

Abstract
2-Oxoacid:ferredoxin oxidoreductases (OFORs) are essential enzymes in microbial one-carbon metabolism. They use thiamine pyrophosphate to reversibly cleave carbon-carbon bonds, generating low potential (~ −500 mV) electrons. Crystallographic analysis of a recently discovered OFOR, an oxalate oxidoreductase (OOR), has provided a second view of OFOR architecture and active site composition. Using these recent structural data along with the previously determined structures of pyruvate:ferredoxin oxidoreductase, structure-function relationships in this superfamily have been expanded and re-evaluated. Additionally, structural motifs have been defined that better serve to distinguish one OFOR subfamily from another and potentially uncover novel OFORs.
Introduction
Enzymes in the 2-oxoacid:ferredoxin oxidoreductase (OFOR) superfamily perform essential chemistry in microbial metabolism [1]. This enzyme superfamily oxidizes 2-oxoacids (α-ketoacids) by splitting the CO2-Cα bond, producing electrons that reduce ferredoxins (Figure 1a–c). These electrons typically have a potential of less than −500 mV [1–3], and are capable of “challenging” reductions, such as that of CO2, N2, H+, SO42−, and aromatic compounds [4–8]. A subset of OFORs are also capable of catalyzing the reverse reaction, fixing CO2 to make a 2-oxoacid [9]. The best studied member of this superfamily is pyruvate:ferredoxin oxidoreductase (PFOR), which catalyzes the transformation of acetyl-CoA to pyruvate, a reaction that is required by all organisms utilizing the reductive tricarboxylic acid (rTCA) cycle [10,11], the Wood-Ljungdahl pathway for acetogenesis [9], and the dicarboxylate/4-hydroxybutyrate cycle [2]. 2-oxoglutarate oxidoreductase (OGOR) is also required by the rTCA cycle [12,13], whereas 2-ketoisovalerate oxidoreductase (VOR) and indolepyruvate oxidoreductase (IOR) play important roles in amino acid metabolism, degrading aliphatic amino acids and aromatic amino acids, respectively [14].
Figure 1.

Reactions, cofactors and architecture of OFORs. a) OFORs (with the exception of OOR) catalyze the reversible oxidation of a 2-oxoacid to generate acyl-CoA, CO2 and two electrons. b) OOR catalyzes the oxidation of oxalate to form CO2 and two electrons, independent of CoA. The reversibility of the reaction has not been established. c) The low potential electrons liberated in (a) and (b) are transferred to ferredoxins for transport throughout the cell. d) The crystal structure of MtOOR (PDB ID 5C4I) is shown as a ribbon diagram with one catalytic monomer colored white, and the other monomer colored and labeled by domain: domain I – green; II – blue; III – yellow; V – wheat; VI – red. The inset shows the arrangement of redox cofactors, TPP-Mg2+ (stick model) and three [4Fe-4S] clusters (ball and stick model), with carbon atoms colored white; nitrogen – blue; oxygen – red; phosphorous – light orange; magnesium – green; sulfur – yellow; and iron – orange. e) Regardless of connectivity, OFORs of all types maintain a basic composition of domains I, II, III, and VI, with domain V also present in most OFORs. Domain IV is present only in dimeric (α2) PFORs, and domain VII, shaded with stripes, is found only in DaPFOR. The scheme for OORs is colored by domain as in (d) with the α, γ, and β chains labeled. The domains that bind the TPP and [4Fe-4S] cluster domains are indicated at the bottom. f) Cartoon of TPP is shown with the pyrimidine in the relevant tautomer and C2 of the thiazole ring deprotonated to form the active ylide. g) The structure of DaPFOR with one monomer colored by domain, highlighting domain VII, and the other monomer in blue.
Oxalate oxidoreductases (OORs), newly described OFORs, have brought additional chemistry and metabolic function to this versatile superfamily of enzymes. OOR oxidizes oxalate to generate two molecules of CO2, recovering the two released electrons [15]. Although little is known about the breadth of OOR use in nature, many microorganisms make use of oxalate-degrading enzymes [16–18]. Oxalate is a waste product of the metabolism of various carbohydrates as well as Vitamin C [19], and is abundant in soil and plants, which produce it as a defense against both predators and microbial infection [20]. The one instance in which OOR has been studied is within the context of the Wood-Ljungdahl pathway in Moorella thermoacetica. Here, OOR funnels both of its products, CO2 and low-potential electrons, into the Wood-Ljungdahl pathway, allowing M. thermoacetica to live on oxalate [15]. OOR is therefore positioned in a special place in cellular metabolism, with its primary function being the assimilation of an exogenous nutrient, rather than conversion and recycling of cellular metabolites, as is the case for the other OFORs.
With a second crystal structure of an OFOR finally determined a decade and a half after the first [21,22], we can now examine structure-function relationships in this important enzyme superfamily. Here we review what is known about the molecular basis of OFOR activity, focusing on PFOR and OOR.
The cofactors of OFORs
In order to capture and transfer low potential electrons one at a time, OFORs make use of one or more [4Fe-4S] clusters (Figure 1d). The one-electron nature of [4Fe-4S] cluster redox chemistry, as opposed to FAD or NAD(P)+, which undergo two-electron redox cycles, means that OFORs must make use of a catalytic cofactor that is both capable of activating the substrate 2-oxoacid, as well as undergoing one-electron radical transfer. This cofactor is thiamine pyrophosphate (TPP; Figure 1f), an organic cofactor that in its active form contains a nucleophilic carbanion [23] and is capable of stabilizing a 1-electron oxidized radical species. These properties make TPP well suited for converting the two-electron currency of bond oxidation into the one-electron currency recognized by [4Fe-4S] clusters.
The overall structure of OFORs
Crystal structures have been determined of two members of the OFOR superfamily: the PFOR from Desulfovibrio africanus (DaPFOR) [24–26] and the OOR from M. thermoacetica (MtOOR) [21,22]. Both enzymes have similar overall structures despite the fact that DaPFOR is an α2 homodimer and MtOOR is a dimer of trimers (αγβ)2 (Figure 1d,e,g). In both instances, the proteins form a multi-domain structure that have domains I, II, III, V, and VI in common, with domains I and VI forming the binding sites for TPP, substrate and one [4Fe-4S] cluster. Domain V houses two additional [4Fe-4S] clusters through which the electrons generated by the oxidation of 2-oxoacids may travel one at a time to the protein surface, where they can be transferred to an external electron acceptor, which is generally a ferredoxin. This common domain architecture extends to all OFORs, though the monomeric unit may comprise anywhere from 1–5 polypeptide chains, and some enzymes are missing domain V and the vast majority lack domains IV and VII (Figure 1e).
The general reaction scheme of OFORs
2-oxoacid oxidation by OFORs begins with nucleophilic attack by TPP on the 2-carbonyl, forming a TPP-substrate covalent adduct and driving decarboxylation of the substrate (Figure 2) [27]. For all OFORs except OOR, this step is followed by a one-electron oxidation [28]. The resulting radical acyl-TPP species is attacked by CoA, which drives the second electron transfer [29]. Finally, elimination of the acyl-CoA product regenerates the enzyme. OOR, on the other hand, must first activate an otherwise inert carboxylic carbon on oxalate before nucleophilic attack by TPP [15]. Following the initial decarboxylation, two subsequent oxidation steps are required to form carboxy-TPP, which is the final intermediate in the cycle. Unlike the other OFORs, OOR makes use of a CoA-independent mechanism to eliminate the final CO2 and regenerate the enzyme.
Figure 2.
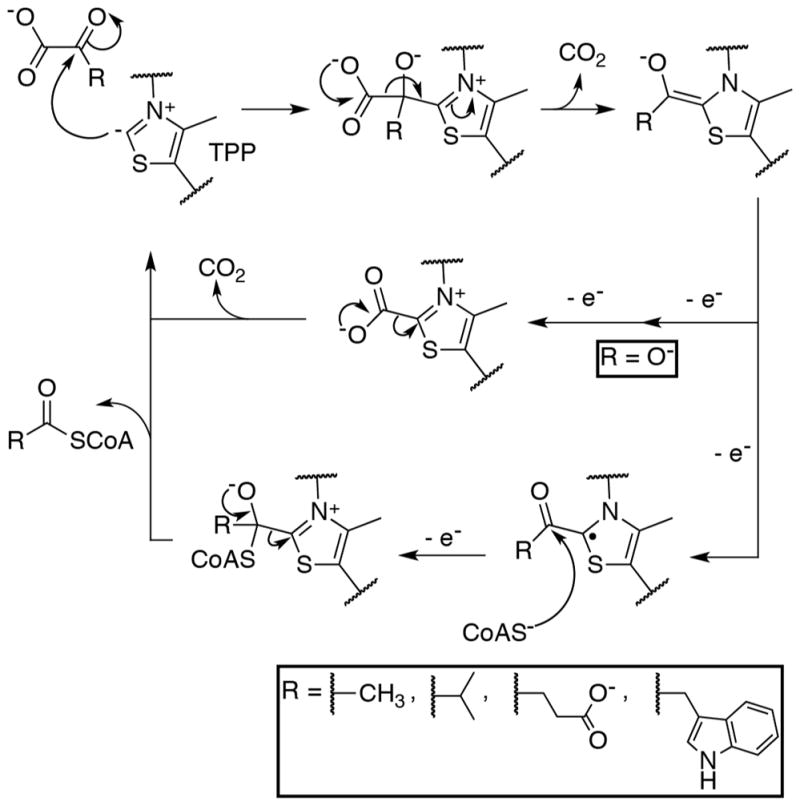
The general reaction scheme for OFORs. Along the top, TPP performs a nucleophilic attack on the α-carbon of the substrate 2-oxoacid, forming a covalent adduct that subsequently undergoes decarboxylation to form a hydroxyalkylidene-TPP (ROH-TPP) intermediate. When the substrate is oxalate (OOR case, shown in the middle), the dihydroxymethylidene-TPP intermediate is oxidized by two one-electron transfers to form carboxy-TPP. Carboxy-TPP is then decarboxylated to release product and regenerate the TPP catalyst. When the 2-oxoacid is not oxalate (PFOR, VOR, OGOR, and IOR cases, shown along the bottom), a one-electron oxidation from the ROH-TPP intermediate forms a stable ROH-TPP radical intermediate. CoA then attacks this radical species, driving the second electron oxidation, and eliminating TPP to form the acyl-CoA product.
Structure and function of pyruvate:ferredoxin oxidoreductase (PFOR)
Previous structural work on DaPFOR has provided insight into substrate binding, and in particular, into how pyruvate is positioned for catalysis through interactions with active-site residues (Figure 3a) [24]. Arg114 provides one positive charge to balance the negatively charged pyruvate substrate. In addition to interactions with Arg114, the carboxylate end of pyruvate hydrogen bonds with the backbone amide nitrogen, the hydroxyl group of Thr31, and the amide nitrogen of the Asn996 sidechain. The methyl-group of pyruvate, in turn, makes hydrophobic interactions with Leu121 and Ile123. Finally, the carbonyl oxygen of pyruvate accepts hydrogen bonds from Arg114 and the imine nitrogen of TPP, which also positions the carbonyl carbon immediately adjacent to the nucleophilic C2 TPP carbon. Once the covalent adduct is formed, the reaction can proceed as shown in Figure 2. To completely oxidize the resulting TPP adduct, CoA must bind to the enzyme, positioning its negatively-charged thiolate in the active site – an event which is associated with a 105 fold acceleration of the second electron transfer [29]. It is not known whether CoA carries out nucleophilic attack before or after the second electron is transferred nor is the mechanism responsible for the rate acceleration fully established, and despite crystallization efforts, the CoA binding site also remains a mystery.
Figure 3.

The active sites of DaPFOR and MtOOR reveal enzyme tailoring for the binding of specific substrates. a) The active site of DaPFOR (PDB ID: 2C42) with protein carbons colored light blue; TPP and pyruvate carbons colored white; nitrogen atoms colored blue; oxygen, red; and sulfur, yellow. Hydrogen bonding interactions with pyruvate are indicated with black dashed lines. b) The empty substrate-binding pocket of OOR (PDB ID: 5C4I) shows Arg31α situated at the bottom of the active site, where it forms a salt bridge to Glu154γ, indicated with dashed lines. Additionally, the Switch loop is observed in the “Asp-out” conformation, with Phe117α oriented towards the active site. OOR is represented similar to PFOR in (a), but with protein carbons colored light green and pink. The inset shows a model of oxalate (carbons colored black) docked into the empty OOR active site, based on the position of pyruvate in PFOR in (a). c) Active site of OOR after reaction with oxalate to form a carboxy-di-oxido-methyl-TPP (COOM-TPP) intermediate (PDB ID: 5EXD). In this structure, the Switch loop is seen in the “Asp-in” conformation with Asp116α pointing toward the active site. d) Active site of OOR following decarboxylation to form a carboxy-TPP intermediate (PDB ID 5EXE). The Switch loop is still in the “Asp-in” conformation with Asp116α pointing toward the TPP, but in this structural depiction of OOR, Arg31α is pointing away from the active site and Glu154γ is disordered.
Structure and function of oxalate oxidoreductase (OOR)
MtOOR shares many structural similarities with DaPFOR [21]. However, a number of substitutions in active site residues appear to tailor OOR for its unique chemistry. Relative to PFOR, a Thr to Arg substitution (Thr31 in DaPFOR, Arg31α in MtOOR) places an additional positive charge in the active site to balance the additional negative charge on oxalate and facilitate its attachment to TPP (Figure 3b). Following TPP-adduct formation, however, this additional positive charge may hinder the next mechanistic steps, which are decarboxylation and electron transfer from TPP to the [4Fe-4S] clusters. Interestingly, crystal structures of OOR reveal that in the presence of substrate, a conformational change of a loop (aka the Switch loop) occurs that repositions Phe117α out of the active site (Figure 3b) and moves into the active site a negatively charged residue, Asp116α (Figure 3c) [22]. With this new loop conformation, Asp116α is proximal to the TPP-adduct, such that it could drive decarboxylation and/or electron transfer (Figure 3c). The Switch loop represents another sequence change with respect to PFOR; the OOR sequence of DPPGDF is substantially altered from its PFOR analog of STHALSI. Additionally, there is no evidence that the equivalent loop in PFOR changes position. Finally, there is a structural substitution at the bottom of the active site. Domain VII of DaPFOR, a domain that is missing in OOR and other OFORs, contributes Met1202, whereas domain III of OOR contributes Glu154γ to occupy the same site (Figure 3a–b). In some of the crystallographic snapshots of the OOR reaction mechanism, Glu154γ forms a salt-bridge with Arg31α, positioning this positively charged residue near the TPP. In other stages of catalysis, domain III and Glu154γ are swung away from the active site and Arg31α is also displaced (Figure 3d). In contrast, all DaPFOR structures show domain VII in the same place, but it is believed that this domain must move for CoA to bind to the active site. In fact, the position of the Met1202 sulfur could represent the binding site for the thiol moiety of CoA.
In summary for OOR, structures reveal residue substitutions compared to PFOR and conformational changes that are capable of altering the electrostatic potential of the active site during catalysis. With Asp116α on a highly flexible loop and the position of Arg31α controlled by Glu154γ on a flexible domain, movements of Asp116α and Arg31α into and out of the active site can change the active site environment to first facilitate the binding of a diprotic substrate (oxalate) and then drive its decarboxylation and the accompanying electron transfer events.
Phylogenetic tree of the OFOR superfamily
Using the X-ray structures of DaPFOR and MtOOR to guide the sequence alignments of fifty-one OFORs, a phylogenetic tree of the OFOR superfamily was constructed (Figure 4). The majority of the fifty-one enzymes considered grouped into 7 monophyletic clades. Group 1 contains MtOOR along with other primarily bacterial enzymes, and so was assigned oxalate as the putative substrate. Group 2 contains mostly archaeal enzymes, none of which have been characterized. Groups 3 and 4 contain PFORs. Group 5 contains 5-subunit ((αβγδε)2) OFORs that have not been thoroughly characterized, but have been found to have either PFOR or OGOR activity in various instances [30,31]. Group 6 contains archaeal IORs. Group 7 contains two putative VORs, though a subset of VORs also exist in group 5 (Figure 4, enzyme 33 [14]). Group 8 contains OGORs.
Figure 4.

An unrooted phylogenetic tree of OFORs. OFORs are classified into 7 monophyletic clades and one paraphyletic group (group 3). Groups are assigned substrate specificity based upon those enzymes within the groups that have been biochemically characterized, which are indicated with a “*”. DaPFOR and MtOOR are indicated with a large |. Group 2 has no biochemically characterized members. The tree was constructed with 51 enzymes from the UniRef90 database (see Supplementary Material for accession codes). An alignment was performed using the PROMALS3D server, with the structures of DaPFOR and MtOOR serving as secondary structure references. The aligned sequences were trimmed around conserved blocks of residues from the OFOR α and β subunits using Gblocks, from which a maximum-likelihood tree was calculated with PhyML, using the LG model of amino acid substitution and 100 bootstrap calculations. Bootstrap values greater than 90 are indicated in parentheses.
It is interesting that PFORs can be separated into three distinct groups within the OFOR superfamily (Figure 4), including two monophyletic clades, (Figure 4, groups 4 and 5), and one paraphyletic group (Figure 4, group 3). These three groups span enzymes found in bacteria, archaea, and eukaryotes, and consist of varying quaternary architectures (Figure 1e). However, the minimal domain architecture of domains I, II, III, V, and VI is conserved (Figure 1d,e). In the active site, nearly all of the above mentioned residues responsible for binding pyruvate are strictly conserved: Arg114, Thr31, Asn996, and Ile123 (Figure 3a). Leu121 is likewise conserved in groups 4 and 5, though it is swapped for an Ile residue in group 3, which nonetheless constitutes a functional conservation. It is unfortunate that only one PFOR, DaPFOR (group 4), has been structurally characterized, and that this enzyme contains domain VII, which other PFORs do not have.
MtOOR is located in group 1 (Figure 4), which forms a phylogenetic clade along with other enzymes that are nearly identical to it and conserve all of the features mentioned above. More intriguing, however, are the enzymes that form group 2, which are closely related to group 1. No enzymes from group 2 have been biochemically characterized and their functions are unknown. Arg31α, a residue known to interact with oxalate from the OOR crystal structures (see above), is conserved in group 1 and group 2 but not in any other OFOR. More specifically, group 1 and 2 OFORs have the motif YPIRP, where Arg31α is the R, whereas all other OFORs contain either YPITP (groups 3–6, and 8) or YPGTP (group 7). However, other features of OORs are not found in group 2 enzymes, such as Asp116α of the Switch loop. Thus, these group 2 enzymes seem to have the residues needed for oxalate attachment to TPP but not for oxalate decarboxylation, raising the possibility that group 2 enzymes are OORs that run in the reverse direction, assembling oxalate from CO2. The reversibility of MtOOR has not been demonstrated but many OFORs are reversible [3,9], and it is tempting to speculate that a different sequence on the Switch loop could strongly favor one direct of the reaction over the other. Unfortunately, OORs cannot yet be recombinantly expressed making predictions like this impossible to experimentally verify.
One might ask why an organism would want to make oxalate, an incredibly difficult molecule to metabolize, and in this light, the identity of organisms that comprise group 2 is interesting. They are primarily metal-leaching archaea, and although these organisms are believed to leach metals primarily through the oxidation of elemental sulfur to sulfuric acid, it is also known that oxalate can serve to leach metals. Thus, an intriguing possibility is that these organisms are using OOR to produce oxalate through CO2 reduction to aid in the retrieval of metal ions from ores. Along these lines, an OFOR from Sulfolobus solfataricus (Uniprot Q7LX68, Q7LX69, Q7LX70), which has 96% identity to subunit α of enzyme 22 in group 2, was found in an operon with a gene for an oxalate/formate antiporter homolog [32].
As with OOR, we expect OGORs, found in groups 5 and 8, to have an additional positively charged residue close the active site to counter balance the additional negative charge that 2-oxoglutarate carries at physiological pH. Unfortunately, without further structural information, we cannot confirm this hypothesis nor predict the location of such a residue. Unlike all other OFORs, OGORs often lack a domain V, the ferredoxin-like domain, (Figure 1e), and so rely on only one [4Fe-4S] cluster to transport electrons to external electron acceptors. The molecular explanation for this domain deletion, if any, is unclear.
Arguably the families of enzymes that have been studied the least are the VORs and IORs. To date, only a handful of VORs and IORs have been isolated and biochemically studied, all of them from archaea [33–35]. The tree shown in Figure 4 has IORs clustered in group 6, which is the group that is most distinct within the OFOR superfamily. The one IOR that has been characterized is from the archaeon Methanothermobacter marburgensis, and all putative IORs are also from archaea. The two characterized VORs, on the other hand, are polyphyletic, with the VOR from M. marburgensis [34] falling in group 7 and the VOR from Thermococcus litoralis [14] belonging to group 3 along with paraphyletic PFORs. It is possible that the two distinct VORs arose by convergent evolution, with some group 3 PFORs and some group 8 OGORs separately acquiring VOR activity, but this idea awaits validation.
Conclusions
The recent structural data on oxalate oxidoreductase from M. thermoacetica has granted insight into what appears to be a unique mechanism for an OFOR, one that involves both loop rearrangements and domain movements during catalysis. These data have also made it possible to propose OOR functionality for a previously uncharacterized phylogenetic clade in the OFOR enzyme superfamily. We look forward to future work in this field to assess the distribution and niches of OORs, especially in regard to group 2 enzymes, but also with respect to oxalate metabolism in the human microbiome that could affect a person’s predisposition for kidney stones. Understanding substrate specificity of OFORs will be further enhanced by structures of enzymes from all eight enzymes subgroups, which will also hopefully reveal a CoA binding site in the CoA-dependent enzymes. Given the surprising features of the two structures characterized to date, we reasonably expect more surprises to come out of further structural studies of these remarkable enzymes.
Supplementary Material
Highlights.
Recent crystal structures shed light on an essential microbial enzyme family.
Conserved structural elements of 2-oxoacid:ferredoxin oxidoreductases revealed.
Molecular basis for oxalate metabolism by ancient enzyme family described.
Phylogeny of enzyme superfamily reveals complex history and unidentified members.
Acknowledgments
This work was supported in part by National Institutes of Health Grants GM069857 (C. L. D.) and by the National Science Foundation (NSF) Graduate Research Fellowship under Grant No. 1122374 (M. I. G.). This research was also made possible by the support of the Martin Family Society of Fellows for Sustainability (M. I. G.). C. L. D. is a Howard Hughes Medical Institute Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
• Of special interest
•• Of outstanding interest
- 1.Ragsdale SW. Pyruvate Ferredoxin Oxidoreductase and Its Radical Intermediate. Chem Rev. 2003;103:2333–2346. doi: 10.1021/cr020423e. [DOI] [PubMed] [Google Scholar]
- 2.Fuchs G. Alternative Pathways of Carbon Dioxide Fixation: Insights into the Early Evolution of Life? Annu Rev Microbiol. 2011;65:631–658. doi: 10.1146/annurev-micro-090110-102801. [DOI] [PubMed] [Google Scholar]
- 3••.Li B, Elliott SJ. The Catalytic Bias of 2-Oxoacid:ferredoxin Oxidoreductase in CO2: evolution and reduction through a ferredoxin-mediated electrocatalytic assay. Electrochimica Acta. 2016;199:349–356. The authors used protein film voltametry to examine the electrochemistry of a PFOR and an OGOR, together with their native ferredoxin redox partners. In addition to demonstrating the reversibility of each of the enzymes they studied, they were also able to record the lowest measured reduction potential for a ferredoxin. [Google Scholar]
- 4.Ragsdale SW, Pierce E. Acetogenesis and the Wood–Ljungdahl pathway of CO2 fixation. Biochim Biophys Acta BBA - Proteins Proteomics. 2008;1784:1873–1898. doi: 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yates MG. Stimulation of the phosphoroclastic system of Desulfovibrio by nucleotide triphosphates. Biochem J. 1967;103:32c–34c. doi: 10.1042/bj1030032c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wahl RC, Orme-Johnson WH. Clostridial pyruvate oxidoreductase and the pyruvate-oxidizing enzyme specific to nitrogen fixation in Klebsiella pneumoniae are similar enzymes. J Biol Chem. 1987;262:10489–10496. [PubMed] [Google Scholar]
- 7.Peck HD., Jr . Bioenergetic Strategies of the Sulfate-Reducing Bacteria. In: Odom JM, Singleton RJ, editors. The Sulfate-Reducing Bacteria: Contemporary Perspectives. Springer; New York: 1993. pp. 41–76. [Google Scholar]
- 8.Dörner E, Boll M. Properties of 2-Oxoglutarate:Ferredoxin Oxidoreductase from Thauera aromatica and Its Role in Enzymatic Reduction of the Aromatic Ring. J Bacteriol. 2002;184:3975–3983. doi: 10.1128/JB.184.14.3975-3983.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furdui C, Ragsdale SW. The Role of Pyruvate Ferredoxin Oxidoreductase in Pyruvate Synthesis during Autotrophic Growth by the Wood-Ljungdahl Pathway. J Biol Chem. 2000;275:28494–28499. doi: 10.1074/jbc.M003291200. [DOI] [PubMed] [Google Scholar]
- 10.Quandt L, Pfennig N, Gottschalk G. Evidence for the key position of pyruvate synthase in the assimilation of CO2 by Chlorobium. FEMS Microbiol Lett. 1978;3:227–230. [Google Scholar]
- 11.Fuchs G, Stupperich E, Eden G. Autotrophic CO2 fixation in Chlorobium limicola. Evidence for the operation of a reductive tricarboxylic acid cycle in growing cells. Arch Microbiol. 1980;128:64–71. [Google Scholar]
- 12.Yamamoto M, Ikeda T, Arai H, Ishii M, Igarashi Y. Carboxylation reaction catalyzed by 2-oxoglutarate:ferredoxin oxidoreductases from Hydrogenobacter thermophilus. Extremophiles. 2010;14:79–85. doi: 10.1007/s00792-009-0289-4. [DOI] [PubMed] [Google Scholar]
- 13.Mai X, Adams MWW. Characterization of a fourth type of 2-keto acid-oxidizing enzyme from a hyperthermophilic archaeon: 2-ketoglutarate ferredoxin oxidoreductase from Thermococcus litoralis. J Bacteriol. 1996;178:5890–5896. doi: 10.1128/jb.178.20.5890-5896.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heider J, Mai X, Adams MWW. Characterization of 2-ketoisovalerate ferredoxin oxidoreductase, a new and reversible coenzyme A-dependent enzyme involved in peptide fermentation by hyperthermophilic archaea. J Bacteriol. 1996;178:780–787. doi: 10.1128/jb.178.3.780-787.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15•.Pierce E, Becker DF, Ragsdale SW. Identification and Characterization of Oxalate Oxidoreductase, a Novel Thiamine Pyrophosphate-dependent 2-Oxoacid Oxidoreductase That Enables Anaerobic Growth on Oxalate. J Biol Chem. 2010;285:40515–40524. doi: 10.1074/jbc.M110.155739. This publication describes the first characterization of an OOR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kotsira VP, Clonis YD. Oxalate Oxidase from Barley Roots: Purification to Homogeneity and Study of Some Molecular, Catalytic, and Binding Properties. Arch Biochem Biophys. 1997;340:239–249. doi: 10.1006/abbi.1997.9896. [DOI] [PubMed] [Google Scholar]
- 17.Tanner A, Bornemann S. Bacillus subtilis YvrK Is an Acid-Induced Oxalate Decarboxylase. J Bacteriol. 2000;182:5271–5273. doi: 10.1128/jb.182.18.5271-5273.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anantharam V, Allison MJ, Maloney PC. Oxalate:formate exchange. The basis for energy coupling in Oxalobacter. J Biol Chem. 1989;264:7244–7250. [PubMed] [Google Scholar]
- 19.Massey LK, Liebman M, Kynast-Gales SA. Ascorbate Increases Human Oxaluria and Kidney Stone Risk. J Nutr. 2005;135:1673–1677. doi: 10.1093/jn/135.7.1673. [DOI] [PubMed] [Google Scholar]
- 20.Franceschi VR, Nakata PA. Calcium Oxalate in Plants: Formation and Function. Annu Rev Plant Biol. 2005;56:41–71. doi: 10.1146/annurev.arplant.56.032604.144106. [DOI] [PubMed] [Google Scholar]
- 21••.Gibson MI, Brignole EJ, Pierce E, Can M, Ragsdale SW, Drennan CL. The Structure of an Oxalate Oxidoreductase Provides Insight into Microbial 2-Oxoacid Metabolism. Biochemistry. 2015;54:4112–4120. doi: 10.1021/acs.biochem.5b00521. The first structure of an OOR is described and compared to the only other structurally characterized OFOR, that of DaPFOR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22••.Gibson MI, Chen PY-T, Johnson AC, Pierce E, Can M, Ragsdale SW, Drennan CL. One-carbon chemistry of oxalate oxidoreductase captured by X-ray crystallography. Proc Natl Acad Sci USA. 2016;113:320–325. doi: 10.1073/pnas.1518537113. The structures of OOR reaction intermediates are described in this paper. These structural data reveal some startling and unprecedented conformational changes in the OOR active site. By examining the changing charge landscape, authors propose a mechanism by which OOR is capable of carrying out the only OFOR reaction that does not require CoA as a substrate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breslow R. On the Mechanism of Thiamine Action. IV.1 Evidence from Studies on Model Systems. J Am Chem Soc. 1958;80:3719–3726. [Google Scholar]
- 24.Chabrière E, Charon M-H, Volbeda A, Pieulle L, Hatchikian EC, Fontecilla–Camps J-C. Crystal structures of the key anaerobic enzyme pyruvate:ferredoxin oxidoreductase, free and in complex with pyruvate. Nat Struct Biol. 1999;6:182–190. doi: 10.1038/5870. [DOI] [PubMed] [Google Scholar]
- 25.Chabrière E, Vernède X, Guigliarelli B, Charon M-H, Hatchikian EC, Fontecilla-Camps JC. Crystal Structure of the Free Radical Intermediate of Pyruvate:Ferredoxin Oxidoreductase. Science. 2001;294:2559–2563. doi: 10.1126/science.1066198. [DOI] [PubMed] [Google Scholar]
- 26•.Cavazza C, Contreras-Martel C, Pieulle L, Chabrière E, Hatchikian EC, Fontecilla-Camps JC. Flexibility of Thiamine Diphosphate Revealed by Kinetic Crystallographic Studies of the Reaction of Pyruvate-Ferredoxin Oxidoreductase with Pyruvate. Structure. 2006;14:217–224. doi: 10.1016/j.str.2005.10.013. This paper describes structural data on PFOR in various bound and liganded states and is the most current structural paper on PFORs. [DOI] [PubMed] [Google Scholar]
- 27.Reed GH, Ragsdale SW, Mansoorabadi SO. Radical reactions of thiamin pyrophosphate in 2-oxoacid oxidoreductases. Biochim Biophys Acta BBA - Proteins Proteomics. 2012;1824:1291–1298. doi: 10.1016/j.bbapap.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mansoorabadi SO, Seravalli J, Furdui C, Krymov V, Gerfen GJ, Begley TP, Melnick J, Ragsdale SW, Reed GH. EPR spectroscopic and computational characterization of the hydroxyethylidene-thiamine pyrophosphate radical intermediate of pyruvate:ferredoxin oxidoreductase. Biochemistry. 2006;45:7122–7131. doi: 10.1021/bi0602516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29•.Furdui C, Ragsdale SW. The Roles of Coenzyme A in the Pyruvate:Ferredoxin Oxidoreductase Reaction Mechanism: Rate Enhancement of Electron Transfer from a Radical Intermediate to an Iron–Sulfur Cluster. Biochemistry. 2002;41:9921–9937. doi: 10.1021/bi0257641. This paper was important in establishing the role of coenzyme A in PFOR chemistry. [DOI] [PubMed] [Google Scholar]
- 30.Yun N-R, Yamamoto M, Arai H, Ishii M, Igarashi Y. A Novel Five-Subunit-Type 2-Oxoglutalate:Ferredoxin Oxidoreductases from Hydrogenobacter thermophilus TK-6. Biochem Biophys Res Commun. 2002;292:280–286. doi: 10.1006/bbrc.2002.6651. [DOI] [PubMed] [Google Scholar]
- 31.Ikeda T, Ochiai T, Morita S, Nishiyama A, Yamada E, Arai H, Ishii M, Igarashi Y. Anabolic five subunit-type pyruvate:ferredoxin oxidoreductase from Hydrogenobacter thermophilus TK-6. Biochem Biophys Res Commun. 2006;340:76–82. doi: 10.1016/j.bbrc.2005.11.155. [DOI] [PubMed] [Google Scholar]
- 32.Peeters E, Albers S-V, Vassart A, Driessen AJM, Charlier D. Ss-LrpB, a transcriptional regulator from Sulfolobus solfataricus, regulates a gene cluster with a pyruvate ferredoxin oxidoreductase-encoding operon and permease genes. Mol Microbiol. 2009;71:972–988. doi: 10.1111/j.1365-2958.2008.06578.x. [DOI] [PubMed] [Google Scholar]
- 33.Kletzin A, Adams MWW. Molecular and phylogenetic characterization of pyruvate and 2-ketoisovalerate ferredoxin oxidoreductases from Pyrococcus furiosus and pyruvate ferredoxin oxidoreductase from Thermotoga maritima. J Bacteriol. 1996;178:248–257. doi: 10.1128/jb.178.1.248-257.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tersteegen A, Linder D, Thauer RK, Hedderich R. Structures and Functions of Four Anabolic 2-Oxoacid Oxidoreductases in Methanobacterium Thermoautotrophicum. Eur J Biochem. 1997;244:862–868. doi: 10.1111/j.1432-1033.1997.00862.x. [DOI] [PubMed] [Google Scholar]
- 35.Mai X, Adams MWW. Indolepyruvate ferredoxin oxidoreductase from the hyperthermophilic archaeon Pyrococcus furiosus. A new enzyme involved in peptide fermentation. J Biol Chem. 1994;269:16726–16732. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.