

Summary
The UAF1-USP1 complex deubiquitinates FANCD2 during execution of the Fanconi anemia DNA damage response pathway. As such, UAF1 depletion results in persistent FANCD2 ubiquitination and DNA damage hypersensitivity. UAF1 deficient cells are also impaired for DNA repair by homologous recombination. Herein, we show that UAF1 binds DNA and forms a dimeric complex with RAD51AP1, an accessory factor of the RAD51 recombinase, and a trimeric complex with RAD51 through RAD51AP1. Two SUMO-like domains in UAF1 and a SUMO-interacting motif in RAD51AP1 mediate complex formation. Importantly, UAF1 enhances RAD51-mediated homologous DNA pairing in a manner that is dependent on complex formation with RAD51AP1 but independent of USP1. Mechanistically, RAD51AP1-UAF1 co-operates with RAD51 to assemble the synaptic complex, a critical nucleoprotein intermediate in homologous recombination, and cellular studies reveal the biological significance of the RAD51AP1-UAF1 protein complex. Our findings provide insights into an apparently USP1-independent role of UAF1 in genome maintenance.
Introduction
Fanconi anemia (FA) is characterized by bone marrow failure, developmental defects, and cancer predisposition (Cohn and D'Andrea, 2008; Longerich et al., 2014). FA patient cells are highly sensitive to DNA interstrand crosslink (ICL)-inducing agents, such as mitomycin C (MMC), and they accumulate chromosomal aberrations upon exposure to these agents (Kee and D'Andrea, 2010). FA proteins from 19 complementation groups and associated factors execute DNA damage response (Longerich et al., 2014; Rickman et al., 2015; Wang et al., 2015). Herein, DNA damage induces the mono-ubiquitination of the FANCI-FANCD2 (ID2) heterodimer, licensing signaling and DNA repair steps, including repair via homologous recombination (HR) mediated by the recombinase RAD51 (also known as FANCR) and its ancillary factors (Cohn and D'Andrea, 2008; Garcia-Higuera et al., 2001).
Timely deubiquitination of FANCD2 is critically important for the FA pathway (Oestergaard et al., 2007). USP1 is the deubiquitinating enzyme (DUB) for monoubiquitinated FANCD2 and PCNA (Huang et al., 2006; Nijman et al., 2005). USP1 stably associates with UAF1/WDR48, and its DUB activity and intracellular stability are dependent on the latter (Cohn et al., 2007; Villamil et al., 2012). UAF1 also interacts with and stimulates other DUBs, such as USP12 and USP46 (Cohn et al., 2009; Sowa et al., 2009).
Similar to USP1 deficiency, depletion of UAF1 results in increased levels of mono-ubiquitinated FANCD2 and PCNA and also leads to hypersensitivity to ICLs (Kim et al., 2009; Park et al., 2013). A more severe impairment of HR occurs in UAF1 mutant cells (Murai et al., 2011; Park et al., 2013). Importantly, heterozygous Uaf1+/- mouse embryonic fibroblasts exhibit a marked hypersensitivity to MMC, etoposide, and other DNA damaging agents and are compromised for HR (Park et al., 2013).
In a large scale proteomic analysis, UAF1-USP1 was found to associate with RAD51AP1 (Sowa et al., 2009), a DNA binding protein and co-factor for RAD51 (Modesti et al., 2007; Wiese et al., 2007). Herein, we demonstrate that UAF1 binds DNA and employs two SUMO-like domains (SLDs) to associate with RAD51AP1 via a SUMO-interacting motif (SIM) in the latter. Importantly, we find that UAF1 synergizes with RAD51AP1 to enhance RAD51-mediated homologous DNA pairing, specifically by promoting the assembly of the synaptic complex, in which ssDNA derived from the nucleolytic processing of a primary lesion is homologously aligned with a duplex molecule (San Filippo et al., 2008). Physical interaction between RAD51AP1 and UAF1 is indispensable for functional synergy in vitro and, accordingly, for protein function in HR and DNA damage repair. Our results shed light on a USP1-independent role of UAF1 in genome maintenance.
Results
Definition of the RAD51AP1-UAF1 interaction interface
We expressed UAF1 in insect cells and devised a method for its purification (see Experimental Procedures), while RAD51AP1 was purified following our published procedure (Wiese et al., 2007). Affinity pulldown verified that RAD51AP1 and UAF1 form a stoichiometric complex (Figure 1A, lane 3).
Figure 1. RAD51AP1-UAF1 complex formation via the SIM and SLD1-SLD2 domains in these proteins.
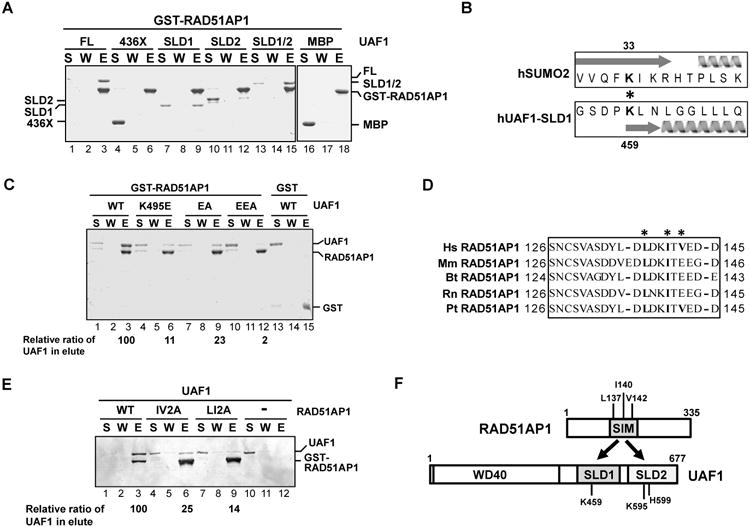
(A) Strep II-tagged UAF1, UAF1-436X, MBP-tagged UAF1-SLD1, UAF1-SLD2, and UAF1-SLD1/SLD2 (SLD1/2) were incubated with GST-tagged RAD51AP1, and protein complexes were captured on glutathione resin and analyzed by SDS-PAGE. S, supernatant containing unbound proteins; W, wash; E, SDS eluate of the glutathione resin.
(B) Alignment of UAF1-SLD1 against SUMO2 is shown. The asterisk highlights the K459 and K33 residues in UAF1 and SUMO2, respectively. Arrow and helix represent beta sheet and alpha helix, respectively.
(C) GST-tagged RAD51AP1 was incubated with Strep II-tagged UAF1 (WT) or the indicated UAF1 mutant, and protein complexes were captured with glutathione resin. Analysis was as in (A).
(D) Sequence analysis reveals a SIM between amino acid residues 137-142 in RAD51AP1. The asterisks highlight the residues targeted for mutagenesis. Hs, Homo sapiens; Mm, Mus musculus; Bt, Bos taurus; Rn, Rattus norvegicus; Pt, Pan troglodytes.
(E) GST-tagged RAD51AP1 (WT) or the indicated RAD51AP1 mutant was incubated with Strep II-tagged UAF1, and protein complexes were captured with glutathione resin. Analysis was as above.
(F) Schematic highlighting the RAD51AP1-UAF1 interaction domains.
See also Figure S1.
UAF1 possesses two SLDs within its C-terminal half, with SLD2 implicated in complex formation with RAD51AP1 (Yang et al., 2011). We purified MBP-tagged SLD1, SLD2, SLD1-SLD2, and the N-terminal 436 residues (436X) to test for RAD51AP1 binding. The results revealed that while the 436X fragment does not interact with RAD51AP1 (Figure 1A, lane 6), both SLD1 and SLD2 are capable of doing so, but SLD1-SLD2 has a higher affinity for RAD51AP1 (Figure 1A, lanes 9, 12 and 15).
Lysine 595 (K595) and histidine 599 (H599) in SLD2 are likely analogous to K33 and H37 in SUMO-2 (Yang et al., 2011), which are important for association with the SUMO interaction motif (SIM) in client proteins (Sekiyama et al., 2008). Using the Phyre2 algorithm (Kelley et al., 2015), we identified K459 in SLD1 to be analogous to K33 in SUMO-2 (Figure 1B). We changed K459 in SLD1 to glutamic acid (K459E) and K595 and H599 in SLD2 individually or together to glutamic acid and alanine (K595E and H599A). By affinity pulldown, we found that K459E mutant is attenuated for RAD51AP1 interaction (Figure 1C, lane 6), while K595E and H599A single mutants are interaction proficient (Figure S1A), although the K595E/H599A double mutant (referred to as the EA mutant) is impaired in this regard (Figure 1C, lane 9). Moreover, we constructed the K459E/K595E/H599A triple mutant (referred to as the EEA mutant) and showed that it is more impaired for RAD51AP1 interaction than are the K459E and EA mutants (Figure 1C, lane 12). Thus, SLD1 and SLD2 of UAF1 both contribute to RAD51AP1 interaction.
By testing RAD51AP1 fragments (Dunlop et al., 2011), we discovered that the UAF1 interaction domain resides within amino acid residues 95-187 of the protein (Figure S1B, lane 9). Since our result differs from a published report suggesting that residues 11-24 of RAD51AP1 are important for UAF1 interaction (Yang et al., 2011), we constructed and tested the ΔN25 mutant missing the N-terminal 25 residues, but found that it is fully capable of UAF1 binding (Figure S1C, lane 6).
We examined additional RAD51AP1 fragments and found that the 1-145 fragment is proficient in UAF1 binding but the 1-132 fragment is not (Figure S1D). Thus, the region within residues 133-145 of RAD51AP1 is indispensable for UAF1 interaction. Consistent with the fact that UAF1 engages RAD51AP1 via two SLDs, we identified a I/L/V-rich sequence between residues 137-142 of RAD51AP1 that fits the SIM signature (Figure 1D) (Hecker et al., 2006; Sun and Hunter, 2012). Indeed, the L137A/I140A (LI2A) and I140A/V142A (IV2A) RAD51AP1 mutants are both compromised for UAF1 interaction (Figure 1E). Thus, the RAD51AP1-UAF1 complex is formed via interaction between SLD1-SLD2 of UAF1 and the SIM motif in RAD51AP1 (Fig. 1F).
We verified that the UAF1 mutants possess wild type affinity for USP1 and FANCI (Figure S1E, F) and that the RAD51AP1 mutants retain the ability to interact with RAD51 (Figure S1G). We also tested the UAF1 mutants for the ability to stimulate the DUB attribute of USP1, using ubiquitin vinyl sulfone (Ub-VS) as activity probe (Borodovsky et al., 2001). The results showed that the UAF1 mutants are as capable as the wild type protein in enhancing USP1 activity (Figure S1H). Thus, the mutants that we have constructed (Table S1) are specifically compromised for RAD51AP1-UAF1 complex formation.
Association of UAF1 with RAD51 via RAD51AP1
RAD51AP1 physically interacts with RAD51 (Modesti et al., 2007; Wiese et al., 2007). We found that UAF1 associates with RAD51 only when RAD51AP1 is present (Figure 2A, lane 18), suggesting a bridging role of RAD51AP1 (Figure 2A). Consistent with this deduction, affinity pulldown showed that SLD1-SLD2 of UAF1 interacts with RAD51 via RAD51AP1, whereas the UAF1-EEA and RAD51AP1-LI2A mutants (Figure 1C, E) fail to form the trimer (Figure 2A). The trimeric complex is species-specific, as yeast Rad51 has no affinity for RAD51AP1-UAF1 (Figure 2B).
Figure 2. Association of UAF1 with RAD51 via RAD51AP1 and DNA-binding activity in UAF1.
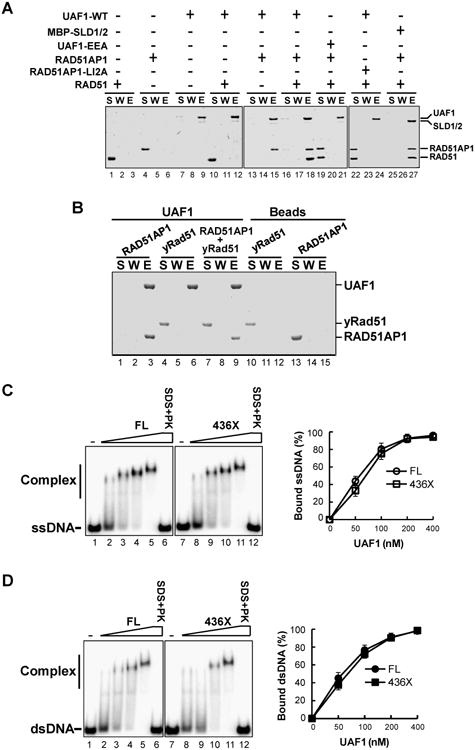
(A) Strep II-tagged UAF1 (WT) or UAF1-EEA was incubated with RAD51AP1, RAD51AP1-LI2A, and RAD51 alone or in combination. Protein complexes were captured on Strep-Tactin resin and the different fractions were analyzed as in Figure 1A. MBP-tagged UAF1 SLD1-SLD2 (SLD1/2) was similarly incubated with RAD51AP1 and RAD51, and amylose resin was used to capture the trimeric protein complex. A control experiment confirmed that RAD51AP1-RAD51 does not bind amylose resin nonspecifically (data not shown).
(B) Strep II-tagged UAF1 was incubated with RAD51AP1, yeast Rad51 or their combination, and Strep-Tactin pulldown was carried out as in (A).
(C) Strep II-tagged UAF1-FL or UAF1-436X was incubated with radiolabeled 80-merss DNA. The mobility shift of the DNA was analyzed. Treatment with SDS and proteinase K (SDS+PK) released the DNA from nucleoprotein complexes. The data were quantified and plotted. The error bars represent mean values ± S.D. of data from three independent experiments.
(D) The ability of UAF1 and UAF1-436X to bind radiolabeled dsDNA was analyzed as in (C).
See also Figure S2.
DNA binding activity in UAF1
RAD51AP1 possesses a DNA binding activity (Modesti et al., 2007; Wiese et al., 2007). Importantly, we found that UAF1 and UAF1-436X both bind ssDNA and dsDNA with nanomolar affinity (Figure 2C, D), and that the UAF1-EEA and RAD51AP1 IV2A and LI2A mutants possess wild type DNA binding activity (Figure S2).
Synergistic action of RAD51AP1 and UAF1 in RAD51 recombinase enhancement
We used the D-loop assay (Wiese et al., 2007) (Figure 3A) to test UAF1 for the ability to enhance the recombinase activity of RAD51. The results showed that UAF1 does not affect D-loop formation (Figure 3B, lanes 3-5), while, as expected (Wiese et al., 2007), RAD51AP1 stimulates the D-loop reaction (Figure 3B, lane 6). Interestingly, the combination of RAD51AP1 and UAF1 enhanced D-loop formation by several fold compared to RAD51AP1 alone (Figure 3B, lanes 8-10). The action of RAD51AP1-UAF1 is species-specific, as the D-loop reaction catalyzed by yeast Rad51 was not affected by it (Figure S3A).
Figure 3. Synergistic action of RAD51AP1 and UAF1 in RAD51-mediated D-loop formation.
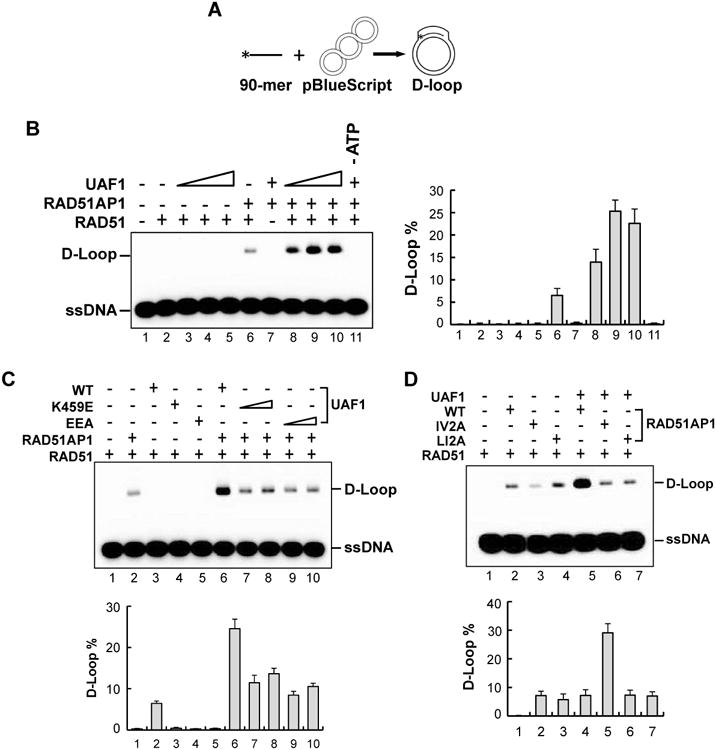
(A) Schematic of the D-loop assay.
(B) RAD51, RAD51AP1, Strep II-tagged UAF1, and the indicated combinations of these proteins were tested for the ability to form D-loops. The percentages of D-loop product from three independent experiments are shown as the mean ± S.D.
(C) The K459E and EEA mutants of UAF1 were tested with RAD51AP1 in the D-loop reaction. Data analysis was as in (B).
(D) The IV2A and LI2A mutants of RAD51AP1 were tested with UAF1 in the D-loop reaction.
See also Figure S3.
Importance of the RAD51AP1-UAF1 complex in RAD51 enhancement
We asked whether synergistic enhancement of the D-loop reaction by RAD51AP1 and UAF1 is contingent upon complex formation between them. Importantly, the results showed that UAF1 mutants (K459E, EA and EEA) compromised for RAD51AP1 interaction (Figure 1C) are less effective in the D-loop reaction (Figure 3C, S3B), whereas the K595E and H599A mutants, which retain the ability to bind RAD51AP1 (Figure S1A), remain proficient in this regard (Figure S3B). Conversely, we found that the RAD51AP1 IV2A and LI2A mutants with impaired UAF1 binding fail to synergize with UAF1 in the D-loop reaction (Figure 3D). We note that these RAD51AP1 mutants are as capable as the wild type counterpart in RAD51 enhancement (Figure 3D). Taken together, the results indicate that complex formation between RAD51AP1 and UAF1 is indispensable for their functional synergy in the D-loop reaction.
We previously described mutants of RAD51AP1 that are either defective in DNA binding (the N-K6RA/C-K7WA mutant) or in RAD51 interaction (the H329A mutant) (Dunlop et al., 2012; Wiese et al., 2007). Affinity pulldown showed that both RAD51AP1 mutants are proficient in complex formation with UAF1 (Figure S3C, D). Importantly, these RAD51AP1 mutants could not stimulate the D-loop reaction even with UAF1 present (Figure S3E). Therefore, the RAD51 interaction and DNA binding attributes of RAD51AP1 are critically important for the functionality of the RAD51AP1-UAF1 complex.
UAF1-USP1 readily associates with RAD51AP1 to form a trimeric complex (Figure S3F). However, like UAF1, neither USP1 nor the UAF1-USP1 complex exerts any influence on the D-loop reaction (Figure S3G; data not shown). Moreover, we verified that (i) UAF1-USP1 is no more effective than UAF1 in the D-loop reaction (Figure S3H), and (ii) USP1 fails to restore functional synergy of the UAF1 EEA mutant with RAD51AP1 (Figure S3I). Thus, USP1 plays no role in the D-loop reaction.
Function of RAD51AP1-UAF1 in synaptic complex assembly
RAD51AP1 enhances synaptic complex assembly that is mediated by the RAD51-ssDNA nucleoprotein filament, commonly referred to as the presynaptic filament (Dray et al., 2010). As reported previously and confirmed here, RAD51AP1 stimulated duplex capture (Figure 4A) by the RAD51 presynaptic filament (Figure 4B, lane 9). As in the D-loop assay, while UAF1 alone had no effect on duplex capture (Figure 4B, lanes 5, 6), its addition with RAD51AP1 enhanced the reaction several fold over the RAD51AP1 alone level (Figure 4B, lanes 10-12). Importantly, the UAF1 K459E and EEA mutants (Figure 1C; Table S1), which are deficient in RAD51AP1 interaction, were less effective in the reaction (Figure S4A). Notably, the RAD51 presynaptic filament failed to capture ssDNA even with RAD51AP1-UAF1 present (Figure S4B).
Figure 4. Duplex DNA capture and synaptic complex assembly by RAD51-RAD51AP1-UAF1.
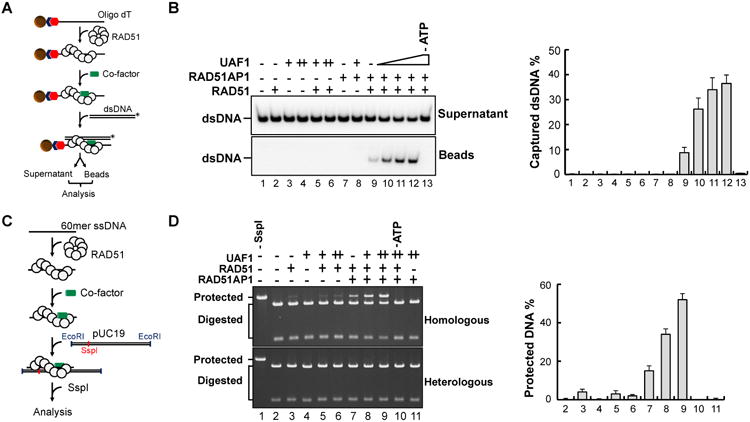
(A) Schematic of the duplex capture assay.
(B) RAD51AP1, Strep II-tagged UAF1, or the combination of these proteins was incubated with the RAD51 presynaptic filament, and the ability to capture dsDNA was analyzed. The percentages of captured dsDNA are shown. The error bars represent mean values ± S.D. of data from three independent experiments.
(C) Schematic of synaptic complex assembly as assayed by protection against SspI digestion.
(D) RAD51AP1, Strep II-tagged UAF1, or their combination was incubated with the RAD51 presynaptic filament, and the protection of dsDNA against SspI digestion was analyzed. Lane 1 was the control without SspI treatment. The protected DNA was quantified and plotted. Error bars are mean ± S.D. of three independent experiments.
See also Figure S4.
We showed previously that RAD51AP1 enhances synaptic complex formation (Figure 4C), and this was confirmed here (Figure 4D, lane 7). While UAF1 alone was unable to promote synaptic complex formation, its co-addition with RAD51AP1 led to strong synergy (Figure 4D, lanes 8, 9). Functional synergy was not observed for the UAF1-EEA and RAD51AP1-VI2A and -LI2A mutants that are compromised for protein complex formation (Figure S4C, D). Taken together, the results unveil a role of the RAD51AP1-UAF1 complex in the synaptic stage of the HR process.
Dependence of DNA damage repair and HR on the RAD51AP1-UAF1 complex
Consistent with affinity pulldown results (Figure 1), while UAF1 co-immunoprecipitated RAD51AP1, the K459E and EEA mutants failed to do so (Figure 5A). In contrast, USP1 co-immunoprecipitated with UAF1 and its mutant forms. A previous study provided evidence that K595 in SLD2 of UAF1 contributes to ELG1 interaction (Yang et al., 2011), which was confirmed by diminished ELG1 that co-immunoprecipitated with the EEA mutant harboring the K595E mutation, although the UAF1 SLD1-K459E mutation had no such effect (Figure 5A). While RAD51AP1 co-immunoprecipitated UAF1 efficiently, the IV2A and LI2A mutants were largely unable to do so (Figure S5A).
Figure 5. Role of the RAD51AP1-UAF1 complex in DNA damage repair and HR.
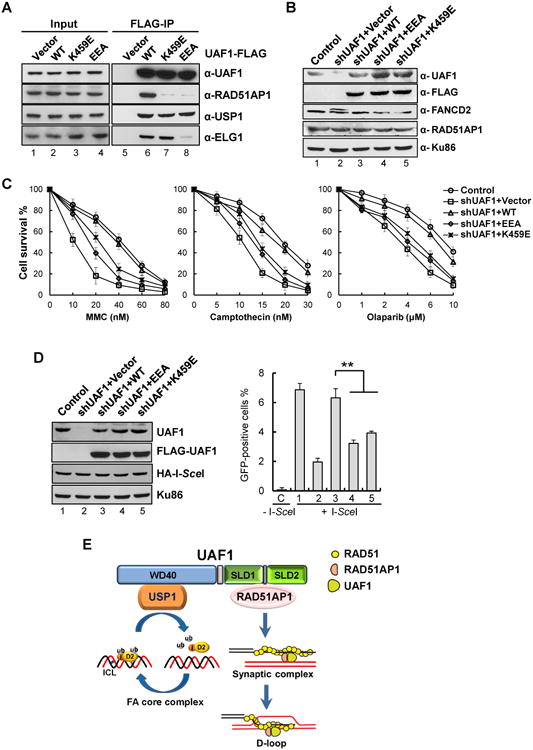
(A) Extracts from HeLa cells expressing wild type (WT) or the mutant form (K459E or EEA) of FLAG-tagged UAF1 were subject to co-immunoprecipitation analysis with anti-FLAG M2 agarose resin. Proteins were revealed by Western blotting.
(B) Protein levels in HeLa cells with constitutive shRNA-mediated knockdown of UAF1 and stably expressing either wild type or the indicated mutant form of UAF1 were determined by Western blotting. Cells without shUAF1 were included as control. Ku86 was used as the loading control.
(C) HeLa cells described in (B) were treated with MMC, CPT or olaparib, and cell numbers were determined after incubation at 37°C for 5 days and normalized to the untreated control. The percentages of surviving cells are shown as the mean ± S.D. from three independent experiments.
(D) U2OS-DR-GFP cells with constitutive shUAF1-mediated knockdown of UAF1 and stably expressing either wild type or a mutant form of UAF1 were transfected with HA-tagged I-SceI plasmid and processed for flow cytometric analysis of GFP. The protein levels are shown in the left panel. The repair efficiency was scored as the percentage of GFP-positive cells. C, Control cells without I-SceI; 1, shRNA control cells; 2, shUAF1 cells; 3,shUAF1 cells with UAF1; 4, shUAF1 cells with UAF1-EEA; 5, shUAF1 cells with UAF1-K459E. Error bars indicate S.E.M. ** indicates P < 0.01, unpaired t-test.
(E) Model for the roles of UAF1 in the FA pathway and HR. UAF1 functions as the substrate targeting subunit of the DUB enzyme central to the FA pathway and also as an important cofactor of RAD51-RAD51AP1 to facilitate the assembly of the synaptic complex during HR.
See also Figure S5.
As expected (Murai et al., 2011; Park et al., 2013), UAF1 depletion in human cells led to hypersensitivity to MMC, camptothecin (CPT), and olaparib (a PARP inhibitor) (Figure 5B, C). Complementation with wild type UAF1 conferred resistance to MMC, CPT and olaparib, but neither the K459E nor EEA mutant could fully restore resistance (Figure 5B, C) even though UAF1-depleted cells and cells expressing the mutants had the wild type level of RAD51AP1 (Figure 5B). Consistent with these data, the RAD51AP1 IV2A and LI2A mutants were less capable than their wild type counterpart in restoring MMC resistance to cells (Figure S5B).
We found that while cells depleted for UAF1 or expressing the UAF1 mutants exhibit WT cell-cycle profile without DNA damage, CPT treatment leads to an increased accumulation of mutant UAF1 cells in the G2/M phase (Figure S5C). We also noted that cells expressing the UAF1 mutants harbor mostly deubiquitinated FANCD2, whereas UAF1 depleted cells show an elevated level of monoubiquitinated FANCD2 (Figure 5B) as previously shown (Murai et al., 2011; Park et al., 2013). These observations are consistent with our finding that complexes of USP1 and the UAF1 mutants retain DUB activity (Figure S1H).
We next inquired if HR is reliant on the RAD51AP1-UAF1 complex. As expected (Park et al., 2013; Yang et al., 2011), UAF1 knockdown caused a 3-4 fold decrease in HR proficiency (Figure 5D, right panel, lane 2). Importantly, while introducing the UAF1 gene restored HR proficiency, the K459E and EEA mutant genes were less capable of complementation (Figure 5D, right panel, lanes 3-5). Similarly, RAD51AP1 knockdown or IV2A and LI2A mutant expression led to HR impairment (Figure S5D).
The findings above reveal that cells expressing UAF1 mutants are less sensitive to DNA damage and remain partially competent for HR when compared with cells depleted for UAF1. Consistent with published results (Murai et al., 2011), genetic data showed that USP1 depletion in human cells by siRNA treatment engenders a lesser degree of CPT sensitivity and HR deficiency than UAF1 depletion, but exerts no additive effect in the UAF1 deficient background (Figure S5E-G). Importantly, combining the UAF1 K459E or EEA mutation with USP1 depletion enhances CPT sensitivity and decreases HR proficiency to the levels in UAF1 deficient cells (Figure S5E-G). These results provide evidence that the DNA repair function of the RAD51AP1-UAF1 complex is likely USP1-independent. However, we cannot exclude the possibility that UAF1 also contributes to HR through other associated DUBs, such as USP12 and USP46 (Cohn et al., 2009; Sowa et al., 2009).
Discussion
We have provided biochemical and genetic data to elucidate the role of the RAD51AP1-UAF1 complex in HR and DNA damage repair (Figure 5E). Specifically, we have shown that (1) UAF1 possesses a DNA binding activity; (2) a SIM motif in RAD51AP1 and the SLD1-SLD2 domain of UAF1 mediate protein complex formation; (3) RAD51AP1 provides a bridging function between UAF1 and RAD51; (4) UAF1 synergizes with RAD51AP1 in the RAD51-mediated D-loop reaction and that functional synergy requires the RAD51AP1-UAF1 complex and also the DNA and RAD51 binding attributes of RAD51AP1; (5) the RAD51AP1-UAF1 complex works in conjunction with the RAD51 presynaptic filament in the capture of the duplex DNA partner and in the assembly of the synaptic complex; and (6) mutants impaired for RAD51AP1-UAF1 complex formation are compromised for the ability to repair DNA damage and to execute HR. Altogether, our work unveils a USP1-independent function of UAF1 in DNA repair and suggests the possibility that RAD51AP1 represents yet another candidate FA gene. We note that even though USP1 has little or no effect on the functional attributes of RAD51AP1-UAF1 in vitro or in cells, it likely contributes to DNA damage repair and HR via its DUB activity (Kim et al., 2009; Murai et al., 2011; this study).
RAD51AP1 deficient human cells are able to assemble RAD51 foci upon DNA damage occurrence (Wiese et al., 2007). Likewise, DT40 UAF1-/-/- chicken cells are proficient in DNA damage-induced Rad51 focus formation (Murai et al., 2011). Consistent with these published results, we have found that HeLa cells depleted of UAF1 remain competent for DNA damage-induced RAD51 focus formation (Figure S5H). Thus, the RAD51AP1-UAF1 complex is likely specific for the synaptic stage of HR.
It will be of interest to determine whether DNA binding by UAF1 is relevant for HR and the integrity of the FA pathway. To accomplish this goal, one would need a mutant of UAF1 that is defective in DNA binding but retains the ability to interact with RAD51AP1, USP1, and other partners. Moreover, it will be important to determine how USP1 influences HR via its DUB activity. Specifically, there are likely unidentified USP1 substrates whose timely deubiquitination helps ensure the proper execution of HR.
RAD51AP1 enhances homologous DNA pairing mediated by the meiosis-specific recombinase DMC1 by facilitating the assembly of the synaptic complex (Dray et al., 2011). Future work will determine the role, if any, of UAF1 in DMC1-dependent HR. We note that SLD2 of UAF1 is involved in ELG1 interaction (Yang et al., 2011; Figure 5A), and it will be of interest to test if ELG1 affects synaptic complex formation mediated by RAD51-RAD51AP1-UAF1.
Experimental Procedures
Mutant construction, protein expression, and protein purification
The details are provided in the Supplemental Information.
Affinity pulldown assay
GST- or MBP-tagged RAD51AP1 (3 μg) and UAF1 (3 μg) were incubated in 30 μl reaction buffer (25 mM Tris-HCl at pH 7.5, 10% glycerol, 0.5 mM EDTA, 0.01% Igepal, 1mM DTT, 150 mM KCl) on ice for 30 min, and then 15 μl glutathione resin (GE Healthcare) or amylose resin (New England Biolabs) was added to capture RAD51AP1 through its GST or MBP tag, respectively. After gentle mixing at 4°C for 1 h, the resin was washed three times with 30 μl of buffer and then treated with 30 μl of 2% SDS to elute bound proteins. The supernatant, last wash, and SDS eluate (10 μl each) were analyzed by 10% SDS-PAGE and Coomassie Blue staining. For affinity pulldown reactions involving Strep II- or MBP-tagged UAF1, Strep-Tactin or amylose resin was used to capture the tagged protein.
DNA mobility shift assay
RAD51AP1 or the indicated mutant (20 to 400 nM) and UAF1 or the indicated mutant (50 to 400 nM) were incubated with radiolabeled ssDNA (2.4 μM nucleotides) or dsDNA (2.4 μM base pairs) (oligonucleotides P1 and P1/P2 in Table S2, respectively) in 10 μl reaction buffer (25 mM Tris-HCl, pH 7.5, 50 mM KCl, 1 mM DTT, 100 μg/ml BSA, 1.5 mM MgCl2) at 37°C for 10 min. The reaction mixtures were resolved in 4% polyacrylamide gels in TBE buffer (40 mM Tris-HCl, pH 8.3, 45 mM boric acid and 1 mM EDTA) at 4°C. After gel drying, the radiolabeled DNA species were visualized and quantified by phosphorimaging analysis.
D-loop assay
The assay was conducted as described previously (Wiese et al., 2007). Briefly, 32P-labeled 90-mer oligonucleotide (2.5 μM nucleotides; see Table S2) and RAD51 (0.8 μM) were pre-incubated in 10.5 μl of reaction buffer, followed by the addition of RAD51AP1 (100 nM), UAF1 (100-400 nM), or the combination of the two proteins in 1 μl and a 5-min incubation. Then, pBlueScript replicative form I DNA (35 μM base pairs) was added in 1 μl to complete the reaction, which was incubated for 10 min before gel electrophoresis and phosphorimaging analysis.
Synaptic complex assembly assay and cell-based experiments
Details on duplex capture, synaptic complex assembly and cell culture, transfection, co-immunoprecipitation, cell survival assay, cell cycle analysis, HR assay and immunofluorescence analysis are provided in the Supplemental Information.
Supplementary Material
Acknowledgments
This work was supported by research grants ES007061, ES015632, ES015252, ES021454, CA168635 and CA92584 from the US National Institutes of Health.
Footnotes
Supplemental Information: This includes five Supplemental Figures and two Supplemental Tables, Extended Experimental Procedures and Supplemental References.
Author Contributions: P.S., G.M.K., Y.X, F.L., C.W., and S.L. conceived the study. F.L., S.L., G.M.K, C.W., and P.S. designed the experiments and analyzed the data. F.L., S.L., A.S.M., C.T., O.B. and D.G.M. executed the experiments. F.L., S.L., C.W., G.M.K. and P.S. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Borodovsky A, Kessler BM, Casagrande R, Overkleeft HS, Wilkinson KD, Ploegh HL. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001;20:5187–5196. doi: 10.1093/emboj/20.18.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn Ma, D'Andrea AD. Chromatin recruitment of DNA repair proteins: Lessons from the Fanconi anemia and double-strand break repair pathways. Mol Cell. 2008;32:306–312. doi: 10.1016/j.molcel.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn Ma, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D'Andrea AD. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell. 2007;28:786–797. doi: 10.1016/j.molcel.2007.09.031. [DOI] [PubMed] [Google Scholar]
- Cohn Ma, Kee Y, Haas W, Gygi SP, D'Andrea AD. UAF1 is a subunit of multiple deubiquitinating enzyme complexes. J Biol Chem. 2009;284:5343–5351. doi: 10.1074/jbc.M808430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray E, Etchin J, Wiese C, Saro D, Williams GJ, Hammel M, Yu X, Galkin VE, Liu D, Tsai MS, et al. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat Struct Mol Biol. 2010;17:1255–1259. doi: 10.1038/nsmb.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray E, Dunlop MH, Kauppi L, San Filippo J, Wiese C, Tsai MS, Begovic S, Schild D, Jasin M, Keeney S, et al. Molecular basis for enhancement of the meiotic DMC1 recombinase by RAD51 associated protein 1 (RAD51AP1) Proc Natl Acad Sci U S A. 2011;108:3560–3565. doi: 10.1073/pnas.1016454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop MH, Dray E, Zhao W, Tsai MS, Wiese C, Schild D, Sung P. RAD51-associated protein 1 (RAD51AP1) interacts with the meiotic recombinase DMC1 through a conserved motif. J Biol Chem. 2011;286:37328–37334. doi: 10.1074/jbc.M111.290015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop MH, Dray E, Zhao W, San Filippo J, Tsai MS, Leung SG, Schild D, Wiese C, Sung P. Mechanistic insights into RAD51-associated protein 1 (RAD51AP1) action in homologous DNA repair. J Biol Chem. 2012;287:12343–12347. doi: 10.1074/jbc.C112.352161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Hecker CM, Rabiller M, Haglund K, Bayer P, Dikic I. Specification of SUMO1- and SUMO2-interacting motifs. J Biol Chem. 2006;281:16117–16127. doi: 10.1074/jbc.M512757200. [DOI] [PubMed] [Google Scholar]
- Huang TT, Nijman SMB, Mirchandani KD, Galardy PJ, Cohn Ma, Haas W, Gygi SP, Ploegh HL, Bernards R, D'Andrea AD. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006;8:339–347. doi: 10.1038/ncb1378. [DOI] [PubMed] [Google Scholar]
- Kee Y, D'Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24:1680–1694. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley La, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, D'Andrea AD. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell. 2009;16:314–320. doi: 10.1016/j.devcel.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longerich S, Li J, Xiong Y, Sung P, Kupfer GM. Stress and DNA repair biology of the Fanconi anemia pathway. Blood. 2014;124:2812–2819. doi: 10.1182/blood-2014-04-526293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modesti M, Budzowska M, Baldeyron C, Demmers Jaa, Ghirlando R, Kanaar R. RAD51AP1 is a structure-specific DNA binding protein that stimulates joint molecule formation during RAD51-mediated homologous recombination. Mol Cell. 2007;28:468–481. doi: 10.1016/j.molcel.2007.08.025. [DOI] [PubMed] [Google Scholar]
- Murai J, Yang K, Dejsuphong D, Hirota K, Takeda S, D'Andrea AD. The USP1/UAF1 complex promotes double-strand break repair through homologous recombination. Mol Cell Biol. 2011;31:2462–2469. doi: 10.1128/MCB.05058-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman SMB, Huang TT, Dirac AMG, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005;17:331–339. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, Ohzeki M, Takata M, Sale JE, Patel KJ. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell. 2007;28:798–809. doi: 10.1016/j.molcel.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Kim JM, Primack B, Weinstock DM, Moreau La, Parmar K, D'Andrea AD. Inactivation of Uaf1 causes defective homologous recombination and early embryonic lethality in mice. Mol Cell Biol. 2013;33:4360–4370. doi: 10.1128/MCB.00870-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman KA, Lach FP, Abhyankar A, Donovan FX, Sanborn EM, Kennedy JA, Sougnez C, Gabriel SB, Elemento O, Chandrasekharappa SC, et al. Deficiency of UBE2T, the E2 ubiquitin ligase necessary for FANCD2 and FANCI ubiquitination, causes FA-T subtype of Fanconi Anemia. Cell Rep. 2015;12:35–41. doi: 10.1016/j.celrep.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Sekiyama N, Ikegami T, Yamane T, Ikeguchi M, Uchimura Y, Baba D, Ariyoshi M, Tochio H, Saitoh H, Shirakawa M. Structure of the small ubiquitin-like modifier (SUMO)-interacting motif of MBD1-containing chromatin-associated factor 1 bound to SUMO-3. J Biol Chem. 2008;283:35966–35975. doi: 10.1074/jbc.M802528200. [DOI] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Hunter T. Poly-small ubiquitin-like modifier (PolySUMO)-binding proteins identified through a string search. J Biol Chem. 2012;287:42071–42083. doi: 10.1074/jbc.M112.410985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villamil Ma, Chen J, Liang Q, Zhuang Z. A noncanonical cysteine protease USP1 is activated through active site modulation by USP1-associated factor 1. Biochemistry. 2012;51:2829–2839. doi: 10.1021/bi3000512. [DOI] [PubMed] [Google Scholar]
- Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, et al. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol Cell. 2015;59:478–490. doi: 10.1016/j.molcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C, Dray E, Groesser T, San Filippo J, Shi I, Collins DW, Tsai MS, Williams GJ, Rydberg B, Sung P, et al. Promotion of homologous recombination and genomic stability by RAD51AP1 via RAD51 recombinase enhancement. Mol Cell. 2007;28:482–490. doi: 10.1016/j.molcel.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Moldovan GL, Vinciguerra P, Murai J, Takeda S, D'Andrea AD. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 2011;25:1847–1858. doi: 10.1101/gad.17020911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.