

Abstract
Fluorogenic dyes such as FlAsH and ReAsH are used widely to localize, monitor, and characterize proteins and their assemblies in live cells. These bis-arsenical dyes can become fluorescent when bound to a protein containing four proximal Cys thiols – a tetracysteine (Cys4) motif. Yet the mechanism by which bis-arsenicals become fluorescent upon binding a Cys4 motif is unknown, and this nescience limits more widespread application. Here we probe the origins of ReAsH fluorogenicity using computational techniques. Our results support a model in which ReAsH fluorescence depends on the relative orientation of the aryl chromophore and the appended arsenic chelate; the fluorescence is rotamer-restricted. Our results do not support a mechanism in which fluorogenicity arises from the relief of ring strain. The calculations identify those As-aryl rotamers that support fluorescence and those that do not and correlate well with experiment. The rotamer-restricted model we propose is supported further by biophysical studies: the excited state fluorescence lifetime of a complex between ReAsH and a high affinity Cys4 motif is longer than that of ReAsH-EDT2, and the fluorescence intensity of ReAsH-EDT2 increases in solvents of increasing viscosity. By providing a higher resolution view of the structural basis for fluorogenicity, these results provide a clear strategy for the design of more selective bis-arsenicals and better-optimized protein targets, with a concomitant improvement in the ability to characterize previously invisible protein conformational changes and assemblies in live cells.
Introduction
Fluorogenic molecules–those that glow only upon interaction with a prescribed protein, lipid, saccharide, or nucleic acid–are essential tools for localizing and monitoring events in live cells, sometimes even in real time.1 Bis-arsenicals, exemplified by the dyes FlAsH2 (fluorescin arsenical hairpin binder) and ReAsH (resorufin arsenical hairpin binder),3 represent one class of fluorogenic molecules (Figure 1).2–4,5 Bis-arsenicals are not fluorescent when coordinated through arsenic to two ethanedithiol ligands (EDT), but can glow brightly when EDT is exchanged for four proximal Cys thiols on a target protein, an arrangement termed a tetracysteine (Cys4) motif.2 The thiols of a Cys4 motif can be close in primary sequence (Figure 2A)2 or distant in sequence but close by virtue of association or conformation (Figure 2B).6 In the latter case, induced bis-arsenical fluorescence can provide a visual readout for structurally defined protein-protein interactions or conformational changes in live cells,7–9 a methodology referred to as bipartite Cys4 display. Applications of FlAsH and ReAsH include studies of connexin trafficking,10 GPCR activation,11 amyloid-beta amyloidogenesis, 12 EGFR activation,7,8 and, most recently, β-arrestin functional dynamics.13
Figure 1.
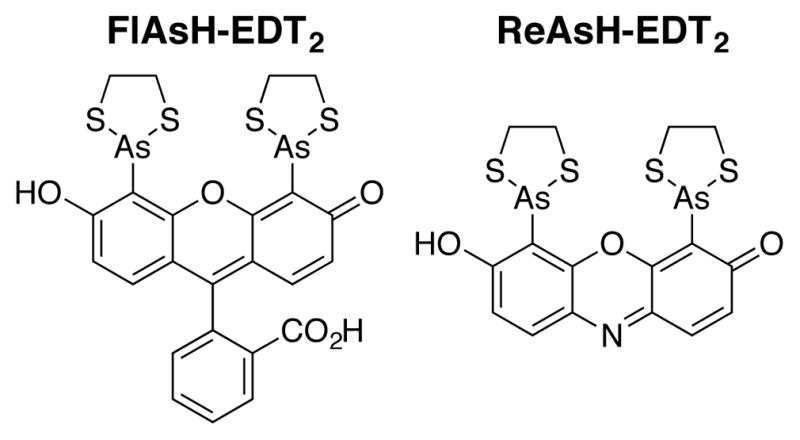
FlAsH-EDT2 and ReAsH-EDT2, two fluorogenic bis-arsenical dyes used for linear2 and bipartite tetracysteine display.7–9
Figure 2.

Tetracysteine (Cys4) (A) and bipartite Cys4 (B) display. Bis-arsenical dyes such as ReAsH are quenched when coordinated to ethanedithiol (EDT) but become fluorescent upon ligand exchange with a (A) single protein carrying a linear Cys4 motif or (B) a protein assembly in which the Cys4 motif is recapitulated upon folding or association.
Despite demonstrated utility for tagging proteins and their assemblies, 7,12,14,15,16 the mechanism by which bis-arsenicals such as ReAsH become fluorescent upon protein association is unknown,14 and this lack of knowledge hinders a more widespread application of both linear2 and bipartite7–9 Cys4 display. Here we probe the origins of ReAsH fluorogenicity using computational techniques. Our results support a model in which ReAsH fluorescence is rotamer-restricted, depending in a critical way on the relative orientation of the aryl chromophore and the appended arsenic chelate. Our results do not support a previously proposed mechanism in which fluorogenicity arises from the relief of ring strain.17 The calculations identify those As-aryl rotamers that support fluorescence and those that do not, and correlate well with published experiments from multiple laboratories. Moreover, the excited state fluorescence lifetime of the complex between ReAsH and a protein-embedded Cys4 motif is longer than that of ReAsH-EDT2, and the fluorescence intensity of ReAsH-EDT2 increases in solvents of increasing viscosity. These observations are in full accord with the rotamer-restricted model. By providing a higher resolution view of the structural basis for fluorogenicity, these results can guide the design of both more selective bis-arsenicals and better-optimized protein targets, with a concomitant improvement in the ability to characterize previously invisible protein conformational changes and assemblies in live cells.
Two mechanisms have been proposed for the gain in fluorescence when ReAsH associates with a linear or bipartite Cys4 motif (Figure 3). Both involve changes in photo-induced electron transfer (PeT), in analogy with current models for the conditional gain in fluorescence (fluorogenicity) of fluorescein and silicon-rhodamine dyes, among others.18,19,20 Both mechanisms posit that ReAsH-EDT2 is quenched by PeT from a high-lying molecular orbital (MO) centered predominantly on arsenic to a lower-lying MO centered on the fluorophore (Figure 3A).2,14 In one mechanism, the energy of the arsenic-centered MO is raised by ring strain (Figure 3B); ligand exchange with a protein Cys4 motif relieves this strain, lowering the energy of the As-centered HOMO to prevent PeT. In another mechanism, the energy of the As-centered HOMO energy depends on the As-aryl dihedral angles Ω and/or Ω’ (Figure 3C)–the relative orientation of the aryl chromophore and the appended arsenic chelate. Here, ReAsH-EDT2 is quenched because the As-aryl bond rotates freely and samples conformations that allow PeT; protein-binding restricts rotation to a low energy conformation in which PeT is prohibited. Regardless of the molecular details, in both mechanisms a change in structure or dynamics lowers the energy of the As-centered orbital to block PeT quenching (Figure 3D).
Figure 3.

Two mechanisms to account for the increase in ReAsH fluorescence upon binding a tetracysteine (Cys4) motif. (A) In both, ReAsH-EDT2 is quenched by PeT from a high-lying molecular orbital (MO) centered on arsenic to a lower-lying MO centered on the fluorophore. (B) Fluorescence induced by relief of ring strain: In this mechanism, ReAsH-EDT2 fluorescence is quenched by PeT from a As-centered orbital whose energy is raised by strain in the As-EDT chelates; the relief of strain when EDT exchanges for a protein Cys4 motif lowers the energy of this orbital to disfavor PeT and allow fluorescence. (C) In this mechanism, fluorescence induced by restricted rotation: ReAsH-EDT2 fluorescence is quenched by PeT in only some As-aryl bond rotamers; exchange of EDT for a protein Cys4 motif restricts rotation to disfavor PeT and allow fluorescence. (D) In both mechanisms, a change in structure or dynamics lowers the energy of the As-centered orbital to block PeT quenching (Figure 3D).
Results and Discussion
We performed Hartree-Fock calculations at the 6–31+G(d) level using Gaussian 0921 to investigate the effect of ring strain and restricted As-aryl rotation on the molecular orbital energy landscape of ReAsH-EDT2. First, we evaluated whether ring strain relief alone could sufficiently stabilize the arsenic-centered HOMO to disfavor fluorescence quenching via PeT.17 To model the effect of ring strain, we computed the energies of the arsenic- and fluorophore-centered HOMOs of both ReAsH-EDT2 (with two 5-membered EDT chelates) and ReAsH-PDT2 (with two 6-membered PDT chelates). In simple cycloalkanes, this difference in ring size accounts for over 5 kcal•mol−1 of strain energy.22 Structures were minimized using the Hartree-Fock method and the basis set 6–31+G(d). All calculations were performed with water solvent using the Polarizable Continuum Model (PCM). Molecular orbitals were assigned to As or the fluorophore core by visual inspection and validated using GaussSum.23 In ReAsH-EDT2, 97% of the orbital denoted the fluorophore-centered orbital was composed of atomic orbitals from fluorophore; the analogous value for the As-centered orbital was 88%.
The results of these calculations are shown in Figures 4 and 5. As expected, in their lowest energy conformations, the As-centered HOMO of ReAsH-EDT2 (Figure 5A) lies above (ΔE = 0.01217 Ha) the As-centered HOMO of ReAsH-PDT2 (Figure 5B) with energies of −0.31055 and −0.32272 Ha, respectively (Figure 4). An energy difference of 0.01217 Ha is significant: In azidofluorescein dyes, as well as those related to Tokyo Green, for example, a 0.01 Ha difference in quencher HOMO energy dictates whether a molecule is quenched or fluorescent.19,24 An even smaller energy difference of 0.001 Ha was invoked to rationalize the behavior of panel of potential methylglyoxal sensors.25 But despite the difference in ring size and calculated orbital energy, in neither ReAsH-EDT2 nor ReAsH-PDT2 is the As-centered HOMO energetically appropriate for PeT: in both cases, the As-centered HOMO lies below the fluorophore-centered HOMO, and PeT is disfavored (Figure 4). These calculations imply that although ring strain may exist in ReAsH-EDT2, it is insufficient to raise the As-centered HOMO energy to facilitate PeT. More importantly, since the As-centered HOMO in the minimum energy structure of ReAsH-EDT2 cannot support PeT, the calculations imply that ReAsH-EDT2 fluorescence must be quenched through a transient, high-energy conformation.
Figure 4.

Absolute and relative energies of fluorophore- and As-centered HOMOs in minimized structures of ReAsH–EDT2 and –PDT2. See Figure 5 for images of minimized structures and the As- and fluorophore-centered MOs.
Figure 5.

Images of As- and fluorophore-centered highest occupied molecular orbitals (HOMO) corresponding to the lowest energy conformations of (A) ReAsH-EDT2 and (B) ReAsH-PDT2; these conformations correspond to states A and B in Figure 7 and are expected to be fluorescent. Also shown are images of the As- and fluorophore-centered highest occupied molecular orbitals (HOMO) of (C) ReAsH-EDT2 and (D) ReAsH-PDT2 corresponding to the lowest energy quenched conformation. These conformations correspond to states C and D in Figure 7.
To investigate whether this transient, high-energy conformation could be one or more As-aryl rotamers, we calculated orbital energies for 1,296 different As-aryl bond rotamers of ReAsH-EDT2 and ReAsH-PDT2. We began with the minimized structures of ReAsH-EDT2 or ReAsH-PDT2 (Figure 5), and systematically rotated the As-aryl dihedral bond angles Ω and Ω’ (C-C-As-S) between −180° and 180° at 10° intervals (Figure 6). Energy calculations (performed at the HF 6–31+G(d) level in water with the PCM solvent model in Gaussian)21 were used to evaluate the resulting structures, and the difference in energy (ΔE) between the As and fluorophore-centered HOMOs was plotted as a function of Ω and Ω’ (Figure 7A). Plots of absolute energies are shown in Figure 7B. In each case, a number of ReAsH-EDT2 and ReAsH-PDT2 rotamers with severe steric clashes (16% and 22%, respectively) were excluded from the analysis.
Figure 6.

Structure of ReAsH-EDT2 and ReAsH-PDT2 illustrating the atoms used to define the As-aryl dihedral bond angles Ω and Ω’.
Figure 7.

Plots illustrating the effect of As-Aryl bond rotation on the (A) energy differences (ΔE) between the As- and fluorophore-centered MOs and the (B) the total energy of ReAsH-EDT2 and ReAsH-PDT2. Points A and B represent the lowest energy conformations of ReAsH-EDT2 and ReAsH-PDT2, respectively. Points C and D represent the lowest energy conformations were PeT is permitted for ReAsH-EDT2 and ReAsH-PDT2, respectively. Point O represents the ReAsH rotamers observed in the NMR structure of the complex with a linear, optimized Cys4 motif (FLNCCPGCCMEP). Points E, F, and G represent the values of Ω and Ω’ in models of the ReAsH complexes of p53-2, p53-3, and EmGFP-1, respectively (shown in Figure 8).
The calculations indicate that the difference in energy (ΔE) between the As and fluorophore-centered HOMOs depends critically on the As-aryl dihedral angles Ω and Ω’ (Figure 7). The value of ΔE varies between ± 0.2; positive values correspond to orbital arrangements that support PeT (As-centered HOMO higher in energy than fluorophore-centered HOMO) and states that are expected to be quenched, whereas negative values correspond to orbital arrangements that do not support PeT (As-centered HOMO lower in energy than fluorophore-centered HOMO) and states that are expected to be fluorescent. Overall, almost half (42% and 47%, respectively) of the evaluated ReAsH-EDT2 and ReAsH-PDT2 rotamers would support PeT and should be quenched; the remaining rotamers should be fluorescent. In particular, the calculations predict that ReAsH rotamers with Ω and Ω‘ values between approximately −70° and 150° and between approximately −150° and 70°, respectively, will glow, whereas rotamers in which either Ω or Ω’ lies outside this range will not. In general, ReAsH-EDT2 rotamers in which even one As-aryl bond is rotated at least 100° from the ideal conformation will be quenched. Because of steric hindrance, the overall difference in rotamer energy is larger for ReAsH-PDT2 (Figure 7).
In addition to the observation that the set of predicted fluorescent ReAsH-EDT2 rotamers includes the minimum energy structure (Figure 5) (Ω/Ω’ = 46.64°/−46.64°), the predicted relationship between As-aryl bond rotation and ReAsH fluorescence is largely in line with previously reported analyses of ReAsH-protein interactions. First, the set of predicted fluorescent ReAsH rotamers includes those observed in the NMR structure of ReAsH bound to a peptide containing a linear, optimized Cys4 motif (FLNCCPGCCMEP).26 Here, the values of Ω and Ω’ (averaged across all 30 structures) were −57.1 ± 6.09° and 54.5° ± 47.8°; this combination lies at the very center of the rotamer plot (Figure 7A, point O) along with the minimum energy structure (Ω/Ω’ = 46.64°/−46.64°) (Figure 7A, point A).
The set of predicted fluorescent ReAsH rotamers is also largely in accord with previous studies that evaluated whether ReAsH became fluorescent when bound to diverse bipartite motifs in two widely studied and important proteins, p53 and EmGFP. In these cases, three of the four variants (p53-2, p53-3, and EmGFP-1) formed fluorescent complexes with ReAsH, whereas one (EmGFP-2) did not.27 Two different p53 variants were evaluated: one contained Cys residues at positions 107, 108, 148, and 149 (p53-2), while the other contained Cys residues at positions 116, 117, 123, and 124 (p53-3) (Figure 8). In both cases, the Ω and Ω’ values for the predicted ReAsH complexes fell within the range associated with fluorescent conformations (p53-2: 133.3, 24.5; p53-3: −37.4, 57.1) (Figure 8A and B). Two variants of EmGFP were also studied: one contained Cys residues at positions 19, 21, 26, and 28; the other at 34, 36, 41, and 43. Again, in both cases the Ω and Ω’ values for the predicted ReAsH complexes fell within the range associated with fluorescent conformations (EmGFP-1: −52.97, −60.13; EmGFP-2: 76.2, 58.6 (Figure 8C and D). Although this analysis would suggest that both EmGFP-1 and EmGFP-2 should form fluorescent ReAsH complexes, EmGFP-2 binds ReAsH poorly (Kd > 500 μM in the absence of EDT; the value for EmGFP-1 is 5 μM).27 Indeed, modeling suggests that ReAsH binding to EmGFP-2 (but not EmGFP-1) disrupts the GFP β-strand network, suggesting that lack of fluorescence results from weak binding rather than from a non-fluorescent conformation.
Figure 8.

Models of ReAsH in complex with previously reported Cys4 motifs within p53 and EmGFP and the associated values of Ω and Ω’: (A) p53-2; (B) p53-3; (C) EmGFP-1 and (D) EmGFP-2. Three of these proteins (p53-2, p53-3, and EmGFP-1) formed fluorescent complexes with ReAsH, while one (EmGFP-2) did not. Hydrogen bonding networks are not shown for clarity. In each case, the minimized structure of the indicated protein variant bound to ReAsH is shown in teal and aligned with the native structure shown in green. As discussed in the text, although the Ω and Ω’ angles for the minimized EmGFP-2 structure fall within the fluorescent range, EmGFP-2 binds ReAsH poorly and the minimized structure contains a disrupted β-strand network.
The set of predicted fluorescent ReAsH rotamers is also in accord with previous studies of Gierasch and coworkers, which evaluated whether the related fluorophore FlAsH became fluorescent when bound to Cys4 variants of cellular retinoic acid-binding protein I (CRABP I), another protein rich in β-sheet structure.16 Several CRABP I variants were evaluated; the FlAsH complex of variant St1’-10, which was stable to the highest concentration of EDT,16 possessed a Cys4 arrangement closest to the ideal, with Ω and Ω’ values of 20.17 and −75.17. In contrast, the FlAsH complexes of variants St1–2 and St1–10, which formed less stable complexes, possessed a Cys4 arrangement further from the minimum energy conformation (Ω and Ω’ values of −176.48 and −110.94 for St1–10 and −40.64 and −65.25 for St1–2) (Figure 9). St1–10, which has Ω and Ω’ outside the ideal range, had the lowest quantum yield of all three complexes.
Figure 9.

Models of ReAsH in complex with previously reported bipartite motifs within CRABP I and the associated values of Ω and Ω’: (A) CRABP I St1-10; (B) CRABP I St1-2; and (C) CRABP I St1’-10. The FlAsH complex of variant St1’-10, which was stable to the highest concentration of EDT,16 possessed a Cys4 arrangement closest to the ideal, with Ω and Ω’ values of 20.17 and -75.17. In each case, the minimized structure of the indicated protein variant bound to ReAsH is shown in teal and aligned with the native structure shown in green.
The rotationally restricted model we propose demands that rotation about the As-Aryl bond occurs on a time scale that allows a population of excited ReAsH-EDT2 molecules to access a high energy conformation where quenching can occur before a photon is emitted. Although few aryl-As rotational barriers are known, in general, aryl-X rotational barriers decrease as the atomic radius of X increases.28 The rotational barriers calculated for aryl-P bonds are low (1 to 3.74 kcal•mol−1 depending on aryl substituent),29 suggesting that the As-aryl rotational barrier in ReAsH is accessible at RT, with a substantial number of molecules (15.6% - 0.2%, assuming a Boltzmann distribution of states) populating even the least favorable rotamer.29,30 But more importantly, the rotationally restricted model predicts that the lifetime of the ReAsH-EDT2 excited state will be shorter than that of ReAsH bound to a protein where little or no rotation (and thus little or no quenching) can occur. Although the excited state lifetime of ReAsH-EDT2 has not been reported, the reported excited state lifetimes of FlAsH bound to the α2A adrenergic receptor or the peptide FLNCCPGCCMEP are between 4 and 5 ns.5,31
To test whether coordination of ReAsH to a protein tetracysteine motif would increase the excited state lifetime, we expressed and purified a variant of maltose binding protein (MBP) modified with a C-terminal, optimized, tetracysteine motif, FLNCCPGCCMEP (MBP-C4).32 MBP-C4 binds ReAsH in the micromolar concentration range in the presence of 1 mM EDT (Kd = 33.7 ± 8.1 μM); fluorescence lifetime measurements were performed under conditions where > 75% of the ReAsH-EDT2 was complexed with MBP-C4. Under these conditions, the time-dependent decay of ReAsH-EDT2 fluorescence and ReAsH•MBP-C4 fluorescence (Figure 10A) fit best to a double exponential function, with an optimal fit to equation (1):
Figure 10.

ReAsH fluorescent lifetimes and the effect of solvent viscosity support a rotamer-restricted model for fluorogenicity. (A) The fluorescence decay of ReAsH-EDT2 and ReAsH-MBP-C4 as a function of time. Wavelength-dependent fluorescence emission of 450 μM (B) resorufin or (C) ReAsH-EDT2 in 100 mM NaOH at room temperature in the presence of the indicated amounts of glycerol. (D) Plot of the % change in the maximum emission of resorufin or ReAsH-EDT2 as a function of % added glycerol.
In this equation, Y represents the observed number of photon counts; i is time, B1 and B2 are coefficients giving the relative contribution of each decay, and T1 and T2 are the excited state lifetimes. In ReAsH-EDT2, the shorter lifetime (T1 = 0.092 ± 0.006 ns) dominates significantly over the longer lifetime (T2 = 3.393 ± 0.008 ns) with coefficients of B1 = 0.2732 ± 0.0005 and B2 = 0.00751 ± 0.00001, respectively. In ReAsH•MBP-C4, however, both species contribute significantly; the shorter lifetime (T1 = 0.13 ± 0.01 ns) and the longer lifetime (T2 = 3.67 ± 0.005 ns) are associated with coefficients of B1 = 0.0984 ± 0.0003 and B2 = 0.01706 ± 0.00002. The 7-fold increase in contribution of the longer lifetime when ReAsH is bound to MBP-C4 is fully consistent with a rotamer-restricted model for fluorogenicity, in which protein binding hinders As-aryl rotation and the resulting PeT quenching.
The rotationally restricted model we propose also predicts that an increase in solution viscosity that hinders As-aryl rotation should increase fluorescence, as under these conditions fewer molecules will access a quenched conformation during the lifetime of the excited state. Indeed, the fluorescence of the BODIPY-based dyes, BV-1 BoMe, and dCbdp, the benzonitrile, DMABN, the benzylidene, DCVJ, the stilbene, p-DASMI, and crystal violet all of which are quenched internally by virtue of bond rotation, increase in solutions of increased viscosity.20,33 For example, the fluorescence intensity of the distorted-BODIPY dye BV-1 increases by 335% between pure water and pure glycerol.20 Dyes that are not affected by rotation-induced quenching show little of no change in fluorescence intensity as the solution viscosity increases.20 Consistent with this trend, the fluorescence intensity of resorufin at the emission maximum (582 nm) was unaffected by addition of between 0 and 10% glycerol (Figure 10B). By contrast, the fluorescence intensity of ReAsH-EDT2 increased by 175% under comparable conditions. The observation that ReAsH-EDT2 fluorescence is sensitive to solution viscosity is fully in accord with the rotamer-restricted model, in which its fluorescence quenched by virtue of As-aryl bond rotation.
Conclusions
In summary, here we report calculations that support a novel mechanism for the observed binding-induced fluorogenicity of the bis-arsenical dye ReAsH. The observation that fluorescence is possible only in certain As-aryl rotational states has two important ramifications. First, it provides a clear strategy for the design of new bis-arsenicals that are even more conformationally restricted, becoming fluorescent in even fewer rotational states: these new derivatives should display lower background fluorescence. It also provides all the information necessary for the algorithmic identification of ideal ReAsH binding sites in proteins of known structure.
Methods
Calculations
All calculations were performed using Gaussian (2009-D.01) and either a PC (Dell with Windows 7 Pro) or the Yale High Performance Computing Cluster. Molecular orbitals and ball-and-stick models of ReAsH (Figure 5) were generated using Gauss View.34 Structures of ReAsH-EDT2, ReAsH-PDT2, and ReAsH-MMT4 (Figure S1) were minimized using Hartree-Fock theory (6–31+G(d) basis set) with water solvent using the Polarizable Continuum Model (PCM).21 Calculations were performed with the following input options: opt rhf/6–31+g(d) scrf=(solvent=water) geom=connectivity. The calculations were performed using the deprotonated form of ReAsH as input, as ReAsH should be > 97.5% deprotonated at physiological pH (pH 7.4) according to the pKa of resorufin, the parent fluorophore (5.8);35 the pKa of ReAsH bound to the FLNCCPGCCMEP peptide is even lower (4.18).36 ReAsH-EDT2 and ReAsH-PDT2 rotamers were generated by opening the minimized structures in Gauss View and using the dihedral angle editor tool to change the SAs-C-C dihedral angles. Thus, the rotation was rigid: the orbitals were calculated after each rotation without any further minimization of the structure. To ensure that the molecular orbitals associated with the lowest energy PET-permitting structure (Ω = −160°, Ω′ = 51°) remained quenched after relaxation of the rest of the molecule, two carbon atoms and all four sulfur atoms were frozen and the structure was minimized in Gaussian (HF 6–31+G(d)) basis set water modeled by PCM). Figure S2 illustrates which atoms were frozen during the minimization. The resulting structure still permitted PeT.
Description of molecular modeling procedures
Models of ReAsH bound to previously studied Cys4 motifs (p53-2, p53-2, EmGFP-1, EmGFP-2,27 CRABP-I St1–10, CRABP-I St1–2, CRABP-I St1’-10)16 were generated by first performing in silico mutagenesis in Pymol37 to alter the appropriate amino acids to cysteine. The following PDB files were used as starting points: p53 (1TUP), EmGFP (1EMA), CRABP-I (1CBI). Cys4 motif models of ReAsH bound to each Cys4 site were generating by including the atoms within each Cys4 motif plus three residues on either side of each cysteine. Gauss View was then used to attach ReAsH to the Cys4 motif model and Gaussian molecular mechanics was used to minimize the structure. Images of the minimized models were generated in Pymol.37 Each minimized conformation is described uniquely by the six distances (Dactual) between each sulfur-sulfur pair. To evaluate the extent to which each modeled site deviated from an optimal geometry, we compared the sum of these six distances in each modeled complex to those calculated for the minimum energy conformation of ReAsH-MMT4 complex (Dideal) (Figure S3) using equation 1; smaller values of the summed score represent complexes that better approximate the minimum energy conformation.
Cloning, expression, and purification of MBP-C4
DNA encoding an FLNCCPGCCMEP ReAsH binding tag32 was appended to the C-terminus of the MBP gene using Gibson Assembly (NEB). The Gibson Assembly product was transformed into XL1-Blue Electroporation-Competent cells (Agilent) and plated on agar plates containing 50 mg/mL kanamycin (AmericanBio). Resulting colonies were picked, grown overnight, and plasmids were purified using a Miniprep Kit (Qiagen). The sequences of the plasmids were confirmed by DNA sequencing. A plasmid containing the correct sequence (pMBP-C4) was transformed in to BL21 (DE3) cells (Agilent) and plated on agar plates containing 50 mg/mL kanamycin. A colony was chosen from that plate, inoculated into a 30 mL starter culture with 50 mg/mL kanamycin, and grown overnight. The starter culture was then diluted into 1 L of LB with 50 mg/mL kanamycin. The culture was grown until it reached an OD600 of 0.8 at which time 1 mM IPTG (AmericanBio) was added to induce protein expression. The cells were grown overnight and spun down the next morning at 4,100 × g for 15 min (Beckman Allegra R centrifuge). Cells were re-suspended in lysis/wash buffer (200 mM NaCl, 20 mM Tris, 10 mM imidazole, 1 mM EDTA (pH 8)) and lysed by sonication. A cOmplete EDTA-free protease inhibitor tablet (Roche Applied Science) was added before lysis. The lysate was spun at 14,000 × g for 15 min. The supernatant of the lysate was incubated with Ni-NTA resin (Qiagen) for 15 min and the resin was loaded onto a column. The column was washed with approximately 20 mL of lysis/wash buffer. and MBP-C4 was eluted in four 3 mL fractions with elution buffer (200 mM NaCl, 20 mM Tris, 250 mM imidazole, 1 mM EDTA (pH 8). Protein identity was confirmed by LC-MS and purity was assessed at ≥ 96% by analysis of a Coomassie-stained SDS-PAGE gel. The protein was dialyzed into Assay Buffer (100 mM Tris, 75 mM NaCl, 3.5 mM TCEP (Sigma) (pH 7.8). The protein was concentrated to 672 μM using an Amicon Ultra-15 Centrifugal Filter (Millipore), aliquoted, and frozen at −80°C.
Viscosity and ReAsH Emission
A 50% glycerol stock solution was prepared by mixing equal volumes of glycerol and MilliQ water and used in all subsequent experiments. Measurements were performed by diluting 0.5 μL of ReAsH-EDT2 in DMSO (Invitrogen) into 1 mL of 100 mM NaOH. A small amount of resorufin (Sigma-Aldrich) was suspended in DMSO and then diluted into 100 mM NaOH. The ReAsH-EDT2 and resorufin concentrations were then quantified on the basis of UV-absorbance (ε for ReAsH-EDT2 = 63000 M−1cm−1 (reference 26); ε for resorufin = 73000 M−1cm−1 (reference38) and diluted to a concentration of 900 nM in 100 mM NaOH. Solutions with 0%, 2%, 5%, 10%, 15%, and 20% (by volume) of glycerol were prepared from the 50% glycerol stock. Glycerol solutions were then mixed 1:1 with the 900 nM solution of ReAsH- EDT2 or resorufin. The emission spectra from 555 nm to 630 nm using an excitation wavelength of 540 nm was measured with a PTI fluorimeter. Five replicates were done for each dye.
Measurement of ReAsH binding to MBP-FLNCCPGCCMEP
Solutions of MBP-C4 with concentrations ranging from 0 to 100 μM were prepared by diluting a frozen stock of MBP-C4 (672 μM) with Assay Buffer. These solutions were incubated at 4° C overnight. Separately, a small quantity of ReAsH-EDT2 (Thermo Scientific) (0.5 μL of a 2 mM stock) was diluted into 100 mM NaOH and its concentration determined by UV spectroscopy (ε = 63,000 cm−1M−1). The ReAsH was then diluted into Assay Buffer supplemented with 2 mM EDT to a final concentration of 100 nM. The ReAsH–EDT2 solution was immediately added in a 1:1 ratio to the MBP-C4 solutions. ReAsH was incubated with the protein for 90 mins. The emission between 555 nm and 640 nm (excitation= 540 nm) of each sample was measured using a PTI fluorimeter. A plot of the average emission between 575 nm and 585 nm vs [MBP-C4] was fit with the following function:
Where Fobs was the observed emission and P was the concentration of protein. The function was fit using Graphpad Prism to give a Kd of 33.71 ± 8.107 μM (Figure S4).
Measurement of fluorescence excited state lifetimes
Fluorescence lifetimes were measured by time-correlated single photon counting using a TD-Fluor Horiba Fluorolog 3 with a 566 nm LED. A frozen stock of MBP-C4 (672 μM) was diluted with Assay Buffer to a final concentration of 200 μM. Separately, a small amount of ReAsH-EDT2 (0.5 μL of a 2 mM stock) was diluted into 100 mM NaOH and the precise ReAsH concentration determined using UV spectroscopy (ε = 63,000 cm−1•M−1). The ReAsH was then diluted with Assay Buffer supplemented with 2 mM EDT to a final concentration of 2 μM. Samples used for fluorescence lifetime measurements were prepared by adding 25 μL of ReAsH-EDT2 solution to 25 μL of MBP-C4 solution. The final concentrations of ReAsH-EDT2 and MBP-C4 were 1 and 100 μM, respectively. As the Kd of the ReAsH•MBP-C4 complex is 33.7 ± 8.1 μM, the fraction of ReAsH bound under these conditions is 75%. Fluorescence lifetimes were measured using a TD-Fluor Horiba Fluorolog 3 with a 566 nm LED. The internal response function (IRF) of the LED was measured using a solution of Ludox (Sigma). The data was fit using the Data Station software. The decay curves of neither ReAsH-EDT2 nor ReAsH•MBP-C4 fit well to a single exponential function (χ2 values of 45.7 and 7.71 for ReAsH-EDT2 and ReAsH-protein solutions, respectively). Therefore the equation was fit with a two exponential function. The bi-exponential function resulted in much better fits with χ2 values of 1.23 and 1.17 for ReAsH-EDT2 and ReAsH-protein solutions, respectively.
Supplementary Material
Acknowledgments
The authors are grateful to the NIH (GM 83257) and the NSF (DGE-1122492 to ASW) for support of this work.
Footnotes
ASSOCIATED CONTENT
Additional data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors.
References
- 1.Lukinavicius G, Johnsson K. Curr Opin Chem Biol. 2011;15:768. doi: 10.1016/j.cbpa.2011.10.015. [DOI] [PubMed] [Google Scholar]; Nadler A, Schultz C. Angew Chem Int Edit. 2013;52:2408. doi: 10.1002/anie.201209733. [DOI] [PubMed] [Google Scholar]; Sletten EM, Bertozzi CR. Accounts of chemical research. 2011;44:666. doi: 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffin BA, Adams SR, Tsien RY. Science. 1998;281:269. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 3.Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. J Am Chem Soc. 2002;124:6063. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 4.Pomorski A, Adamczyk J, Bishop AC, Krezel A. Org Biomol Chem. 2015;13:1395. doi: 10.1039/c4ob02256d. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nakanishi J, Maeda M, Umezawa Y. Anal Sci. 2004;20:273. doi: 10.2116/analsci.20.273. [DOI] [PubMed] [Google Scholar]; Cao H, Chen B, Squier TC, Mayer MU. Chem Commun (Camb) 2006:2601. doi: 10.1039/b602699k. [DOI] [PubMed] [Google Scholar]; Cao H, Xiong Y, Wang T, Chen B, Squier TC, Mayer MU. J Am Chem Soc. 2007;129:8672. doi: 10.1021/ja070003c. [DOI] [PubMed] [Google Scholar]; Fu N, Xiong Y, Squier TC. Bioconj Chem. 2013;24:251. doi: 10.1021/bc300619m. [DOI] [PubMed] [Google Scholar]; Rutkowska A, Plass T, Hoffmann JE, Yushchenko DA, Feng S, Schultz C. Chem Biochem. 2014;15:1765. doi: 10.1002/cbic.201402189. [DOI] [PubMed] [Google Scholar]; Fujii T, Shindo Y, Hotta K, Citterio D, Nishiyama S, Suzuki K, Oka K. J Am Chem Soc. 2014;136:2374. doi: 10.1021/ja410031n. [DOI] [PubMed] [Google Scholar]; Stafforst T. Chem Biochem. 2012;13:505. doi: 10.1002/cbic.201100787. [DOI] [PubMed] [Google Scholar]; Tour O, Adams SR, Kerr RA, Meijer RM, Sejnowski TJ, Tsien RW, Tsien RY. Nat Chem Biol. 2007;3:423. doi: 10.1038/nchembio.2007.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spagnuolo CC, Massad W, Miskoski S, Menendez GO, Garcia NA, Jares-Erijman EA. Photochem Photobiol. 2009;85:1082. doi: 10.1111/j.1751-1097.2009.00565.x. [DOI] [PubMed] [Google Scholar]
- 6.Luedtke NW, Dexter RJ, Fried DB, Schepartz A. Nat Chem Biol. 2007;3:779. doi: 10.1038/nchembio.2007.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scheck RA, Lowder MA, Appelbaum JS, Schepartz A. ACS Chem Bio. 2012;7:1367. doi: 10.1021/cb300216f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doerner A, Scheck R, Schepartz A. Chem Bio. 2015;22:776. doi: 10.1016/j.chembiol.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowder MA, Doerner AE, Schepartz A. J Am Chem Soc. 2015;137:6456. doi: 10.1021/jacs.5b02326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Science. 2002;296:503. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmann C, Gaietta G, Bunemann M, Adams SR, Oberdorff-Maass S, Behr B, Vilardaga JP, Tsien RY, Eisman MH, Lohse MJ. Nat Meth. 2005;2:171. doi: 10.1038/nmeth742. [DOI] [PubMed] [Google Scholar]
- 12.Lee J, Culyba EK, Powers ET, Kelly JW. Nat Chem Biol. 2011;7:602. doi: 10.1038/nchembio.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee M-H, Appleton KM, Strungs EG, Kwon JY, Morinelli TA, Peterson YK, Laporte SA, Luttrell LM. Nature. 2016 doi: 10.1038/nature17154. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, Lohse MJ, Hoffmann C. Nature. 2016 doi: 10.1038/nature17198. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheck RA, Schepartz A. Accts Chem Res. 2011;44:654. doi: 10.1021/ar2001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffin BA, Adams SR, Tsien RY. Meth Mol Biol. 2015;1266:1. doi: 10.1007/978-1-4939-2272-7_1. [DOI] [PubMed] [Google Scholar]; Irtegun S, Wood R, Lackovic K, Schweiggert J, Ramdzan YM, Huang DCS, Mulhern TD, Hatters DM. ACS Chem Bio. 2014;9:1426. doi: 10.1021/cb500242q. [DOI] [PubMed] [Google Scholar]; Cornell TA, Fu J, Newland SH, Orner BP. J Am Chem Soc. 2013;135:16618. doi: 10.1021/ja4085034. [DOI] [PubMed] [Google Scholar]; Tsytlonok M, Itzhaki LS. Chem Biochem. 2012;13:1199. doi: 10.1002/cbic.201200012. [DOI] [PubMed] [Google Scholar]; Stafforst T. ChemBiochem. 2012;13:505. doi: 10.1002/cbic.201100787. [DOI] [PubMed] [Google Scholar]; Kapurniotu A. ChemBiochem. 2012;13:27. doi: 10.1002/cbic.201100631. [DOI] [PubMed] [Google Scholar]; Davis OB, Bishop AC. Bioconj Chem. 2012;23:272. doi: 10.1021/bc200562y. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rutkowska A, Haering CH, Schultz C. Angew Chem Int Ed. 2011;50:12655. doi: 10.1002/anie.201106404. [DOI] [PubMed] [Google Scholar]; Pace CJ, Huang Q, Wang F, Palla KS, Fuller AA, Gao J. ChemBiochem. 2011;12:1018. doi: 10.1002/cbic.201000736. [DOI] [PubMed] [Google Scholar]; Irtegun S, Ramdzan YM, Mulhern TD, Hatters DM. JOVE. 2011 doi: 10.3791/2857. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hu Y, Su B, Kim CS, Hernandez M, Rostagno A, Ghiso J, Kim JR. ChemBiochem. 2010;11:2409. doi: 10.1002/cbic.201000435. [DOI] [PubMed] [Google Scholar]; Guy J, Castonguay R, Pineda NBCR, Jacquier V, Caron K, Michnick SW, Keillor JW. Mol Biosyst. 2010;6:976. doi: 10.1039/b918205e. [DOI] [PubMed] [Google Scholar]; Chen VL, Bishop AC. Chem Comm. 2010;46:637. doi: 10.1039/b919815f. [DOI] [PMC free article] [PubMed] [Google Scholar]; Coleman BM, Nisbet RM, Han S, Cappai R, Hatters DM, Hill AF. Biochem Biophys Res Comm. 2009;380:564. doi: 10.1016/j.bbrc.2009.01.120. [DOI] [PubMed] [Google Scholar]
- 16.Krishnan B, Gierasch LM. Chem Biol. 2008;15:1104. doi: 10.1016/j.chembiol.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen B, Cao H, Yan P, Mayer MU, Squier TC. Bioconj Chem. 2007;18:1259. doi: 10.1021/bc0603900. [DOI] [PubMed] [Google Scholar]
- 18.Miura T, Urano Y, Tanaka K, Nagano T, Ohkubo K, Fukuzumi S. J Am Chem Soc. 2003;125:8666. doi: 10.1021/ja035282s. [DOI] [PubMed] [Google Scholar]; Carlson JC, Meimetis LG, Hilderbrand SA, Weissleder R. Angew Chem Int Ed. 2013;52:6917. doi: 10.1002/anie.201301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shieh P, Hangauer MJ, Bertozzi CR. J Am Chem Soc. 2012;134:17428. doi: 10.1021/ja308203h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu L, Yuan Z, Simmons JT, Sreenath K. RSC Adv. 2014;4:20398. doi: 10.1039/C4RA00354C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frisch MJT, GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam MJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 22.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science; Sausalito, CA: 2006. [Google Scholar]
- 23.O’Boyle NM, Tenderholt AL, Langner KM. J Comput Chem. 2008;29:839. doi: 10.1002/jcc.20823. [DOI] [PubMed] [Google Scholar]
- 24.Urano Y, Kamiya M, Kanda K, Ueno T, Hirose K, Nagano T. J Am Chem Soc. 2005;127:4888. doi: 10.1021/ja043919h. [DOI] [PubMed] [Google Scholar]
- 25.Wang T, Douglass EF, Fitzgerald KJ, Spiegel DA. J Am Chem Soc. 2013;135:12429. doi: 10.1021/ja406077j. [DOI] [PubMed] [Google Scholar]
- 26.Madani F, Lind J, Damberg P, Adams SR, Tsien RY, Graslund AO. J Am Chem Soc. 2009;131:4613. doi: 10.1021/ja809315x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodman JL, Fried DB, Schepartz A. ChemBiochem. 2009;10:1644. doi: 10.1002/cbic.200900207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amatatsu Y. J Phys Chem A. 2008;112:8824. doi: 10.1021/jp8043972. [DOI] [PubMed] [Google Scholar]; Johnson RD. NIST Standard Reference Database. 2013;101 [Google Scholar]
- 29.Peng HL, Payton JL, Protasiewicz JD, Simpson MC. J Phys Chem A. 2009;113:7054. doi: 10.1021/jp810119k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naumov VA, Tafipol’skii MA, Samdal S. Russ J Gen Chem. 2003;73:896. [Google Scholar]; Noble-Eddy R, Masters SL, Rankin DW, Wann DA, Robertson HE, Khater B, Guillemin JC. Inorg Chem. 2009;48:8603. doi: 10.1021/ic901018s. [DOI] [PubMed] [Google Scholar]
- 31.Spille JH, Zürn A, Hoffmann C, Lohse MJ, Harms GS. Biophys J. 2011;100:1139. doi: 10.1016/j.bpj.2010.08.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin BR, Giepmans BN, Adams SR, Tsien RY. Nat Biotechnol. 2005;23:1308. doi: 10.1038/nbt1136. [DOI] [PubMed] [Google Scholar]
- 33.Dziuba D, Jurkiewicz P, Cebecauer M, Hof M, Hocek M. Angew Chem Int Ed. 2016;55:174. doi: 10.1002/anie.201507922. [DOI] [PubMed] [Google Scholar]; Haidekker MA, Theodorakis EA. J Biol Eng. 2010;4:11. doi: 10.1186/1754-1611-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kuimova MK. Phys Chem Chem Phys. 2012;14:12671. doi: 10.1039/c2cp41674c. [DOI] [PubMed] [Google Scholar]; Haidekker MA, Theodorakis EA. Org Biomol Chem. 2007;5:1669. doi: 10.1039/b618415d. [DOI] [PubMed] [Google Scholar]; Ferri G, Nucara L, Biver T, Battisti A, Signore G, Bizzarri R. Biophys Chem. 2016;208:17. doi: 10.1016/j.bpc.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 34.Dennington R, Keith T, Millam J. Semichem Inc; Shawnee Mission, KS: 2009. [Google Scholar]
- 35.Coleman DJ, Kuntz DA, Venkatesan M, Cook GM, Williamson SP, Rose DR, Naleway JJ. Anal Biochem. 2010;399:7. doi: 10.1016/j.ab.2009.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pomorski A, Adamczyk J, Bishop AC, Krezel A. Organic & biomolecular chemistry. 2015;13:1395. doi: 10.1039/c4ob02256d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The PyMOL Molecular Graphics System, Version 1.7.4. Schrodinger, LLC; [Google Scholar]
- 38.Klotz AV, Stegeman JJ, Walsh C. Anal Biochem. 1984;140:138. doi: 10.1016/0003-2697(84)90144-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.