encoding Sequences
Because most of 5′ UTRs (untranslated region) and 3′ UTRs of genes are not available in the annotated genomes of the 231 fungal species, we counted the number of exons within the MAPK cascade component-encoding sequences (CDSs). In this study, the number of exons was used as a parameter to display the exon–intron structure of a gene. For all of the MAPK, MAPKK, and MAPKKK subfamilies, Basidiomycota fungi consistently had a greater number of exons than Ascomycota fungi (fig. 6A ). Among the five Ascomycota classes with at least seven species each, the genes from the Saccharomycetes had the fewest exons, most of which (fig. 6B ) had one exon. There were no obvious differences in the exon–intron structure among the other four classes of Ascomycota (fig. 6B ).
Fig. 6.—

The mean number of exons in each of the four MAPK, three MAPKK, and three MAPKKK subfamilies at the phylum level (A) and class level within the Ascomycota (B).
The number of exons in all members within each subfamily was plotted against the above-modified phylogenetic tree (fig. 7A ). For each of the ten subfamilies, the degree of exon number dispersion (demonstrated by the standard error of the exon number among members of a subfamily) among the genes appeared to be consistent with the taxonomic position at the phylum level. All of the MAPK cascade subfamilies in Basidiomycota fungi displayed greater exon number dispersion compared with the Ascomycota fungi (supplementary table S5, Supplementary Material online). In this study, a small number of species from each of early diverging fungal phyla were sampled, which were thus not considered in the analysis of exon number dispersion.
Fig. 7.—

Diversification of the gene structure of MAPK cascade CDSs. (A) The number of exons in all MAPK cascade components. In each subfamily, the 231 fungal species were organized according to their lineage status shown in the legend. For example, a red dot shows the number of exons in a gene from a Saccharomycetes fungus. (B) Stacked bar charts show the gene structure diversity among members of a subfamily. The digit within a bar shows the number of fungal species that demonstrate a difference (blue) or no difference (red) in gene structure among multiple subfamily members. (C) The diversity in gene structure among members within the Fus3/Kss1 or Slt2/Smk1-MAPK subfamily from Saccharomycete fungi (upper panel) and non-Saccharomycete fungi (down panel). The digits within the pie denote the number of fungal species with a difference (blue) or no difference (red) in gene structure among members of the Fus3/Kss1 or Slt2/Smk1-MAPK subfamily.
For the species with one member in a subfamily, the Basidiomycota fungi also had a greater number of exons than the Ascomycota (supplementary fig. S2, Supplementary Material online), and the degree of exon number dispersion in the Basidiomycota was greater than that in the Ascomycota (supplementary table S6, Supplementary Material online).
Regarding the species with multiple members in a subfamily, approximately half of them demonstrated diversity in exon–intron structure among the members within a subfamily, with the exception of the Hog1-MAPK and Bck1-MAPKKK subfamilies (fig. 7B ). Differences in exon–intron structure were identified among the multiple Hog1-MAPK subfamily members within each of the 17 out of 23 fungal species. Similarly, there were differences in exon–intron structure among the multiple Bck1-MAPKKK subfamily members within each of the 11 species (out of 17).
The Fus3/Kss1-MAPK and Slt2/Smk1-MAPK subfamily expanded in a significantly greater number of species compared with the other subfamilies, half of which showed differences in the number of exons among the members. However, the Saccharomycetes fungi exhibited distinct exon–intron structures compared with the other fungi. For the Fus2/Kss1-MAPK subfamily, there were no differences in exon number among the members within each of all 36 Saccharomycetes fungi; in contrast, a difference was identified in 78% of the other 46 species. Similarly, 74% of the Saccharomycetes fungi showed no exon–intron structure difference among the members of the Slt2/Smk1 family, but 77% of the other 27 species demonstrated a difference (fig. 7C ).
Discussion
In this study, we retrieved the full complement of MAPK cascade components from 231 fungal species encompassing 9 phyla. Using the largest set of data to date, we characterized the evolutionary origin of fungal MAPK cascade components, and the conservation and diversity of MAPK cascades across the fungal kingdom. The phylogenetic analysis of MAPKK proteins from fungi, plants, and animals revealed that fungal MAPKK family members might have one ancestor that underwent two duplications to produce the Ste7, Mkk, and Pbs2-MAPKK subfamilies. The presence of the Ste7 and Pbs2-MAPKK subfamilies in the basal phylum Cryptomycota indicates that the first duplication could have occurred in the very early stage of the emergence of the fungal kingdom, and the second duplication could have taken place after the divergence of the Cryptomycota from other fungal phyla. Similarly, the phylogenetic analysis showed that fungal MAPKKK family members had one ancestor that duplicated twice to produce the Bck1, Ssk2, and Ste11-MAPKKK subfamilies during an early stage of the emergence of the fungal kingdom. For MAPK family, ML and Bayesian trees had different topology in the clade containing Fus3-MAPKs, Slt2/Smk1-MAPKs, and Hog1-MAPKs and their plant and animal homologs, but they consistently had two major clades with high statistical support values. Therefore, both phylogenetic analysis methods showed that fungal MAPK family members might have two ancestors. The Fus3, Hog1, and Slt2-MAPK subfamilies might share one ancestor; the Ime2-MAPK subfamily arose separately with its own ancestor. Only one Hog1-MAPK protein and one Ime2-MAPK protein were identified in the basal phylum Cryptomycota. Excluding the highly reduced parasites (17 Microsporidia and 1 Neocallimastigomycota fungus) that had completely lost MAPK cascades, all of the other 212 species carried at least 3 MAPK subfamilies. Therefore, it is possible that the duplication of the ancestor of the Fus3/Kss1, Hog1, and Slt2/Smk1-MAPK subfamilies occurred after the divergence of the ancestor of Cryptomycota from that of other fungal phyla. Notably, only one genome (R ozella allomycis) was available for Cryptomycota in this study, so the lack of some MAPK cascade components could have resulted from the independent loss of the components in the fungus or from poor sequencing; additional genomes from this phylum are needed to better evaluate the timing of the duplication of the ancestors of MAPK cascade components.
The subfunctionalization that produced the Fus3/Kss1, Hog1, and Slt2/Smk1-MAPK subfamilies involves the diversification of TXY motif sequences in the activation loop (Fig. 5). Almost all of the Hog1-MAPK subfamily members carried the TGY motif, while most of the members of the Fus3/Kss1- and Slt2/Smk1-MAPK subfamilies had the TEY motif. Excluding the basal phylum (Cryptomycota) and two highly reduced parasites (Microsporidia and Neocallimastigomycota), all of the other phyla had the Hog1-MAPK subfamily and at least one of the Fus3/Kss1- and Slt2/Smk1-MAPK subfamilies, suggesting that diversification of the TXY motifs might also occur in the early stage during the emergence of the fungal kingdom. Taken together, all of the currently known MAPK, MAPKK, and MAPKKK subfamilies resulted from gene duplications followed by subfunctionalization during the early stage of the emergence of the fungal kingdom. Thus, MAPK cascades are remarkably conserved in the fungal kingdom.
Nevertheless, we identified significant diversity in the exon–intron structure of genes encoding MAPK cascade components. The degree of exon–intron structure diversification in MAPK cascade component-encoding genes in Basidiomycota was greater than Ascomycota. All of the MAPK, MAPKK, and MAPKKK subfamilies displayed a similar pattern of gene structure diversification between the Ascomycota and Basidiomycota, suggesting that these functionally related families share the same evolutionary history. Functionally related genes involved in other biological processes also share the same evolutionary history (Li et al. 2014). Notably, Hog1- and Fus3/Kss1-MAPK subfamily members are extremely conserved in the TXY motifs and under strong purifying selection, but they show significant diversification in exon–intron structure. This finding suggests that diversification of the exon–intron structure could be a major mechanism underlying the diversity in the Hog1- and Fus3/Kss1-MAPK subfamilies. The exon–intron structure is involved in virtually every step of mRNA processing and thus fulfills a broad spectrum of functions. The diversity of the exon–intron structure of genes encoding MAPK cascade components in fungi should provide great plasticity to the evolutionarily conserved MAPK cascades to regulate diverse developmental processes, different lifestyles, and adaptation to various ecological niches.
Gene expansion and loss are other important mechanisms in the diversification of MAPK cascades in the fungal kingdom. The Fus3/Kss1 and Slt2/Smk1-MAPK subfamilies showed greater expansion than the other MAPK subfamilies and the MAPKK and MAPKKK families; their expansions were clearly lineage specific. However, it remains to be elucidated how these expansions contributed to the evolution of the specific lineages of fungi. In contrast, no clear relationship was identified between the loss of MAPK cascade components and fungal phylogeny, with the exception of the two lineages of highly reduced parasites (Microsporidia and the Neocallimastigomycota fungus Piromyces sp.). Because no MAPK cascade components were identified in all 17 species of Microsporidia, the complete loss of MAPK cascades could be universal in Microsporidia. The 17 microsporidia were restricted to different animals, including mammals, fish, and insects; therefore, the loss of MAPK cascades could have occurred prior to the development of host specificity. Microsporidia have the smallest known (nuclear) eukaryotic genomes because their parasitic lifestyle has led to the loss of many genes (Keeling and Fast 2002). In this study, we found that signaling conferred by MAPK cascades was also not required for Microsporidia to form host–parasite relationships. We also found that such signaling was not required for the Neocallimastigomycota fungus Piromyces sp. (an obligate anaerobic symbiont of herbivorous mammals) to develop symbiotic relationship with its host because complete loss of the MAPK cascade also occurred in this fungus.
Compared with other fungal taxa, the MAPK cascades of the Saccharomycetes fungi appear to have undergone a unique evolutionary trajectory. First, genes encoding MAPK cascade components in the Saccharomycetes fungi have a distinct exon–intron structure because they all lack introns in their CDSs, in contrast to other fungal classes that display significant diversity in exon–intron structure. The lack of introns in genes encoding MAPK cascade components is similar to many other genes in Saccharomycete yeasts (Juneau et al. 2007). Second, only the Saccharomycetes fungi have Smk1-MAPK proteins. Although Smk1-MAPK proteins have been grouped into the Slt2/Smk1-MAPK subfamily based on current data, they have different TXY motifs in the activation loop. Finally, all of the MAPK, MAPKK, and MAPKKK subfamilies expanded in Saccharomycetes, which demonstrated the second largest number of MAPK cascade components (13 per species) among all of the examined classes, indicating a strong redundancy among the components. This is consistent with the whole-genome duplication in the Saccharomycetes lineage (Piskur 2001).
In conclusion, using the largest collection of fungal MAPK cascade components to date, we characterized the evolutionary history of fungal MAPK cascades. The currently existing MAPK, MAPKK, and MAPKKK subfamilies resulted from duplications and subsequent subfunctionalizations during the early stage of the emergence of the fungal kingdom; the fungal MAPK cascades are thus remarkably conserved across the fungal kingdom. However, we found that the diversification of gene structure and gene expansion and loss are important mechanisms underlying the significant diversity observed in the core components of fungal MAPK cascades. These diversifications correlate with the evolution of fungal species and lifestyles; thus, they could be important mechanisms facilitating evolutionarily conserved MAPK cascades to regulate a great diversity of fungal output responses that are involved in different development processes, lifestyles, and adaptation to diverse ecological niches.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was funded by the National Natural Science Foundation of China (31272097 and 31471818) and Zhejiang Provincial Natural Science Foundation of China (LR13C010001) and “1000 Young Talents Program of China.”
Literature Cited
- Altekar G, Dwarkadas S, Huelsenbeck JP, Ronquist F. 2004. Parallel metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics 20:407–415. [DOI] [PubMed] [Google Scholar]
- Basse CW, Steinberg G. 2004. Ustilago maydis, model system for analysis of the molecular basis of fungal pathogenicity. Mol Plant Pathol. 5:83–92. [DOI] [PubMed] [Google Scholar]
- Bayram Ö, et al. 2012. The Aspergillus nidulans MAPK module AnSte11-Ste50-Ste7-Fus3 controls development and secondary metabolism. PLoS Genet. 8:e1002816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke DL, Woodlee GL, McClelland CM, Seymour TS, Wickes BL. 2001. The Cryptococcus neoformans STE11alpha gene is similar to other fungal mitogen-activated protein kinase kinase kinase (MAPKKK) genes but is mating type specific. Mol Microbiol. 40:200–213. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes L, et al. 2005. Cell signaling pathways in Paracoccidioides brasiliensis—inferred from comparisons with other fungi. Genet Mol Res. 4:216–231. [PubMed] [Google Scholar]
- Furukawa K, Hohmann S. 2013. Synthetic biology: lessons from engineering yeast MAPK signalling pathways. Mol Microbiol. 88:5–19. [DOI] [PubMed] [Google Scholar]
- Gustin MC, Albertyn J, Alexander M, Davenport K. 1998. MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 62:1264–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel LP, Nicole MC, Duplessis S, Ellis BE. 2012. Mitogen-activated protein kinase signaling in plant-interacting fungi: distinct messages from conserved messengers. Plant Cell 24:1327–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM. 1991. Protein kinase catalytic domain sequence database: identification of conserved features of primary structure and classification of family members. Methods Enzymol. 200:38–62. [DOI] [PubMed] [Google Scholar]
- Hawksworth DL. 2001. The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res. 105:1422–1432. [Google Scholar]
- James TY, et al. 2006. Reconstructing the early evolution of fungi using a six-gene phylogeny. Nature 443:818–822. [DOI] [PubMed] [Google Scholar]
- Jia LJ, Tang WH. 2015. The omics era of Fusarium graminearum: opportunities and challenges. New Phytol. 207:1–3. [DOI] [PubMed] [Google Scholar]
- Jin K, Han L, Xia Y. 2014. MaMk1, a FUS3/KSS1-type mitogen-activated protein kinase gene, is required for appressorium formation, and insect cuticle penetration of the entomopathogenic fungus Metarhizium acridum. J Invertebr Pathol. 115:68–75. [DOI] [PubMed] [Google Scholar]
- Juneau K, Palm C, Miranda M, Davis RW. 2007. High-density yeast-tiling array reveals previously undiscovered introns and extensive regulation of meiotic splicing. Proc Natl Acad Sci U S A. 104:1522–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, Fast NM. 2002. MICROSPORIDIA: biology and evolution of highly reduced intracellular parasites. Annu Rev Microbiol. 56:93–116. [DOI] [PubMed] [Google Scholar]
- Li Y, Calvo SE, Gutman R, Liu JS, Mootha VK. 2014. Expansion of biological pathways based on evolutionary inference. Cell 158:213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park G, Pan S, Borkovich KA. 2008. Mitogen-activated protein kinase cascade required for regulation of development and secondary metabolism in Neurospora crassa. Eukaryot Cell. 7:2113–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskur J. 2001. Origin of the duplicated regions in the yeast genomes. Trends Genet. 17:302–303. [DOI] [PubMed] [Google Scholar]
- Rispail N, et al. 2009. Comparative genomics of MAP kinase and calcium-calcineurin signalling components in plant and human pathogenic fungi. Fungal Genet Biol. 46:287–298. [DOI] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. 2006. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34:609–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 30:2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. 1999. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 79:143–180. [DOI] [PubMed] [Google Scholar]
- Xu JR. 2000. MAP kinases in fungal pathogens. Fungal Genet Biol. 31:137–152. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, et al. 2009. Mitogen-activated protein kinase hog1 in the entomopathogenic fungus Beauveria bassiana regulates environmental stress responses and virulence to insects. Appl Environ Microbiol. 75:3787–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Li J, Yu J. 2006. Computing Ka and Ks with a consideration of unequal transitional substitutions. BMC Evol Biol. 6:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.