

Abstract
Approximately 35% of the human genome can be identified as sequence devoid of a selected-effect function, and not derived from transposable elements or repeated sequences. We provide evidence supporting a known origin for a fraction of this sequence. We show that: 1) highly degraded, but near full length, ribosomal DNA (rDNA) units, including both 45S and Intergenic Spacer (IGS), can be found at multiple sites in the human genome on chromosomes without rDNA arrays, 2) that these rDNA sequences have a propensity for being centromere proximal, and 3) that sequence at all human functional rDNA array ends is divergent from canonical rDNA to the point that it is pseudogenic. We also show that small sequence strings of rDNA (from 45S + IGS) can be found distributed throughout the genome and are identifiable as an “rDNA-like signal”, representing 0.26% of the q-arm of HSA21 and ∼2% of the total sequence of other regions tested. The size of sequence strings found in the rDNA-like signal intergrade into the size of sequence strings that make up the full-length degrading rDNA units found scattered throughout the genome. We conclude that the displaced and degrading rDNA sequences are likely of a similar origin but represent different stages in their evolution towards random sequence. Collectively, our data suggests that over vast evolutionary time, rDNA arrays contribute to the production of junk DNA. The concept that the production of rDNA pseudogenes is a by-product of concerted evolution represents a previously under-appreciated process; we demonstrate here its importance.
Keywords: degraded rDNA, vestigial centromere, genome evolution, concerted evolution
Introduction
For decades, molecular biologists have worked towards resolving the origin of the excessive size of most eukaryotic genomes. It is now recognized that much of the genome is junk DNA, which is consistent with contemporary genome evolution theory (Lynch 2007; Doolittle 2013; Graur et al. 2015). Efforts made to determine the fraction of the human genome demonstrated to fall into the functional category, using differing methods, provide values of 2.56–3.25% as the most conservative estimate (Lunter et al. 2006), to a more liberal estimate of 10–15% (Ponting and Hardison 2011). We use the term “functional” in the sense of a “selected effect function” (Doolittle 2013; Graur et al. 2015), which would include protein coding, RNA specifying, and nontranscribed control regions. Much of the remaining fraction of the genome can be defined as junk DNA, that is, DNA that does not encode information promoting the survival and reproduction of the organisms that bear it (Doolittle 2013), but is not under negative selection. Transposable elements (TEs), the major source of junk DNA in the human genome, have been determined to account for one half or more of human DNA (De Koning et al. 2011). There remains within the human genome a fairly large fraction, a minimum of 35%, which is devoid of a selected-effect function, and derived neither from transposable elements nor from highly repeated sequences. Herein, we characterize a portion of this DNA, and show that a sizable fraction is related to ribosomal DNA (rDNA) sequence.
Concerted Evolution of rDNA
Concerted evolution of rDNA clusters brought about by unequal cross over (UXO) is a well-documented process (Brown et al. 1972; Szostak and Wu 1980; Averbeck and Eickbush 2005). Proposed in 1970s (Smith 1974; Ohta 1976), UXO can readily account for the observation that the many rDNA gene copies within an individual and within a species remain identical or nearly so in sequence, while between closely related species the sequence can vary widely in the nontranscribed intergenic spacer region which is under less stringent selection than the 18S, 5.8S and 28S mature rRNA specifying genes. Stults et al. (2008) have demonstrated large scale, dynamic length variation occurring at rDNA clusters in human, the direct result of UXO occurring both inter- and intrachromosomally. The recombination rate observed by Stults et al. (2008), a conservative estimate of 11% per generation, meets the requirements stated by Smith (1974) and by Ohta (1976) in their theoretical calculations of the necessary rate to maintain homogeneity in rDNA copies. In humans, the rDNA repeat unit is 43 kb in length (Gonzalez and Sylvester 1995) and there are five rDNA repeat arrays per haploid genome, which are found on each of the short arms of the acrocentric chromosomes, these are HSA13–15 and HSA21–22 (Henderson et al. 1972). Due to the crucial role of rDNA in establishing the region within nuclei where ribosomes assemble, these regions also go by the name of nucleolus organizer regions (NOR).
Although it had been suggested previously that rDNA units at the extreme edges of arrays may be prevented from participating in the homogenizing events of concerted evolution (Brownell et al. 1983), no previous study has addressed the evolutionary fate of extreme edge units in eukaryotes with large genomes. Our work addresses this topic and falls into three broad examinations: extreme rDNA array edges, displaced rDNA throughout the human genome, and a low-level rDNA genetic signature.
The first component we will present is a detailed analysis of rRNA specifying gene copies present at the extreme edges of the rDNA arrays. Based on two criteria: (1) the total number of nucleotide variants observed compared with expected and (2) the ratio of mature rRNA specifying to spacer sequence variants observed compared with what would be expected for sequences undergoing purifying selection, we categorize all of these rDNA copies as pseudogenes. (Note that throughout the term “rRNA specifying” is used to imply the entire 45S region of an rDNA unit, while the term “mature rRNA specifying” is used to imply only the combined 18S, 5.8S, and 28S rRNA regions of an rDNA unit) These data indicate that rDNA repeat units at the extreme edges of clusters are unable to undergo the homogenizing events of concerted evolution, and yet continuously accumulate in the genome.
The second component we will present is a description of the genomic distribution and identity of rDNA sequences of 500–2,000 bp in length found at multiple sites throughout the human genome and occurring predominantly near centromeres rather than being randomly distributed along chromosomes. All human chromosomes are involved in this analysis. Overall, these data will indicate that by some unknown mechanism, entire units of rDNA are “escaping” the confines of the rDNA arrays and are found in association with centromeres distributed throughout the genome.
The last component of our article will examine a topic that stems from the prior concepts and involves the quantification of a population of even shorter strings of rDNA sequence. However, this population of sequence makes up a larger fraction of the total genome. To this end, we will describe a genome-wide “ribosomal-DNA-like” signal comprised of smaller sequences. The signal is weak but present above levels expected by random chance alone. In some surveyed locations, this signal comprises nearly 2% of the sequence.
The Lack of Available rDNA Sequence
There is currently no complete rDNA sequence array present in genome builds for any genome of a higher eukaryote, this is because of the difficulties of sequencing the large head-to-tail tandemly repeated units of which the rDNA clusters are composed. Instead there are gaps in genome builds where rDNA clusters are known to exist based on cytogenetic work. We are afforded an opportunity to look at sequence close to active rDNA arrays as a result of work done by Floutsakou et al. (2013) in their efforts to find DNA involved in organizing nucleoli. They sought and found junctional sequence that is present between the telomere and the active cluster, termed “distal junction” or DJ, and sequence that is present between the active cluster and the centromere, termed “proximal junction” or PJ. DJ and PJ sequences were demonstrated by Floutsakou et al. (2013) to be present on all rDNA bearing-chromosomes. The BAC clones that are the source of DJ, PJ, and their adjacent sequences have not been connected by contiguous linkage to the chromosomes from which they are derived, but their locations as nearer to active arrays make them interesting and informative for studies of rDNA.
A Model for the Relationship between rDNA and Junk DNA
Based on our combined findings, we propose that a portion of the eukaryotic genome is derived from the accumulation of an excess of degrading rDNA. The acquisition of such sequence is thought to be the result of exceptionally high levels of UXO in rDNA clusters and a reduced capacity of edge-most rDNA units to participate in corrective recombination events. Ribosomal DNA units therefore accumulate in the genome as pseudogenes in near-cluster regions, and in the absence of cellular functions capable of recognizing and removing them, they will persist in the genome. Although this finding is not surprising, it has not yet been documented. It is also important to note that the idea of pseudogenes contributing to the abundance of junk DNA in the genome is not a novel concept (e.g., sequences derived from protein coding genes and from other RNA-specifying genes), however, the impact of concerted evolution on the generation of rRNA pseudogenes reflects a different mechanism for the addition of pseudogenes to the genome. As would be expected for sequence no longer under selection, over vast evolutionary time rDNA pseudogene copies become increasingly less recognizable. By mechanisms that remain to be fully elucidated—but which likely involve centromeres—rDNA units are dispersed from the arrays where they originate into other portions of the genome. Our findings indicate that in eukaryotes some portion, and perhaps a sizable portion of the genome, is most likely composed of both ancient and recent degrading segments of once-active rDNA.
Materials and Methods
Ribosomal DNA Sequence Probes
Sequences used for probing various genomic regions are the entire canonical rDNA unit sequences for human (includes genes for RN28S1, RN18S1, RN5-8S1; GenBank accession: U13369.1) and for mouse (GenBank accession: BK000964.3). Complete rDNA units were segmented into the following differentiated parts: a 5′ external transcribed spacer (ETS), 18S rRNA, internal transcribed spacer 1 (ITS1), 5.8S rRNA, ITS2, 28S rRNA, 3′ ETS, and an intergenic spacer (IGS). This allowed us to keep track of the relationship between each sequence alignment and its respective portion of the canonical rDNA unit. To increase the stringency of probes, sequences known to be present elsewhere in the genome (such as Alu elements and mono-, di- and tri-nucleotide repeats) were manually edited out of probe sequence with the aid of RepeatMasker Open-4.0 (Smit et al. 2013–2015) and Tandem Repeats Finder (Benson 1999). Default parameters were used for both programs; these probes are referred to as “tailored” throughout. If the term “tailored” is not used it means that sequence for the entire rDNA unit was used in an alignment without removal of mobile and repetitive sequence. In some cases, it is better to use the whole unit with repeats to assess similarity to an entire rDNA unit.
Sequences Analysed
Distal Junction and Proximal Junction Clones
Sequences were obtained from GenBank using the following accession codes. For the Distal Junction region, these are: AL353644.34 (Clone AL353644), AL592188.60 (Clone AL592188), NT_187317 (Clone FP671120), CT476837.18 (Clone CT476837), NT_187318 (Clone FP236383). For the Proximal Junction region, these are: CR392039.8 (Clone CR392039), KC876028.1 (Clone LA15_25H3), KC876027.1 (Clone N29M24), KC876029.1 (Clone LA14_101B3), KC876030.1 (Clone LA13_165F6).
Genomic Data Examined for Large Segments of Ribosomal DNA Sequence
All complete chromosome sequences, or subsamples therein (e.g., for analyses of regions closer to the centromere of HSA21), were obtained from reference sequence of the current Human Genome Build, Homo sapiens GRCh38.p8 (Benson et al. 2005). Complete chromosome sequences span the GenBank accession codes: NC_000001–NC_000022 for chromosomes HSA1–HSA22, and NC_000023 for chromosome HSAX.
Synteny Blocks Observed for Low-Level rDNA Signal
Two hundred (200) kilobases of sequence were randomly selected from six human–mouse syntenic genome blocks. Chromosome number and sampling position were randomly selected via an algorithm in R (R Core Team 2015), and the NCBI Homology Maps database was used (Wheeler et al. 2003). The genomic region of each human–mouse syntenic block pair is as follows: [Human = H, Mouse = M] for Block A H is 1..200,000 bp Accession = NT_022221.13, M is 100 kb left and right of FAM110C gene, Accession = NC_000078.6; Block B 100 kb left and right of CNTN5 gene, H Accession = NC_000011.10, M Accession = NC_000075.6; Block C 100 kb left and right of TBX5 gene, H Accession = NC_000012.12, M Accession = NC_000071.6; Block D 100 kb left and right of EDIL3 gene, H Accession = NC_000005.10, M Accession = NC_000079.6; Block E 100 kb left and right of COL11A1 gene, H Accession = NC_000001.11, M Accession = NC_000069.6; Block X 100 kb left and right of TMEM47 gene, H Accession = NC_000023.11, M Accession = NC_000086.7. Mouse Block A is located on a chromosome with an rDNA cluster (Suzuki et al. 1990). Human Block A and Human/Mouse Blocks B through X are on chromosomes without rDNA clusters; these are also located in regions of sequence that do not contain any other annotated genes. Figure 8, which illustrates the position of synteny blocks and rDNA cluster positions was created by modifying the ideograms publically available for noncommercial use from Adler and Willis (1991) and Adler (1992), these are available online at: http://pathology.washington.edu/research/cytopages/ (last accessed January 11, 2017). Ideograms were previously cited by, or adapted from, Harden and Klinger (1985) and Nesbitt and Francke (1973).
Fig. 8.—

Regions of Homo sapiens and Mus musculus genomes that were used in over-representation of rDNA-like sequence analysis. “Synteny blocks” for each organism are indicated on chromosomes (see light blue boxes); a letter to the left indicates the identity we have assigned to a particular block. Blocks with corresponding letters between human and mouse share synteny (e.g., Block A H. sapiens vs. Block A M. musculus). Red circles indicate the position of rDNA clusters. Block A in M. musculus is the only synteny region that comes from a chromosome also harboring an rDNA cluster. Images are not to scale. Ideograms modified from Adler (1992) and Adler and Willis (1991).
Acquisition of Alignment Data
Assessment of Distal Junction and Proximal Junction Pseudogenes
To collect data pertaining to the number of sequence variants between pseudogenes and canonical rDNA, each pseudogene was aligned individually to the tailored rDNA probe using CLUSTAL Omega version 1.2.1 (Li et al. 2015). To accomplish this, we had to concatenate the various segments of tailored rDNA probe that had had repetitive sequence removed to allow for a contiguous alignment to be built between canonical rDNA and putative pseudogenes. The use of the tailored rDNA probe was done to avoid over estimating the number of variants, which would have inevitably increased if the whole rDNA unit were used because of the presence of variable repetitive sequences. Following pairwise alignment, the number of variants between pseudogene and canonical rDNA, both indels and SNPs, were manually determined. Indels were treated as variants and not missing data points so that our calculations would be comparable to the values stated in Stage and Eickbush (2007), who used the same approach. The multiple sequence alignment presented in figure 1B of all the rDNA pseudogenes was resolved using CLUSTAL Omega version 1.2.1 (Li et al. 2015). The clone placement along DJ and PJ junctions in figure 1A was assembled to scale using the Map to Reference feature in the program Geneious v8.0.5 (Kearse et al. 2012). Geneious was also used to create all of the illustrations in figure 1.
Fig. 1.—

Analysis of pseudogenes located at extreme edges of rDNA arrays (i.e., in Distal Junction [DJ] and Proximal Junction [PJ] regions). (A) Position of rDNA pseudogenes. A diagram of an acrocentric chromosome is provided; the green box represents a centromere and green circles represent telomeres. The insert shown with a dashed line indicates the orientation and size of DJ and PJ regions with associated cosmids and BACs from Floutsakou et al. (2013) shown directly beneath. Cosmid and BAC names are indicated in black, while the chromosome source for the sequence is shown in red. Repeating gray boxes, with a yellow line in the center, indicate the position of functional rDNA relative to DJ and PJ (the word “rDNA” is also given). (B) Visual representation of the degree of dissimilarity of DJ and PJ pseudogenes compared with the canonical rDNA unit. Mature rRNA and spacer regions of the rDNA unit are indicated at the top, and are labelled and coloured blue. DJ and PJ pseudogenes [listed to the left of sequences] are then compared with canonical rDNA, highlighting indels [black horizontal bars] and SNPs [black vertical bars]. All gray regions highlight identical nucleotide positions.
Genome-Wide Searches for Ribosomal DNA
Unless otherwise indicated in the results text, for example, where sometimes only megablast was used, the alignment data was compiled from successive alignment attempts in the online NCBI program BLAST, using each of the default bl2seq parameters available, including: megablast, discontiguous megablast, and blastn. These three parameters have a decreasing amount of nucleotide length and mismatch stringency (Altschul et al. 1990; Geer et al. 2010). Between the three parameters used, the absolute minimum alignment seed length equalled 11 bp, and the expect threshold equalled 10. To avoid double counting hit positions, alignment overlaps were always edited out of the dataset by concatenating overlaps into a single alignment hit string; this was accomplished using an algorithm written in R (R Core Team 2015).
Data Analysis
Ribosomal DNA Pseudogenes
Methods for determining the number of variants for pairwise comparisons of canonical rDNA to pseudogenes are described earlier. In calculating variants/kb, sequence size values were always rounded to the nearest tenths decimal place.
Genome-Wide Searches for Ribosomal DNA
Histograms, boxplots, scatterplots, a cumulative proportion plot, and maps of megablast alignment hit position along human chromosomes were produced in R v3.2.1 (R Core Team 2015) using the package ggplot2 (Wickham 2009). Histograms have bin-widths of 10,000 bp to allow visualization of such a large dataset, that is, positions along millions of nucleotides. Maps of megablast hit position are modified scatterplots without y-axes and jittered data points to allow for the interpretation of data points that would otherwise be too densely overlaid. This approach is particularly useful in resolving trends in regions that had large segments of rDNA-like sequence. The alignment map provided in figure 5B is modified from the graphical output produced during the BLAST alignment run (Altschul et al. 1990; Camacho et al. 2009). Dot plots were produced using the program JDotter v1.0 (Brodie et al. 2004).
Fig. 3.—

Analysis of centromere associated rDNA sequence present on HSA21. (A) A subsample of 2 Mb to the left and right of centromere sequence presented in figure 2; it is therefore a more detailed look at feature “B- -B” (fig. 2). Megablast, discontiguous BLAST and blastn were used to obtain alignment hits. Peaks along histogram indicate regions of sequence with high amounts of rDNA-like sequence. (B) Distribution of alignment hit sizes in the 2-Mb regions that are left [red] and right [green] of the centromere of panel A. The data have been log transformed, Σ = sum of nucleotides, and diamonds denote the means of their respective boxplots.
Test for rDNA Signal in Human and Mouse Synteny Regions and the q-Arm of HSA21
Tailored rDNA probes were aligned to each of their respective synteny blocks. The total quantity of sequence similarity between tailored probes and synteny blocks was calculated and expressed as a percentage of bp within each synteny block. Spurious rDNA to synteny block alignments are unpreventable in this calculation, thus, a baseline abundance of false positive alignments was also calculated. This was achieved by repeating the above with 10 random sequences identical in length and %GC content to each synteny block. GC% was calculated using the online MBCF Oligo Calculator provided at http://mbcf149.dfci.harvard.edu/docs/oligocalc.html (last accessed January 11, 2017) (Dana-Farber Cancer Institute and Harvard University). Random sequences were produced using the Random DNA Generator available online at http://www.faculty.ucr.edu/∼mmaduro/random.htm (last accessed January 11, 2017) (The Maduro Lab, University of California at Riverside, Riverside, CA).
A similar analysis was conducted on the entire q-arm of HSA21. We calculated the true abundance of rDNA-like sequence after calculating the fraction of alignment hits that are most likely caused by random matches. As above, artificial sequence was computer generated to reflect a randomized sequence, however, this time it was with respect to HSA21 q-arm size and approximate %GC along the length of the chromosome arm. To accomplish this, a random nucleotide was generated for each position of the q-arm with probabilities G = C=%GC/2 and A = T = 1–%GC/2. For each position i, %GC was calculated from 200,001 sites of the q-arm sequence, 100,000 sites on either side of position i. Positions at the start or end used the first or last 200,001 sites. This was completed using in-house software.
Results
Ribosomal DNA Copies at Extreme rDNA Array Edges
We used sequence previously identified by Floutsakou et al. (2013) for our initial analysis of rDNA residing nearest to active rDNA arrays. The sequence in Floutsakou et al. (2013) was originally obtained using the Ensemble trace archive of whole genome shotgun sequences (more than 27 million traces). In their search for junctions between rDNA arrays and neighboring non-rDNA, called either distal junction (DJ) if towards telomere (Gonzalez and Sylvester 1997) or proximal junction (PJ) if towards centromere (Sakai et al. 1995), Floutsakou et al. (2013) obtained five clones with rDNA sequence adjoining DJ sequence, and five clones with rDNA sequence adjoining PJ sequence. All five array-bearing human chromosomes are represented in these clones, four from HSA21, three from HSA22 and one from each of HSA13, HSA14 and HSA15. Four of the clones have more than one copy of the rDNA repeat unit, permitting us to have for analysis five full-length copies and nine partial copies of the rDNA unit. In our analysis, we have assessed each rDNA copy’s status as compared with the canonical human rDNA sequence (Gonzalez and Sylvester 1995).
Figure 1A shows a generalized ideogram of an array-bearing acrocentric chromosome. The green rectangle represents the centromere. The gap in the p-arm of the ideogram indicates the position of rDNA loci; the heavy black lines on either side represent DJ and PJ sequence. Although the general structure of the chromosomes is known from cytogenetic work, for all array-bearing chromosomes with the exception of HSA21, there is no contiguous sequence available on the p-arm side of the centromeres. In figure 1A, dashed grey lines indicate an expansion of the rDNA region, also showing are the lengths of DJ and PJ. Below these PJ and DJ lengths, the cosmids and BAC clones that contain rDNA sequence identified by Floutsakou et al. (2013) are shown. Clone names, length, orientation relative to the chromosome, and positions of rDNA copies along DJ and PJ are indicated (see white boxes along clones for rDNA copies in fig. 1A). For simplicity and for reference, in cases where clones have more than one rDNA copy, the unit nearest the junction is labeled “1”, while the unit furthest from the junction is labeled “2”. The white boxes of figure 1A only span 45S rDNA-related sequence. The sequence between them was not analysed in greater detail; however, these regions between rDNA copy 1 and copy 2, and between rDNA copy 2 and the remaining array, are homologous to IGS sequence (fig. 1A and supplementary fig. S1, Supplementary Material online).
Figure 1B shows the results of aligning each of the rDNA copies found in figure 1A clones to the rRNA specifying portion of the canonical rDNA unit, that is, the 45S region of rDNA, which is ∼13 kb in length. The names of rRNA (e.g., 18S) and spacer segments (e.g., ITS1) along the transcribed region are shown in blue. When observing the alignments, solid grey regions indicate 100% pairwise identity to the canonical sequence, while black vertical bars and black horizontal dashes indicate nucleotide differences and indels, respectively. Sequence differences between the rDNA copies derived from clones and the canonical rDNA unit can be observed in all individual rRNA segments and spacer regions of the multi-gene rRNA specifying unit (i.e., all parts of the 45S region). As an example of the numbers and kinds of differences observed, for the sequence of CT476837 (1) there are 106 mismatches, 52% of which are 1–10 bp indels, while 48% are SNPs; again these mismatches are distributed across the entire length of the sequence. This sequence is typical of the others and was chosen as an example because it is named as the defining DJ rDNA edge sequence in Floutsakou et al. (2013).
We wished to determine whether the degree of sequence difference observed in figure 1B warranted designating any of these rDNA copies as pseudogenes. We assessed two criteria: (1) the total number of sequence variants observed, and (2) the ratio of mutations found in mature rRNA specifying regions compared with spacer regions (ETS + ITS). The second criterion was meant to test whether or not the sequence is under purifying selection. By testing these two properties, one can determine if rDNA sequence falls within the range of nucleotide variation to be expected, or if it falls outside of the expected range, and therefore may be classified as a pseudogene. We rely on the work of Stage and Eickbush (2007) for providing the expected range of values for both of these criteria. It is the most thorough work done to date on these questions. Stage and Eickbush (2007) attempted to directly measure the nucleotide diversity between rDNA units within a species and did so for 12 species of Drosophila. They used original sequencing reads for unassembled sequence available as trace archives, which allowed them to locate nucleotide sequence variants (both indels and SNPs) and to quantify levels of variation present within a species in the 7.8–8.2 kb rRNA specifying portions of rDNA units. Stage and Eickbush (2007) found, on average, 10 variants in each species (ranging from 3 in Drosophila willistoni to 18 in D. grimshawi). The average for all species was about 1 variant/kb (ranging from 0.38 to 2.3 variants/kb). In addition, they found that variants were 10–20× more frequent in spacer regions compared with mature rRNA specifying regions (the mean number of variants/kb was 0.21 for mature rRNA specifying regions and 2.59 for spacer regions; or if expressed as a ratio, ∼1:12 for mature rRNA specifying:spacer regions). Table 1 shows our observations for each rDNA copy (or part thereof in the case of incomplete units) in PJ and DJ clones aligned to canonical rDNA sequence using a CLUSTAL W alignment algorithm (Thompson et al. 1994; Li et al. 2015). In keeping with the methodology in Stage and Eickbush (2007), indels, regardless of their length, are assessed as a single variant, the same rule applied to substitutions. The variants/kb for all rDNA units exceeds the expectation of ∼1/kb (average of 10.8/kb observed, range of 3–28/kb; table 1). When variants/kb of mature rRNA specifying and spacer sequence are calculated, the values clearly exceed the thresholds of 0.21 for mature rRNA specifying and 2.59 for spacer regions. In rDNA copies from clones of figure 1B the mature rRNA specifying mean = 6.36 variants/kb and spacer region mean = 16.93 variants/kb. Additionally, the mature rRNA specifying:spacer region variant/kb ratio is much less than the 1:12 difference calculated by Stage and Eickbush (2007). It is also important to note that it is unlikely that the values above are influenced by sequencing errors. Sequences are of a high quality. Nine of the ten clones have Phred scores ≥ 30 as indicated in the supplementary data from Floutsakou (2013) and the associated GenBank records for clones (available from accession codes provided in Methods and Materials; Wheeler et al. 2003). The tenth clone sequence has a > 7.5× coverage in Q20 bases (Wheeler et al. 2003).
Table 1.
Sequence Variation between DJ and PJ Pseudogenes and the Canonical rDNA Unit
| rDNA Pseudogene [Chromosome] | Size (bp) | Total Variants | Variants/kb (95% CI) |
Ratio (95% CI for S) | ||
|---|---|---|---|---|---|---|
| Total | M | S | M: S | |||
| CT476837 Ψ1 [21] | 12,047 | 106 | 9 (7,10) | 7 (5,10) | 12 (9,15) | 1:2 (1,3) |
| FP671120 Ψ1 [21] | 12,047 | 147 | 12 (10,14) | 8 (6,11) | 17 (14,21) | 1:2 (2,3) |
| FP671120 Ψ2 [21] | 9,543 | 272 | 28 (25,32) | 16 (13,20) | 40 (35,46) | 1:3 (2,3) |
| FP236383 Ψ1 [21] | 12,050 | 210 | 17 (15,20) | 11 (9,14) | 25 (21,30) | 1:2 (2,3) |
| FP236383 Ψ2 [21] | 12,048 | 140 | 12 (10,14) | 9 (7,12) | 16 (13,20) | 1:2 (1,2) |
| AL592188 Ψ1 [22] | 12,051 | 107 | 9 (7,11) | 6 (4,8) | 12 (9,15) | 1:2 (1,3) |
| AL592188 Ψ2 [22] | 11,100 | 89 | 8 (6,10) | 6 (4,9) | 10 (8,13) | 1:2 (1,3) |
| AL353644 Ψ1 [22] | 12,055 | 103 | 9 (7,10) | 6 (4,8) | 11 (9,14) | 1:2 (1,3) |
| AL353644 Ψ2 [22] | 11,105 | 90 | 8 (6,10) | 6 (4,9) | 10 (8,13) | 1:2 (1,3) |
| CR392039 [21] | 1,985 | 20 | 10 (6,14) | 2 (1,6) | 14 (9,22) | 1:8 (1,61) |
| LA15_25H3 [15] | 2,115 | 14 | 7 (3,10) | 3 (1,16) | 21 (12,39) | 1:7 (2,20) |
| N29M24 [22] | 5,343 | 93 | 17 (14,21) | 3 (2,6) | 24 (19,30) | 1:8 (3,17) |
| LA14_101B3 [14] | 5,337 | 48 | 9 (6,12) | 3 (2,6) | 12 (9,16) | 1:4 (2,9) |
| LA13_165F6 [13] | 5,365 | 51 | 10 (7,12) | 3 (2,6) | 13 (10,17) | 1:4 (2,9) |
Note.—Confidence intervals are calculated under the assumption that variants occur independently at sites or not. The confidence intervals for PJ pseudogene M:S ratios are larger than those for DJ ratios because small differences in the mature rRNA specifying rate can lead to large differences in 1/rate; nevertheless, because all PJ pseudogenes are truncated in a way that would cause them to lack functional rRNA specifying portions of the entire 45S rRNA transcript, there is no questioning their identity as pseudogenes. Variants (substitutions + indels) calculated after aligning sequences using CLUSTALW matrix. Ψ1 and Ψ2 are used to denote pseudogenes 1, and pseudogenes 2, referred to in figure 1A. Mature rRNA specifying = M, spacer (ETS + ITS) = S.
Taken together, the above values suggest that the rDNA copies of DJ and PJ clones are under relaxed selection; therefore not behaving as if they were experiencing the purifying selection of concerted evolution that would occur if they were still functional. Consequently, we conclude that all the rDNA copies adjacent to DJ and PJ presented in figure 1A are pseudogenes. As both PJ and DJ regions are the closest confirmed sequence to active rDNA clusters thus far reported, the finding that rDNA pseudogenes are present in DJ and PJ regions indicates that extreme array ends are most likely not undergoing the same degree of sequence homogenization typical of more central rDNA array regions.
Genome-Wide In Silico Search for rDNA-Related Sequence
We have further scanned sequence from Human Genome Build GRCh38.p2 (Wheeler et al. 2003) using BLAST functions. The next set of analyses includes HSA21 detailed surveys and surveys of the entire human genome.
HSA21 is the shortest of the human chromosomes and has the most deposited sequence arising from the p-arm of any of the five rDNA-array-bearing chromosomes. Using a “tailored rDNA probe” that consists of the 43-kb canonical rDNA repeat sequence (45S and IGS) with all repetitive sequences removed (including Alu sequences, LINEs, SINEs, and low complexity regions caused by mono-, di- and tri-nucleotide repeats) the length of HSA21 was assessed for sequence matching rDNA. The use of a tailored probe insures that alignment matches are likely derived from similarities to rDNA sequence itself and not merely from the presence of commonly occurring mobile and repetitive elements/motifs.
Figure 2 shows a histogram of the number of rDNA-like alignment hits with respect to nucleotide position along HSA21 when the tailored rDNA probe is aligned to the entire HSA21 build. Figure 2 data reveals three HSA21 regions of interest. The first, labeled “A” (fig. 2) corresponds to the DJ/rDNA junctional BACs FP671120 and FP236383 found in the scan done by Floutsakou et al. (2013), and described as part of the pseudogene analysis presented in figure 1 and table 1. They were placed in the build as unassembled scaffolds consisting of multiple overlapping cloned sequences and aligned by FISH to the p-arm of HSA21 by Hattori et al. (2000). In the current Human Genome Build GRCh38.p2, they are erroneously placed on the centromeric side of the gap left for the rDNA array. The second region of interest, labeled “B” (fig. 2) is sequence 2 Mb to the left and right of the centromere. The third, labeled “C” (fig. 2) shows an “rDNA-like signal” that is present at varying levels along the remaining q-arm of HSA21. The latter two regions will be described in more detail below.
Fig. 2.—

rDNA sequence present along HSA21. The number of nucleotides within alignment hits with respect to position along Chromosome 21 is plotted. Centromere-related sequence is found in the region where histogram bars drop to zero after position 10 Mb. Alignment hits were found using megablast, Discontiguous BLAST and blastn algorithms in BLAST. Three features are shown: “A” indicates pseudogene sequence placed along the HSA21 build from DJ/PJ contigs, “B- -B” indicates centromere associated rDNA hits, and “C-…-C” shows the q-arm of HSA21 and how the level of rDNA-like sequence does not drop to zero (compare to centromere position after 10 Mb).
Ribosomal DNA Sequence 2 Mb Left and Right of HSA21 Centromere
We have examined 2 Mb on both sides of HSA21’s centromere (feature “B” of fig. 2). These 2-Mb regions include long discontiguous sequences derived from “orphan” scaffolds from HSA21 beginning on the HSA21 p-arm, continuing through the centromere, which itself cannot yet be contiguously sequenced (Aldrup-MacDonald and Sullivan 2014), and then proceeds to include sequence from the beginning of the long contiguous scaffold of the HSA21 q-arm. With this HSA21 centromere proximal sequence, and using our tailored rDNA probe, we searched for evidence of rDNA-like sequence near the HSA21 centromere. The results showing the number of rDNA-like alignment hits relative to position (2 Mb left or right of HSA21 centromere) are shown in the histogram in figure 3A. The centromere itself does not have any rDNA-like sequence (as determined by absence of hits; the value drops to zero in fig. 3A at ∼11–13 Mb); otherwise hits are present across the entire centromere proximal region with peaks occurring at 9 and 14 Mb. To quantify the abundance of rDNA-like hits and to classify the range of hit sizes, boxplots were produced for each centromere side (fig. 3B). We observe a similar abundance of rDNA-like sequence on either side of the centromere. This is apparent from the distribution of the boxplots, see spread of the interquartile range and max | min whisker values (fig. 3B). Although the q-arm side has a greater total number of hits (Σ = 6.8 kb) than the p-arm side (Σ = 4.8 kb), the p-arm side has larger alignment sizes than the q-arm side (see sizes of outliers in fig. 3B). The outlier hits on the q-arm side, however, are more numerous. The above observations suggest that positions on the left of the centromere, and thus nearer to the rDNA array, might have sequence more recently acquired and therefore less degraded (i.e., occurring as fewer, larger and continuous sequences). This finding can be interpreted as indicating that rDNA arrays are acting as the source of this near centromere sequence.
Distribution of rDNA-Related Sequence throughout the Human Genome
To determine if rDNA fragments are more concentrated near centromeres, or if HSA21 is unique in this regard, we further surveyed the distribution of rDNA-related sequence present throughout the human genome. The initial search conducted above for solely HSA21 used all three BLAST algorithms available, that is, megablast, discontiguous megablast, and blastn. To facilitate the search of an even larger sequence dataset, such as the entire human genome, we used only the more stringent “megablast” algorithm provided by the BLAST software, rather than using megablast in combination with discontiguous megablast and blastn algorithms. The megablast algorithm requires a word size seed of 28 bp of match before beginning an alignment recognition, and is less tolerant of mismatch by using a [+1 reward, −2 penalty] match/mismatch score and a linear gap cost. The use of such scoring parameters generates a smaller alignment dataset where regions identified as rDNA are larger and more nearly perfect matches. The position of megablast hits along the length of each human chromosome can be seen in figure 4; note that centromere regions are located internally to the dashed lines with red dots. Hits are not evenly distributed; some chromosomes have many hits, others have very few. For example, HSA15 had no megablast hits. Prior to displaying, the hits on individual chromosomes were scanned to remove any occurrence of overlap in the megablast alignment results, so each hit is a unique site. The vertical position of hits along the y-axis in figure 4 varies only to provide greater resolution of hits where they are clustered, thus the y-axis has no numerical value. Even after scattering (or “jittering”) hits vertically, some regions contain such a dense assemblage of hits that together they appear as a solid block. Note that the sequence is highly compressed due to the very long length of chromosomes; hits that appear to be in an identical position could be many kilobases apart on the chromosome.
Fig. 4.—

Large megablast alignment hits that match canonical human rDNA plotted along all human chromosomes. X-axes differ to allow all graph panels to be the same size; the y-axes have no value, data points are only vertically spread to make the data more legible. Megablast hits are indicated in blue, while the start/stop position of centromeres are highlighted in red [with their thresholds shown as dashed vertical lines].
The assemblage of hits to the left of the centromere on HSA21 is derived from the sequences described above as DJ/PJ junctional regions and the near-centromere sites described above in figure 3. We wanted to obtain more information for other high-rDNA-density assemblages. Since HSA20 has the assemblage with the most hits, a dot plot analysis of the 1.6 Mb contributing to the dense assemblage of hits on HSA20 was completed (fig. 5A). Four regions within this 1.6 Mb region showed a resemblance to an entire rDNA unit (see spread of megablast hits along the 1.6-Mb sequence in fig. 5B). We further tested, using a dot plot, if megablast hit order reflected canonical positioning in the rDNA unit. This would then indicate that a large and highly degraded rDNA copy was present. The results of the dot plot indicate that such is the case, we found evidence for four large and degraded rDNA copies along HSA20, one of these copies located at position 1.2 Mb in figure 5B is shown in figure 5C as an example (see supplementary fig. S2, Supplementary Material online, for dot plots of all four HSA20 rDNA pseudogenes). In the dot plot of figure 5C, the vertical axis has sequence of the entire 43 kb human canonical unit [45S and IGS; no repeats removed]. The horizontal axis has sequence located at and around the 1.2-Mb position within the 29,290,000–30,922,000 bp region [1.6 Mb total] of HSA20 that has many rDNA alignment hits (fig. 5B). The parameters for marking a region are relaxed such that 50/100 bp of match results in one dot. If there is an overall match between sequence on the vertical line and sequence on the horizontal line, a diagonal line is formed in the graph. Figure 5C shows the presence of two unmistakable diagonal lines. The lines are interrupted by small gaps and displacements that are characteristic of indels and SNPs showing that the rDNA unit present at that site is highly degenerate in comparison to the canonical sequence, yet diagonal lines cover a large portion of the transcribed region [approximately at positions 35,000–43,000 bp; therefore ∼8 kb of the 13-kb 45S rDNA region that is transcribed] and of the IGS [approximately at positions 43,000–65,000 bp; therefore ∼22 kb of the 30-kb IGS] (fig. 5D). The extraneous, pattern-forming dots in the region of the IGS are due to repeated sequence within IGS (recall the probe is unedited in fig. 5C). When sequence is repeated tandemly a rectangle forms on the diagonal line. When repeated sequence is displaced across the IGS, matches show above and below the line. Such extraneous dots are not observed above or below the transcribed portion, as there are no large repeating sequences within it. There are approximately 20 megablast hits ranging in size from 65 to 2,173 bp that combine to form the diagonal lines in figure 5C. As mentioned above, the remainder of the megablast hits in figure 5A when observed as dot plots revealed three more regions with similar segments of degenerating rDNA (see supplementary fig. S2, Supplementary Material online). The conclusive presence of the IGS means that this result could not be obtained by a mechanism reliant on reverse transcription, it is only possible if entire chromosomal segments of multiple units have been incorporated into the chromosome, or perhaps if this is a region of a former active rDNA array.
Fig. 5.—

Identifying highly degraded rDNA units in regions far removed from rDNA arrays. (A) A closer look at the HSA20 data shown in figure 4; blue dots indicate megablast hits, red dots with accompanying dashed vertical bars indicate centromere thresholds. (B) The 1.6 Mb that contributes most to the assemblage of megablast hits present near the centromere at HSA20. The image is modified from the output produced by BLAST (Altschul et al. 1990; Camacho et al. 2009). Megablast hits are indicated along the 1.6-Mb and color coded according to size. The genomic coordinates for 1.6 Mb of sequence are also provided. (C) A closer look at the megablast hits present at the 1.2-Mb position of the entire 1.6 Mb region using a dot plot. The ∼80 kb of sequence at 1.2 Mb is compared with the canonical rDNA unit. Using a sliding window size of 50/100 bp, there is a strong indication that this region along the 1.6 Mb of HSA20 contains a highly degraded rDNA unit [see diagonal lines that are highlighted in blue, and which represent matches for the rRNA specifying and spacer region (IGS) of canonical rDNA]. (D) The dot plot shows two diagonal lines as an artefact of which base in the repeated array is counted as “zero”. Since we use the standard base numbering scheme, the real sequence is artificially split. The diagram illustrates where the sequences align within a repeated array. Array is shown in black/gray. Pseudogene is shown in blue. The diagram in (D) is not to scale.
In figure 4, the lengths of alignment hits vary from <100 to >2,000 bp. To observe the distribution of number and size of hits from the centromere outwards, we plotted the distance from the centromere versus the size of each hit. Results of this are shown in figure 6A. Hits are more numerous nearer the centromere, and most hits are <500 bp long. There are two peaks of large hits, one very near the centromere and one at a distance of ∼40 Mb.
Fig. 6.—
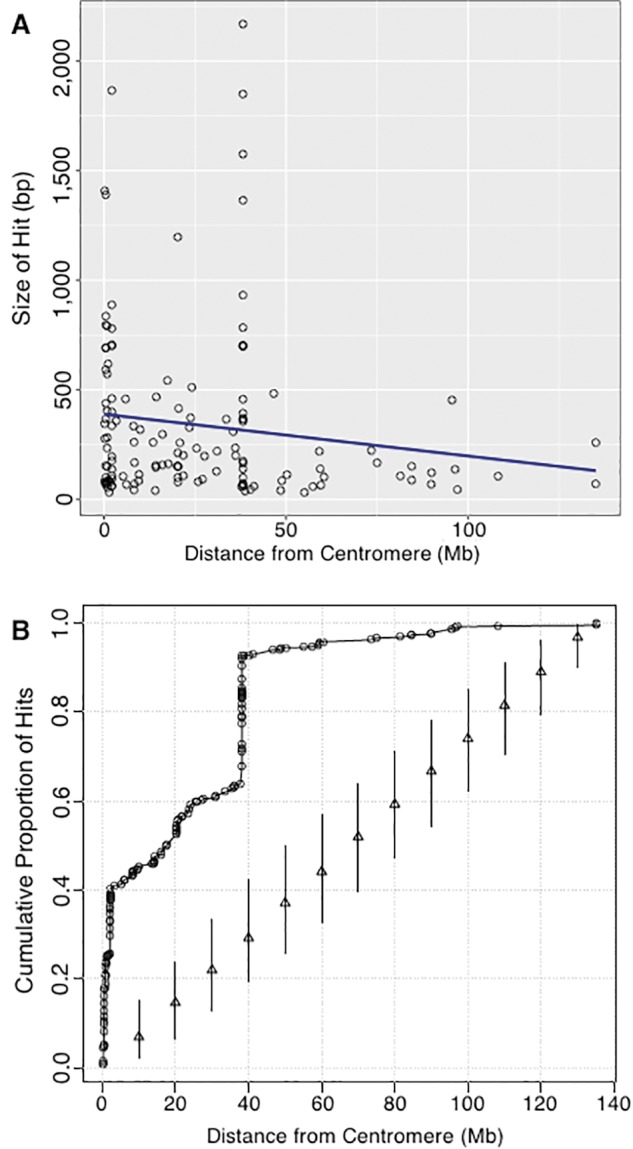
Determining if rDNA megablast hits occur closer to centromeres. (A) Size of megablast hits presented in figure 4 relative to their distance from centromeres. Black open circles represent Megblast hits and a linear trend-line is indicated in blue. (B) The cumulative proportion of megablast hit sites as a function of distance from centromere [black open circles]. For comparison, the cumulative proportions were calculated for 1,000 random assignments of the observed hit counts to locations randomly generated from a uniform distribution. Triangles indicate median cumulative proportions and vertical lines give the 2.5th and 97.5th percentiles of the cumulative proportions over random draws.
This observation of hits at ∼40 Mb away from a centromere prompted a closer look at HSA2, the chromosome primarily responsible for the peak at ∼40 Mb (see large block of hits present to the right of centromere in HSA2 of fig. 4). The evolutionary history of HSA2 is interesting and relevant to this work. HSA2 is an evolutionarily recent fusion of two smaller, acrocentric chromosomes that are currently still present as such in all other great apes. This was first suggested following studies of chromosomal banding patterns and more recently confirmed by direct sequence comparison between HSA2 and the Pan troglodytes chromosomes PTR12 and PTR13, as well as comparison with other great ape patterns (Baldini et al. 1993; Stanyon et al. 2008). When the fusion occurred, only one of the two centromeres could remain active if the chromosome was to be stabilized. When sequence of HSA2 is examined in detail, the presence of the inactive, vestigial centromere derived from one of the two chromosomes in the fusion event is detectable, and it is near the position of the high density of hits on HSA2, as can be seen in figure 7A. A dot plot analysis of the region containing the high density of hits on HSA2 (fig. 7B) shows a result similar to that seen for HSA20 (fig. 5C). Clearly, there is a remnant of a full-length rDNA unit at this genomic position. With this explanation for the cluster of hits on HSA2, we can score these hits as “centromere associated” because they are next to the vestigial HSA2 centromere, and can therefore conclude that the majority of hits, and all of the largest hits found are centromere associated. A plot of the cumulative proportion of hits against distance from centromeres further confirms that more rDNA hits occur closer to centromeres throughout the human genome (fig. 6B).
Fig. 7.—

Highly degraded rDNA unit present near vestigial centromere on HSA2. (A) Detail of the HSA2 panel from figure 4. The region shown is a close up of sequence to the right of the active centromere and extending to the edge of the vestigial centromere. Megablast hits [blue dots], HSA2’s active centromere [red dot with dashed gray vertical bars] and HSA2’s vestigial centromere threshold [dashed green vertical bars] are indicated along HSA2. The vestigial centromere threshold corresponds to 2q21.3 to 2q22.1 loci (Avarello et al. 1992; Baldini et al. 1993). Because this threshold is a best estimate of cytogenetic banding locations, two gene positions are also plotted as controls: HNMT [Gene ID: 3176] that occurs in 2q22.1 and ACMSD [Gene ID: 130013] that occurs in 2q21.3 [according to the Entrez Gene Database at NCBI]. (B) Megablast hits located at “*” in panel A, and which are associated with a degrading rDNA unit. About 107 kb have been subsampled from the 1 Mb surrounding the high number of hits in panel A. Similar to figure 5C, diagonal lines produced at a sliding window size of 50/100 bp indicate matches to rRNA specifying and IGS rDNA sequence.
Genome-Wide Low-Level “rDNA-Like” Signal
Returning to feature C displayed in figure 2; although there are no matches of a size comparable to those observed in feature A and feature B, there is still a constant low level of matching to the tailored-rDNA-probe. If there were no matches, the signal would drop to zero, as can be observed in the centromere region. When summed, the total number of alignments matching the tailored probe is 93,085 bp. Many of the matches are short (the shortest are 11 bp); thus the possibility of a match by chance alone, not reflecting an ancestral rDNA origin, must be considered. In total 31,649,825 bp of HSA21, the length of the q-arm scaffold, was scanned to obtain the results seen in figure 2. Ten randomized control sequences of the same length were computer generated, matching the %GC content every 200 kb to better reflect the true %GC composition of the HSA21 q-arm. A search for alignments to the tailored probe was conducted using these 10 in silico sequences. The in silico sequences returned a value for the total sum of nucleotides aligned by chance alone of 11,615 bp ± 435 bp SD. Using this control value as a correction, we can obtain a corrected estimate of the total sum of 81,469 bp along the HSA21 q-arm that are true matches to rDNA, reflecting an evolutionary origin to that sequence. The value is equal to 0.26% of the q-arm of HSA21. The total fraction of the genome contributed by the ∼300 copies of rDNA in functional arrays is ∼0.4% (Stults et al. 2008), so this value reflects a fairly significant contribution to the genome of sequence derived from, but no longer part of rDNA arrays.
At this scale of observation, it is difficult to discern the pattern of hits; there are no long stretches that fall to zero along the HSA21 q-arm length as can be seen at the site of the centromere, a region of ∼0.4 Mb (Hattori et al. 2000), but compression of the sequence makes a visible assessment of quantity unreliable. Megablast rDNA hit data demonstrate that the distribution of long segments derived from rDNA across the genome is uneven but the distribution brought to light by combining information from megablast, discontiguous megablast and blastn search methods for HSA21 seem to be more even. We attempted to obtain a better view of the distribution of smaller hits by choosing shorter portions of the genome in which to make a detailed scan, and by including analysis from the Mus musculus genome for augmenting confidence in the results. Six blocks of sequence 200 kb long were chosen from dispersed regions in the human genome, and were matched with the syntenic regions of the same size from the M. musculus genome. Figure 8 provides ideograms showing human and mouse karyotypes; the positions of the rDNA clusters relative to the location of each 200 kb block analysed is indicated. For all but one block (Mouse Synteny Block A), the regions were taken from chromosomes without an rDNA cluster. With the exception of Human Synteny Block A, each syntenic region was “anchored” by a GenBank annotated gene that was centered in the block. Human Block A is also the only block that is near the telomere of the chromosome from which it is derived. Sequence 100 kb in length to the left of the first nucleotide in the first exon and to the right of the last nucleotide in the last exon of the anchoring gene was sampled; synteny regions B through X contained no additional GenBank annotated genes besides the anchoring gene. The %GC for each of the six regions was determined, and 10 random sequences identical in length and % GC content to each synteny block were generated. These data allowed us to calculate the baseline abundance or “noise” that can be attributed to false positive alignments generated by BLAST (bl2seq) [see black circles in fig. 9]. Sequence alignments were determined for the combined transcribed 45S and IGS portions of the rDNA unit (i.e., an entire tailored repeat unit) against each of the randomized controls and against each synteny block. The probability of observing such a large (standardized) difference between the genomic percentage and the average random percentage, had the genomic percentage come from the same distribution, was always <0.005 (fig. 9). The standardized difference is where Y is the genomic percentage, is the mean random percentage and Sxis the corresponding sample standard deviation. Probability calculations assumed a normal model for the data. This statistically significant over-abundance cannot be due to reverse-transcription; IGS sequences are present in the alignments. The dot plot results in figure 5 showing the distinct presence of the IGS sequence supports this conclusion. With these findings, we conclude that for all the cases we have analysed there is a small but statistically significant general abundance of rDNA like sequence in blocks derived from genome sequence devoid of protein coding sequence. Although the value fluctuates between genomic locations, among the regions we have scanned the highest fraction of rDNA-like sequence equates to ∼2% of the sequence analysed (Block C for both human and mouse; control value is subtracted from the observed value for a chromosomal segment to obtain the percent; fig. 9). In general, the “ribosomal-DNA-like” signal is comprised of rDNA alignment matches ranging from 20 to 117 bp [average = 36 bp and mode = 32 bp].
Fig. 9.—
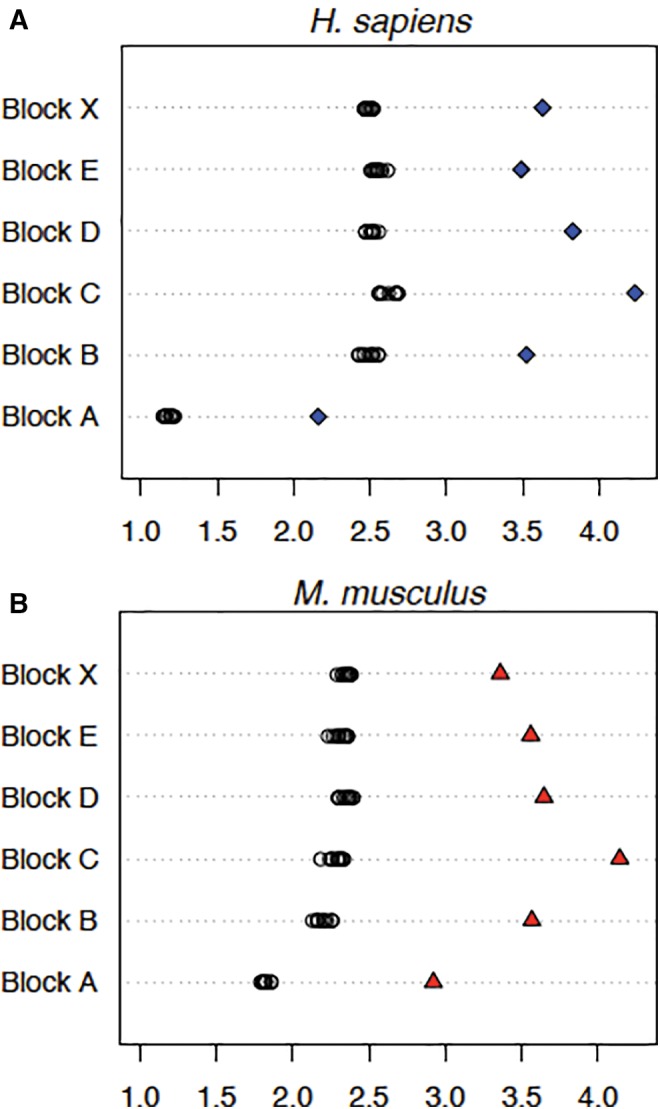
Over-representation of ribosomal DNA-like sequence [45S + IGS] distributed in human and mouse genomes. For each synteny block (A–E and X) both rDNA to genomic sequence similarity [see blue diamonds (A) and red triangles (B)] and rDNA to random sequence similarity [see open circles; replicated 10 times per synteny block (n = 10)] were calculated. The X-axis in both (A) and (B) represents the percentage of nucleotides that are rDNA-like. Results from probing with combined mature rRNA specifying and spacer (ETS + IGS) sequences are shown (the tailored probe was used). Each synteny block is 200 kb long, with the exception of mouse synteny block A, which is 205,973 bp long. This block is longer because we included the anchoring gene’s sequence in the block. The difference between genomic sequence similarity and random sequence similarity can be interpreted as the “true” over-abundance of rDNA-like sequence (e.g., this value for block C in both human and mouse would be ∼2%).
Discussion
Our major contention is that sequence initially derived from active rDNA is present at varying degrees of decay throughout the eukaryotic genome. We propose that a limited ability to participate in the unequal crossing over associated with concerted evolution is responsible for continuously generating rDNA pseudogenes at array edges. This process comes about as a by-product of the selective pressure to maintain high numbers of functional and near-identical copies of rDNA at more central regions in active rDNA arrays. We further propose that units of rDNA initially present in an active cluster are dispersed into the genome by a mechanism that involves centromeres. Following this dispersal, mutation and rearrangement events in the displaced rDNA copies accumulate until the sequence bears little resemblance to the former entire functional rDNA units. Over time, this process will add to the pool of junk DNA.
We presented evidence for our hypotheses from three different perspectives. The first was an analysis of rDNA pseudogenes in cloned sequence containing PJ and DJ junctions, and which represent the only currently available sequence in the rDNA arrays themselves. From these clones, rDNA sequence was analysed from each of the five acrocentric array-bearing chromosomes; 14 full or partial copies of the rRNA specifying region were analysed in total. For all of these, in spite of a high percent identity to the canonical rDNA sequence, there is strong evidence that they are pseudogenes based on the following: (1) divergence from canonical rDNA sequence and (2) reduced difference in the accumulation of mutations in mature rRNA specifying sequence to spacer region (ETS + ITS) sequence. As terminal copies of the rDNA unit are at a disadvantage for participating in the homogenizing process of concerted evolution, this is not a surprising result, but it is the first time that it has been documented.
How far into the array do pseudogenes extend? We cannot know from sequencing approaches, but other work may shed light on this question. Evidence of pseudogene presence within tandemly repeated rDNA comes from the work of Caburet et al. (2005), in which molecular combing was used to visualize, by hybridization of rDNA probes, individual stretches of rDNA from human. This approach demonstrated the existence of palindromic re-organization of the rDNA unit, revealing sequence that is often missing crucial rDNA components and differs in overall length from the canonical unit. Caburet et al. (2005) state that the work “reveals a totally unanticipated degree of complexity” in rDNA sequence arrangement. One-third of the molecules analysed by Caburet et al. (2005) had noncanonical configuration of rDNA elements. The precise location of these structures with respect to active rDNA cannot be determined, but the long stretches of tandem repeats observed show that they are much nearer the active cluster than the edge sequence we analysed, and yet they are pseudogenes.
The second perspective supporting our hypothesis comes from a genome-wide survey for relatively long rDNA-derived segments. The longest of these are ∼2 kb (∼5% the length of the full unit). We made observations of their size range and their distribution pattern. The majority of fragments, and all of the largest fragments are found near centromeric sequence, including a dense aggregation near the vestigial centromere on HSA2. A close examination of the genomic context of the aggregations found on HSA2 and HSA20, accomplished using dot plot analyses at high resolution, revealed that there are five full length, near 43 kb rDNA units present in these regions (both transcribed 45S and IGS portions are present); four on HSA20 and one on HSA2. These are undeniably derived from formerly functioning rDNA, but have at this time accumulated so many mutational changes that only the most sensitive methods can recognize their former identity.
The third perspective is based on attempts to determine an over-all proportion of sequence in the genome that can be attributed to former rDNA sequence, now dispersed away from functioning arrays and highly degraded. This analysis was merited by our initial finding of displaced whole rDNA units in the genome. We used a match length of 11 bp as the shortest permitted match, and employed multiple searches to detect any matching sequence to a “tailored” probe, that is, removing all sequence not uniquely attributable to rDNA. This was done for the entire length of the q-arm of HSA21, and for six blocks of 200 kb each elsewhere in the genome. In addition, six blocks of syntenically identical regions from the genome of Mus musculus were scanned. We reasoned that observing a similar over-abundance of sequence in two matched regions, one from mouse and one from human, would further support the claim that alignment matches are anciently acquired rDNA sequences having undergone enough degradation that only short segments remain recognizable. Because the degradation has been independent since their divergence, we would not expect to see the same sequences present, but would expect to see similar levels of rDNA-related sequence. Such is the case, as can be seen by observing that Block C in figure 9 contains the highest rDNA-like signal in both human and mouse. Because mutational rates vary in lineages and along the length of chromosomes within a single lineage (Hardison et al. 2003), some differences are expected when two regions are compared between species, but overall they are quite similar. By allowing segments as short as 11 bp to be counted as a match, we needed to be cognizant that some segments would be detected by chance alone and therefore needed to design controls to correct for false positive matches. This was done by generating random sequence of the same %GC, assessing it for potential matches to the tailored probe, and then subtracting the level of control matches found from the level found in genomic sequence. It is the amount of matching over and above that expected to be the random level that is called “true over-representation”. Average values of 0.26% of total sequence for HSA21 q-arm, 1.2% of total sequence for the six human blocks and 1.3% of total sequence for the six mouse blocks were the “true over-representation” values measured.
The crucial link unifying all of our data comes from detailed observations of the genomic context in which the multiple-hit megablast assemblages (presented in fig. 4) were found. The detection at five sites of nearly entire 43 kb rDNA units, in such a state of degradation that they are not detectable without very close scrutiny, provides us with confidence in making the suggestions that they (1) are originally derived from functioning arrays of rDNA, and (2) represent a “middle” age class that is “younger” in evolutionary age than rDNA from small fragments composing the “rDNA-like” (over-representation) data presented in figure 9, and are of an evolutionarily “older” age then PJ and DJ rDNA pseudogenes. These complete but highly degraded and displaced rDNA pseudogenes are composed of sequence strings that intergrade in length from very short segments to longer strings, that when combined clearly resemble an entire rDNA unit. The level of mutational differences found in these displaced rDNA copies suggests that the pseudogenes are at an evolutionary time point that provides a glimpse of what the small fragments found in the “over-representation” data presented in figure 9 would have been in the evolutionary past. It is noteworthy that the small fragments making up the data in figure 9 come from all parts of the 43-kb unit, including IGS. This argues that reverse-transcription is not the main source of “over-representation” sequence, and argues this even more strongly in light of the full length IGS component seen in the dot plot alignments.
The dual nature of the rDNA pseudogenes found near centromeres, highly similar to complete rDNA units and yet degraded enough to share properties of the small “rDNA-like” (over-representation) fragments, provides the evidence that what we are uncovering with our observations is a dynamic process. Our documentation of the current abundances and locations of rDNA fragments depicts a transitory state. The process requires the generation of rDNA pseudogenes by a mechanism other than reverse-transcription, distribution of them into the genome at sites far removed from functional rDNA arrays, and their eventual degradation to sequence almost unrecognizable as rDNA.
Because the generation of rDNA pseudogenes at array edges is an inherent feature of the concerted evolution of functional multi-copy rDNA, it would have been occurring since the advent of tandem repeat array nucleolar organizers (NORs). It is inseparable from their existence. The nucleolus, dependent for its existence on NORs, is considered a feature of the very earliest cells in the eukaryotic lineage (Pittis and Gabaldón 2016). We contend that the accumulation of sequence originally functioning as rDNA but no longer used as such has participated in producing a fraction of sequence that falls under the umbrella of eukaryotic junk DNA. This fraction is actually degraded rDNA that is almost unrecognizable. Parts of displaced rDNA could, over time, be co-opted into functioning in new ways; the rDNA harbors a variety of “selected domains” with potentially useful functionality. But much of it could also, and more likely, just continue to accumulate, degrade and contribute to the background sequence of eukaryotic chromosomes.
Extant eukaryotic lineages differ widely in the abundance of junk DNA they harbor. Some, such as S. cerevisiae have such compact genomes that they have likely evolved mechanisms to prevent accumulating junk DNA in large excess. Others are clearly able to function in the presence of a huge excess of DNA. The marbled lungfish (Protopterus aethiopicus), for example, has a genome size 44× that of human, on the order of 132 billion bp (Gregory et al. 2007). Prokopowich et al. (2003) analysed the genome sizes and rDNA content of 162 different species, 94 of them animals, 68 of them plants, and found a strong positive correlation between genome size and rDNA content (r = 0.68, P< 0.0001, α = 0.05). The largest of these has a genome size of ∼100 billion nucleotides and contains ∼20,000 rDNA units. Humans have a genome size of ∼3 billion nucleotides and contain ∼300 rDNA units. These observations are suggestive of a role of rDNA excess in the excessive size of eukaryotic genomes. The rDNA numbers are generally determined by hybridization techniques, which are incapable of distinguishing between functional rRNA specifying sequence and pseudogenes. Based on our results, a survey of eukaryotic genomes of a range of sizes, completed using our methodology would likely provide more information about the processes we have begun to uncover.
For pseudogenes of rDNA to spread in the genome, two components at a minimum are required: sequence must be generated and then must be distributed. UXO in itself is capable of generating excess sequence, but a mechanism of distribution is not obvious. We had not anticipated the centromeric localization of the majority of large rDNA fragments in the genome, but that result suggests a possibility for at least one mode of distribution. Centromeres themselves are subject to their own concerted evolution (Warburton and Willard 1995; Aldrup-MacDonald and Sullivan 2014). They are composed of very long stretches of tandem repeats ∼171 bp in length, and the sequence of the repeats is more similar within a species than it is between species, hallmarks of sequence undergoing concerted evolution. Homologous centromere pairs have been shown to have characteristic patterns of repeating arrays (Aldrup-MacDonald and Sullivan 2014), hence exchanges between nonhomologous centromeres are unlikely to be frequent, but they must occur at some level to exhibit concerted evolution characteristics. During a nonhomologous exchange, if rDNA sequence adjacent to the centromere sequence was included in the exchange, rDNA could be moved to other chromosomes. There still remains to be envisioned a mode of moving rDNA away from the active unit and nearer to centromeres on rDNA bearing chromosomes. The data shows that it happens, but by what mechanism can only be speculated. Perhaps the position of rDNA arrays on the short arm of acrocentric chromosomes contributes to this mechanism, or perhaps PJ sequence plays a role. PJ sequence has an odd distribution, being found on all rDNA bearing chromosomes (adjacent to the clusters themselves) but also elsewhere in the genome (Floutsakou et al. 2013). Additionally, over longer evolutionary times, chromosome breakage and rejoining is an ongoing process that can move sequence to new genomic neighborhoods. Given the known extent of chromosomal reorganization over time, see Misceo et al. (2008) for example, all segments of a chromosome, at some point in time, can have been a neighbor to an rDNA cluster and may have taken rDNA with them after a breakage.
Conclusion
We think our study of rDNA sequence throughout the genome has provided important insight. Regarding our attempt at measuring an rDNA-like signal, we do recognize that the regions we surveyed are a small fraction of the total genome, and also that the distances between the edge sequences we studied and the active portion of the rDNA array remains unknown—as currently there is no way of measuring their absolute proximity to the functioning sequences. Although the latter problem is an inescapable one, we feel that if an approach could be developed that incorporated the expectations of our model into a more automated survey of the entire genome a more precise measure of an rDNA-signal would be feasible. Our report highlights that rDNA pseudogenes are produced at rDNA array edges, portions of the rDNA array can get displaced into genomes, and over evolutionary time these displaced genes degrade and contribute to junk DNA.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the Acadia Biology Department [AGS, AGA and other departmental student summer research scholarships to B.M.R. and A.H.]. We thank Brian Hill and David H. Parma for early discussions on the approach, Michael Gray and W. Ford Doolittle for encouragement and advice on the manuscript. We thank F. Keyser and P. Porskamp for technical assistance in R coding. Author contributions: M.S. developed model; B.M.R. and M.S. designed research; B.M.R. performed research; B.M.R., E.S., A.M.H. and M.S. analysed data; B.M.R., E.S., and M.S. wrote the article. We also thank two anonymous reviewers for helpful comments on a previous version of the manuscript.
Literature Cited
- Adler D. 1992. Idiogram album: mouse. http://pathology.washington.edu/research/cytopages/.
- Adler D, Willis M. 1991. Idiogram album: human. http://pathology.washington.edu/research/cytopages/.
- Aldrup-MacDonald M, Sullivan B. 2014. The past, present, and future of human centromere genomics. Genes 5:33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Avarello R, Pedicini A, Caiulo A, Zuffardi O, Fraccaro M. 1992. Evidence for an ancestral alphoid domain on the long arm of human chromosome 2. Hum Genet. 89:247–249. [DOI] [PubMed] [Google Scholar]
- Averbeck KT, Eickbush TH. 2005. Monitoring the mode and tempo of concerted evolution in the Drosophila melanogaster rDNA locus. Genetics 171:1837–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldini A, et al. 1993. An alphoid DNA sequence conserved in all human and great ape chromosomes: evidence for ancient centromeric sequences at human chromosomal regions 2q21 and 9q13. Hum Genet. 90:577–583. [DOI] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. 2005. GenBank. Nucleic Acids Res. 33:D34–D38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie R, Roper RL, Upton C. 2004. JDotter: a Java interface to multiple dotplots generated by dotter. Bioinformatics 20:279–281. [DOI] [PubMed] [Google Scholar]
- Brown DD, Wensink PC, Jordan E. 1972. A comparison of the ribosomal DNA’s of Xenopus laevis and Xenopus mulleri: the evolution of tandem genes. J Mol Biol. 63:57–73. [DOI] [PubMed] [Google Scholar]
- Brownell E, Krystal M, Arnheim N. 1983. Structure and evolution of human and African ape rDNA pseudogenes. Mol Biol Evol. 1:29–37. [DOI] [PubMed] [Google Scholar]
- Caburet S, et al. 2005. Human ribosomal RNA gene arrays display a broad range of palindromic structures. Genome Res. 15:1079–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koning APJ, Gu W, Castoe TA, Batzer MA, Pollock DD. 2011. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 7:e1002384.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF. 2013. Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci U S A. 110:5294–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floutsakou I, et al. 2013. The shared genomic architecture of human nucleolar organizer regions. Genome Res. 23:2003–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer LY, et al. 2010. The NCBI BioSystems database. Nucleic Acids Res. 38:D492–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez IL, Sylvester JE. 1995. Complete sequence of the 43-kb human ribosomal DNA repeat: analysis of the intergenic spacer. Genomics 27:320–328. [DOI] [PubMed] [Google Scholar]
- Gonzalez IL, Sylvester JE. 1997. Beyond ribosomal DNA: on towards the telomere. Chromosoma 105:431–437. [DOI] [PubMed] [Google Scholar]
- Graur D, Zheng Y, Azevedo RBR. 2015. An evolutionary classification of genomic function. Genome Biol Evol. 7:642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory TR, et al. 2007. Eukaryotic genome size databases. Nucleic Acids Res. 35:D332–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden DG, Klinger HP, editors. 1985. ISCN 1985: an international system for human cytogenetic nomenclature. New York: Karger. [Google Scholar]
- Hardison RC, et al. 2003. Covariation in frequencies of substitution, deletion, transposition, and recombination during Eutherian evolution. Genome Res. 13:13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M, et al. 2000. The DNA sequence of human chromosome 21. Nature 405:311–319. [DOI] [PubMed] [Google Scholar]
- Henderson AS, Warburton D, Atwood KC. 1972. Location of ribosomal DNA in the human chromosome complement. Proc Natl Acad Sci U S A. 69:3394–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:12:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, et al. 2015. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 43:W580–W584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G, Ponting CP, Hein J. 2006. Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput Biol. 2:e5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2007. The origins of genome architecture. Sunderland: Sinauer Associates, Inc. Publishers. [Google Scholar]
- Misceo D, et al. 2008. Tracking the complex flow of chromosome rearrangements from the Hominoidea ancestor to extant Hylobates and Nomascus gibbons by high-resolution synteny mapping. Genome Res. 18:1530–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbitt MN, Francke U. 1973. A system of nomenclature for band patterns of mouse chromosomes. Chromosoma 41:145–158. [DOI] [PubMed] [Google Scholar]
- Ohta T. 1976. Simple model for treating evolution of multigene families. Nature 263:74–76. [DOI] [PubMed] [Google Scholar]
- Pittis AA, Gabaldón T. 2016. Late acquisition of mitochondria by a host with chimaeric prokaryotic ancestry. Nature 531:101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP, Hardison RC. 2011. What fraction of the human genome is functional?. Genome Res. 21:1769–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokopowich CD, Gregory TR, Crease TJ. 2003. The correlation between rDNA copy number and genome size in eukaryotes. Genome 46:48–50. [DOI] [PubMed] [Google Scholar]
- Sakai K, et al. 1995. Human ribosomal RNA gene cluster: identification of the proximal end containing a novel tandem repeat sequence. Genomics 26:521–526. [DOI] [PubMed] [Google Scholar]
- Smit AF, Hubley R, Green P. 2014. RepeatMasker Open-4.0. 2013–2015. http://www.repeatmasker.org.
- Smith GP. 1974. Unequal crossover and the evolution of multigene families. Cold Spring Harb Symp Quant Biol. 38:507–513. [DOI] [PubMed] [Google Scholar]
- Stage DE, Eickbush TH. 2007. Sequence variation within the rRNA gene loci of 12 Drosophila species. Genome Res. 17:1888–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanyon R, et al. 2008. Primate chromosome evolution: ancestral karyotypes, marker order and neocentromeres. Chromosome Res. 16:17–39. [DOI] [PubMed] [Google Scholar]
- Stults DM, Killen MW, Pierce HH, Pierce AJ. 2008. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 18:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Kurihara Y, Kanehisa T, Moriwaki K. 1990. Variation in the distribution of silver-staining nucleolar organizer regions on the chromosomes of the wild mouse, Mus musculus. Mol Biol Evol. 7:271–282. [DOI] [PubMed] [Google Scholar]
- Szostak JW, Wu R. 1980. Unequal crossing over in the ribosomal DNA of Saccharomyces cerevisiae. Nature 284:426–430. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2015. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton P, Willard H. 1995. Interhomologue sequence variation of alpha satellite DNA from human chromosome 17: Evidence for concerted evolution along haplotypic lineages. J Mol Evol. 41:1006–1015. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, et al. 2003. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 31:28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2009. ggplot2: elegant graphics for data analysis. New York: Springer Science & Business Media. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.