

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease whose prevalence has reached global epidemic proportions. Although the disease is relatively benign in the early stages, when severe clinical forms, including nonalcoholic steatohepatitis (NASH), cirrhosis and even hepatocellular carcinoma, occur, they result in worsening the long-term prognosis. A growing body of evidence indicates that NAFLD develops from a complex process in which many factors, including genetic susceptibility and environmental insults, are involved. In this review, we focused on the genetic component of NAFLD, with special emphasis on the role of genetics in the disease pathogenesis and natural history. Insights into the topic of the genetic susceptibility in lean individuals with NAFLD and the potential use of genetic tests in identifying individuals at risk are also discussed.
Keywords: NASH, alcoholic liver disease, gene variants, PNPLA3, TM6SF2
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD), whose prevalence has reached global epidemic proportions, is the most common chronic liver disease [1]. Although the disease is relatively benign in the early stages, when severe clinical forms such as nonalcoholic steatohepatitis (NASH), cirrhosis and even hepatocellular carcinoma (HCC) occur, the long-term prognosis worsens [2]. The most dramatic event in the natural history of the disease is the incidence of NAFLD-related HCC [3]. Hence, knowledge of the disease pathogenesis and predisposing factors is crucial for understanding the disease biology and making decisions on diagnostic or therapeutic interventions, the latter being the main goal of Precision Medicine.
A growing body of evidence indicates that the disease develops as a result of a complex process in which many factors, including genetic susceptibility and environmental insults, are involved [2,4]. Furthermore, NAFLD severity and progression is modulated by epigenetic factors, including liver-specific DNA-methylation changes and microRNAs that significantly modulate the liver transcriptome [5-11].
In this review, we focused on the genetic component of NAFLD, with special emphasis on the role of genetics in the disease pathogenesis and natural history. Insights into the topic of the genetic susceptibility in lean individuals with NAFLD and the potential use of genetic tests in identifying individuals at risk are also discussed.
NAFLD IS A POLYGENIC AND HERITABLE DISEASE
NAFLD is a heritable and complex trait, the genetic component of which has been explored by a myriad of studies that used different approaches illustrated in Figure 1.
Figure 1.
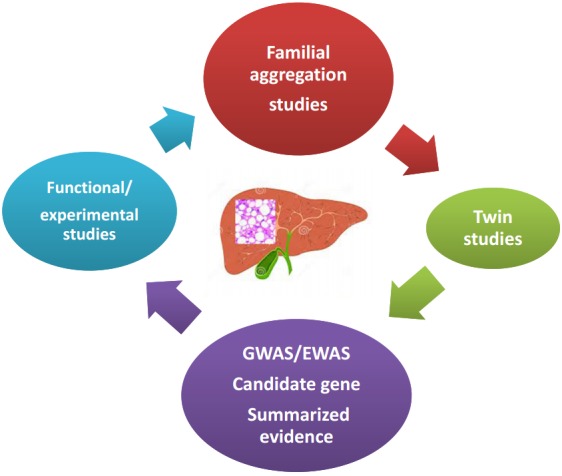
NAFLD, is a polygenic and heritable disease. Picture summarizes different approaches that have been used to date to explore the genetic component of NAFLD. Familial aggregation studies are typically conducted in order to identify the genetic component of complex diseases; the goal of this strategy is to assess clustering of the disease within families. Twin genetic studies have also contributed to the knowledge of the genetic susceptibility of complex diseases; large twin registries are highly beneficial resources for comparing the genetic risk of a disease between monozygotic and dizygotic twins. GWAS and EWAS, which include a global survey of sequence variants across the entire genome or variants in the coding regions, respectively, have shed light onto the genetic component of NAFLD, as well as other complex diseases in the last decade. Candidate gene association studies are focused on loci selected on the basis of their known or presumed function or on their biological plausibility in the disease pathophysiology. Finally, functional and experimental studies aim to provide mechanistic insights into the role of a variant or locus of interest in the explored phenotype. NAFLD, nonalcoholic fatty liver disease; GWAS, genome-wide association study; EWAS, exome-wide association study.
Robust evidence from population-based and familial-aggregation studies, as well as twin-studies, has provided in-depth knowledge on NAFLD or NAFLD-related outcomes. According to the available data, the heritability estimates range from 20 to 70%, depending on the study design, ethnicity and the methodology used—including imaging technology—to characterize the phenotype (Fig. 2). While some imaging technologies employed are highly affordable and easy to implement in clinical setting, others are highly sensitive and specific but expensive. Among those, liver ultrasound (US), abdominal computed tomography (CT) and magnetic resonance spectroscopy (MRS) are most widely utilized for measuring either qualitatively or quantitatively the amount of liver fat infiltration.
Figure 2.

Heritability estimates of NAFLD. Picture summarizes results of studies that estimated the heritability of NAFLD. NAFLD, nonalcoholic fatty liver disease; MRI-PDFF, magnetic resonance imaging−proton-density fat fraction; MRE, magnetic resonance elastography; Liver US, liver ultrasound.
Struden and colleagues provided the first evidence on the heritability of NAFLD by examining familial forms of cryptogenic cirrhosis [12]; the study included 18 members of eight kindreds containing two or more afflicted members. The authors observed that NASH coexisted within four kindreds, whereby the afflicted patients formed mother-daughter, sister-sister, sister-brother, fatherdaughter, and male-female cousin dyads [12].
More recent reports based on large populations have yielded more precise estimations of the magnitude of NAFLD heritability. For example, Speliotes and investigators from the collaborative GIANT, MAGIC and GOLD population-based consortia reported the heritability estimates of hepatic steatosis as 26−27% in a large (n=6,629) sample of subjects of European descent [13]. Wagenknecht and coworkers from the IRAS-Insulin Resistance Atherosclerosis Study Family Study reported the heritability of NAFLD in 795 Hispanic American and 347 African-American adults and found similar figures of about ~31% [14]. An interesting aspect of the Wagenknecht et al.’s study is the large disparity in the NAFLD heritability between cohorts of different ethnicities, which was greater in the Hispanic cohort (33%) compared with the African American cohort (14%) [14].
Palmer and investigators from the Jackson Heart Study (JHS), ARIC (Atherosclerosis Risk in Communities Study), the Insulin Resistance Atherosclerosis Family Study (IRASFS), Genetic Epidemiology Network of Arteriopathy (GENOA), and Family Heart Study (FamHS), which included five African American (n=3,124) and one Hispanic American (n=849) cohort, also reported differences in the heritability estimates between African (20%) and HispanicAmerican (34%) families [15].
Schwimmer et al. performed a familial aggregation study of overweight children with biopsy-proven NAFLD and family members in whom the phenotype was assessed by MRS, and observed that fatty liver was significantly more common in siblings (59%) and parents (78%) of children with NAFLD as compared with obese children without family history of the disease [16]. Recently, results from The Genetics of NAFLD in Twins Consortium illustrated the heritability of hepatic steatosis (based on MRI-proton-density fat fraction) and fibrosis (based on stiffness measured by magnetic resonance elastography), which was estimated at ~50% [17]. Cui et al. from the same group also found in the same cohort of twins a high level (~75.6%) of shared genetic effect between steatosis and fibrosis. However, the authors surprisingly reported almost absent environmental effects [18]. In contrast, results pertaining to another twin cohort that included 208 adult Hungarian twins (63 monozygotic and 41 dizygotic pairs), while providing no support for the heritability of NAFLD, suggest a large shared and unshared environmental effect (74.2% and 25.8%, respectively) [19]. The heritability of NAFLD and its association with abnormal vascular parameters was also assessed in this study [19], showing that NAFLD is concomitant with carotid plaques or carotid intima media thickness. The association of NAFLD and carotid plaques was previously proven by a meta-analysis of 3,497 subjects (1,427 patients and 2,070 controls) [20]. Moreover, genetic co-variance assessment of metabolic syndrome (MetS)-associated traits in The Genetics of NAFLD in Twins Consortium revealed a significant association between hepatic steatosis and body mass index (BMI) and hyperinsulinemia, and between hepatic fibrosis and glycated haemoglobin (HbA1c) [18]. It is noteworthy that the available data mining strategies and systems biology approaches strongly suggest genetic commonality between NAFLD and MetS [21].
GENETIC FACTORS THAT INFLUENCE THE SEVERITY AND NATURAL HISTORY OF NAFLD
The role of genetic variation in NAFLD, specifically single nucleotide polymorphisms (SNPs), has been the focus of extensive research in the last decade, including classical candidate gene association studies [4], as well as novel genome-wide association studies (GWAS) [13,22-26] and exome-wide association studies (EWAS) (Fig. 1) [27].
Authors of the candidate gene association studies have identified several loci associated with the disease susceptibility and progression, including variation in transcription factors involved in the circadian rhythm (CLOCK transcription factor), the signal transducer and activator of transcription 3 STAT3, the multidrug-resistance-associated protein gene (ABCC2), and the nuclear pregnane X receptor (PXR) [28-31]. Authors of other association studies reported variants in genes involved in metabolic and inflammatory pathways [32], whereby the candidate genes for NAFLD were selected either on the basis of their known or presumed function, or due to their biological plausibility in the disease pathophysiology.
Modeling strategies that included system biology approaches, integration of gene-protein interactions and prediction of associated pathways showed that the majority of loci that were reported in association with NAFLD or NASH were not only related with the regulation of lipid homeostasis and the cellular lipid metabolic process, but also pathways involved in cardiovascular system regulation and nuclear receptors [4]. We indeed predicted for the first time the role of NR1H4 or farnesoid X nuclear receptor, a ligandactivated transcription factor that functions as a receptor for bile acids and the RXRA or retinoid X receptor, a nuclear receptor that mediates the biological effects of retinoids by their involvement in retinoic acid-mediated gene activation [4]. This information [4] has been the platform for attractive pharmacological targets that were further assayed and validated as treatment strategies in NASH [33,34].
The results yielded by the first GWAS of NAFLD as a part of which 9,229 nonsynonymous sequence variations were screened have significantly contributed to our knowledge of the genetic component of the disease [26]. The nonsynonymous rs738409 C/G variant in PNPLA3 (patatin-like phospholipase domain containing 3, also known as adiponutrin or calcium-independent phospholipase A2-epsilon), which encodes the amino acid substitution I148M, is regarded as the major genetic component of NAFLD and NASH [35]. The risk effect of rs738409 on developing fatty liver in the context of NAFLD is the strongest ever reported for a common variant modifying the genetic susceptibility of NAFLD (5.3% of the total variance) [35,36]. The rs738409 is not only significantly associated with the accumulation of fat in the liver (the lipid fat content in carriers of the GG homozygous genotype is 73% higher compared with that measured in the carriers of the CC genotype) but also with the histological disease severity and progression of NAFLD (odds ratio-OR 1.88 per G allele; 95% confidence interval-CI 1.03–3.43; GG vs. CC homozygous carriers OR 3.488, 95% CI 1.859–6.545) [35,36]. The rs738409 also explains a fraction of sexual dymorphism associated with NAFLD, as significantly higher effect was demonstrated in women compared with men [35]. Conversely, association analysis of rs738409 and MetS-associated diseases did not reveal any link with obesity or type diabetes, as summarized in a meta-analysis [35].
Extensive knowledge on the role of PNPLA3 in the regulation of liver metabolic functioning was recently gained by employing different strategies, including functional in vitro studies and experimental animals. PNPLA3 is a multifunctional enzyme with both triacylglycerol (TAG) lipase and acylglycerol O-acyltransferase activity that participates in TAG hydrolysis and the acyl-CoA independent transacylation of acylglycerols, as reviewed recently [37]. The promoter activity of PNPLA3 is upregulated by glucose concentrations in a dose dependent manner [38]. The effect of rs738409 variant has also been the subject of extensive research in the last decade, which has led to the consensus that the G-NAFLD-risk allele is associated with a loss of function [39]. Collectively, the available evidence indicates that the variant participates in hepatocyte triacylglycerol remodeling [39-41]. On the other hand, we recently uncovered a novel role of rs738409 in global liver metabolism by performing high-throughput metabolic profiling of PNPLA3 siRNA-silencing and overexpression of wild type and mutant Ile148Met variants (isoleucine/methionine substitution at codon 148) in Huh/7 cells [42]. Of note, silencing of PNPLA3 was associated with a global perturbation of Huh-7 hepatoma cells that resembled a catabolic response associated with protein breakdown [42]. Overexpression of the PNPLA3 Met148 variant was associated with a 1.75-fold increase in lactic acid in comparison with the empty vector, suggesting a shift to anaerobic metabolism and mitochondrial dysfunction. Together, these results might explain the implication of the variant in disease progression.
The GWAS strategy was also employed by other groups in the search for the genetic component of NAFLD, and their respective studies involved different populations, study designs, sample sizes, and approaches employed in the characterization of the liver phenotype. For example, Chalasani et al. focused on female adults with NAFLD diagnosed by liver biopsy [22], while Speliotes and colleagues conducted an exploration of the heritability of hepatic steatosis at the population level with abdominal-CT [13]. On the other hand, Feitosa et al. utilized a combined approach of abdominal-CT and alanine-aminotransferase (ALT) levels as a surrogate of disease severity [23], while Kawaguchi and colleagues focused on establishing the genetic risk in Asian-descent patients [24,25] and DiStefano et al. measured liver fat content in morbidly obese individuals [43]. The GWAS strategy was also used to explore the genetic locus that specifically influenced liver enzyme levels in the population, including alanine aminotransferase (ALT) [44,45].
Collectively, GWAS on NAFLD uncovered not only highly replicated variants such as rs738409, but also variants in loci whose function is diverse in the context of NAFLD. For instance, variants in: 1) PPP1R3B (protein phosphatase 1, regulatory Subunit 3B) that is implicated in the regulation of glycogen synthesis in liver or skeletal muscle; 2) FDFT1 (farnesyl-diphosphate farnesyltransferase) that is involved in cholesterol biosynthesis; 3) ERLIN1 (ER lipid raft associated 1) that mediates the endoplasmic reticulum-associated degradation; 4) LTBP3 (latent transforming growth factor beta) that plays a structural role in the extracellular matrix; 5) PARVB (parvin beta) that plays a role in cytoskeleton organization and cell adhesion and 6) variants in the NCAN/TM6SF2/CILP2/PBX4 multilocus, all of them were reported to be associated with NAFLD.
The results obtained in these first NAFLD-EWAS [27] prompted several replication studies in different populations around the world [46-51]. Their combined findings have definitively confirmed that the causal variant in the multi-gene locus named NCAN/TM6SF2/CILP2/PBX4 is the nonsynonymous rs58542926 variant located in the TM6SF2 (transmembrane 6 superfamily member 2) gene. In the initial study, Kozlitina et al. demonstrated that rs58542926 encoding an amino acid substitution p.Glu167Lys (E167K) was significantly associated with hepatic triglyceride content (HTGC), as measured by proton magnetic resonance spectroscopy (H-MRS) [27]. The authors showed that the effect of rs58542926 on HTGC was independent of the effect mediated by the rs738409, obesity, and insulin resistance, as assessed by HOMA-IR, or alcohol intake [27]. However, authors of the subsequent studies reported conflicting results regarding the association with fatty liver, histological steatosis, NASH or fibrosis [48]. In fact, the association with liver fibrosis [47] remains to be confirmed, as most of the studies cited above showed that the association does not resist adjustment by NASH [46] or is not statistically significant [49,51].
Authors of functional studies on TM6SF2 gene demonstrated that this locus and the mentioned variant are relevant for the NAFLD disease biology. Specifically, Kozlitina et al. showed in vitro that murine hepatoma cells expressing the Lys167-TM6SF2 (E variant) protein have reduced expression levels compared with the wild-type [27], while Mahdessian demonstrated that TM6SF2 is localized in the endoplasmic reticulum and the ER-Golgi intermediate compartment of human liver cells [52]. Available experimental evidence indicates that, while TM6SF2 protein is required to mobilize neutral lipids for VLDL assembly [52], it is not required for secretion of apoB-containing lipoproteins [41].
Our group explored the level of liver TM6SF2 expression in subjects with NAFLD at different stages of disease severity, observing that TM6SF2 protein expression was significantly reduced in the liver of patients with NAFLD [49]. In addition, we found that liver TM6SF2 immunoreactivity was reduced in the NAFLD-risk T-allele (Lys167) carriers. Moreover, allelic-specific expression analysis of cDNA isolated from the liver tissue confirmed that expression levels of rs58542926-T are about 56% of that of the C allele [49]. These findings suggest that the TM6SF2-NAFLD-risk T-allele is associated with decreased gene and protein expression in the liver of affected patients [49].
TM6SF2-rs58542926 presents an interesting clinical paradox, as while the C (Glu167) allele is consistently associated with increased cardiovascular risk by increasing circulating LDL-cholesterol [53], the T allele (Lys167) is associated with NAFLD and NASH [46-51]. In fact, the impact of the variant on the risk of CVD is indeed explained by the protection of the T allele against having elevated blood lipid levels [48]. Finally, summarized evidence of the association of rs58542926 and the level of serum transaminases indicates that ALT (n=94,414) and AST (n=93,809) levels are significantly associated with the variant in NAFLD, but not in other chronic liver diseases, including chronic hepatitis C and B [54]. However, this increase represents -2.5 (9.8%) and 1.2 (5%) IU/L of ALT and AST, respectively, which is relatively small compared with the large effect of the PNPLA3-rs738409 variant [54].
Finally, a variant in GCKR locus (glucokinase regulatory gene), also uncovered by NAFLD-GWAS, has recently gained attention of researchers due to its biological plausibility in the disease pathogenesis. Specifically, the missense variant rs780094 was associated with a modest risk of having a fatty liver [13], whereby summarized evidence demonstrates a ~ 1.2-fold higher risk of developing NAFLD [55]. Interestingly, GCKR mutations have been involved in the maturity-onset diabetes in young individuals [56], given that diabetes/glucose intolerance/insulin resistance is a well-known risk factor for NAFLD.
A missense rs641738 C>T variant formally located in the transmembrane channel-like 4 gene (TMC4), but also at a few hundred bases of the 3’ untranslated region of the MBOAT7 (membrane bound O-acyltransferase domain-containing 7 gene) [57], was recently associated with the risk of NAFLD in European Caucasian population, but not in individuals of other ethnicities. Unfortunately, these findings were not further investigated in studies involving other populations of patients and controls of European descent [58]. Thus, the association with the MBOAT7 should be further confirmed or refuted in other populations.
In summary, available and replicated evidence on the genetic risk of NAFLD suggests presence of at least three missense variants in three different but biologically plausible loci (PNPLA3, TM6SF2 and GCKR) associated with the disease severity and progression. Nevertheless, the NAFLD-associated variants have a quite diverse effect on the susceptibility of NAFLD—from intermediate (~ OR 3.4) to low (OR 1.2). This, in turn results in a rather diverse frequency of the risk allele (MAF) (from ~30% for rs738409 to 7% for rs58542927) (Fig. 3). Collectively, these results support the notion of common variants [59] in the pathogenesis of NAFLD, which are presently deemed the major contributors of the disease risk. This observation also highlights the paucity of information on the role of rare variants, as well as structural variation, gene-by-gene-interaction, and gene-by-environment interaction in the biology of the disease, all of which must be explored further as they likely contribute to the disease heritability (Fig. 3). Of note, genome wide exploration of mitochondrial DNA has revealed a substantial proportion of the missing heritability of NAFLD and the disease severity associated with genetic variation in genes of the oxidative-phosphorylation chain (OXPHOS) [60]. In fact, we have shown that patients with different degrees of fibrosis have an overall enrichment of 1.4-fold mutation rate [60]. It is noteworthy that epigenetic marks not only in nuclear but also mitochondrial DNA-encoded genes, which are by definition transmitted among generations and may explain part of the missing heritability, may be pivotal for the development and progression of NAFLD, as was reported by our group for the first time [7,9,10]. Figure 3 highlights the major milestones in the attainment of knowledge on NAFLD, from the first phenotypic description of the disease [61] to the first scoring system for the histological assessment of NASH by Brunt and coworkers [62] and major findings yielded by GWAS [13,26,27].
Figure 3.

Knowledge gained on the role of common variants in the genetic risk of NAFLD and the missing heritability. Figure illustrates milestones in the knowledge of NAFLD, including the genetic component of the disease. In early 1950, the description of cirrhosis in patients with type 2 diabetes not only provided the first clinical characterization of NAFLD as a disease in which hepatomegaly was primarily explained by fatty infiltration but also introduced the notion of the natural history of the disease, implying that fatty liver can develop into an end-stage outcome. In 1999, Elizabeth Brunt and colleagues provided a thorough characterization of the histological phenotype underpinning future studies on the role of inflammation, ballooning degeneration and fibrosis in the disease biology. The result of the first NAFLD-GWAS was a major breakthrough, as the rs738409 was identified as the largest ever replicated variant in the history of hepatology that not only explains the genetic component of NAFLD but other liver diseases as well, including chronic viral hepatitis and HCC [76]. The picture finally illustrates areas in which knowledge of the genetic component of NAFLD is insufficient or absent, also referred as “missing heritability”, including the role of rare variants, structural variation, and interactions between genes and environment. T2D, type 2 diabetes; NASH, nonalcoholic steatohepatitis; PNPLA3, patatin-like phospholipase domain containing 3; GCKR, glucokinase gene regulator; TM6SF2, transmembrane 6 superfamily member 2; OR, odds ratio; MAF, minor allele frequency; GXG, gene by gene interaction; GXE, gene by environment interaction; mtDNA, mitochondrial DNA. # The association of Laennec's cirrhosis with diabetes mellitus [61]. *Scoring systems described by Brunt et al (1999) [62]
THE GENETIC SUSCEPTIBILITY IN LEAN NAFLD
Although the pathogenesis of NAFLD is not fully understood, a growing body of evidence supports the notion that the disease is strongly associated with MetS and its intermediate phenotypes, including obesity, type 2 diabetes, and cardiovascular disease [2,3,20,63].
Nevertheless, epidemiological studies, particularly those conducted in (but not restricted to) Asia [64-66], highlight the observation that NAFLD can be also seen in lean subjects. In fact, ~10−20% of all NAFLD cases in non-obese Americans and Asians are ascribed to lean NAFLD. As the results yielded by current studies are conflicting and inconclusive, the risk factors associated with lean NAFLD are still being revised. There are few published reports that include well-characterized NAFLD in non-obese patients. For instance, findings of a study involving a large number of lean patients from Asia with NAFLD proven by liver biopsy have been recently published, suggesting that while the risk factors in lean and obese NAFLD are common, lean NAFLD patients tend to have less severe disease form and may have a better prognosis than obese patients [64]. Still, among lean NAFLD patients, hypertriglyceridemia and higher creatinine were significantly associated with advanced liver disease [64]. The most remarkable finding of this study is the lack of difference in the magnitude of the association between the PNPLA3 rs738409 variant and the disease severity in lean NAFLD patients in comparison with non-lean NAFLD patients [64]. However, in their US-based study, Feldman and colleagues have not confirmed this observation, possibly due to the small sample (including non-obese patients) studied [67]. Finally, authors of a population-based study conducted in Hong Kong that included a large population of subjects reported that the rs738409 G-allele was more common in non-obese than in obese NAFLD patients [65].
In conclusion, while the role of genetic factors associated with the risk of NAFLD in lean patients is not fully understood, it is plausible to speculate that variants associated with the risk of type 2 diabetes or insulin resistance [21,60], as well as variants in the mitochondrial DNA, are involved [60]. At any rate, it is reasonable to assume that rs738409, which is not associated with either obesity or type 2 diabetes [35], has no differential influence in the genetic risk of NAFLD in lean individuals. In fact, meta-regression analysis (random effects model, within-study variance estimated with the unrestricted maximum-likelihood method) of BMI, fasting glucose and insulin levels, and HOMA-IR index according to the rs738409 variant (homozygous GG and CC) in pooled estimates including 1404 subjects from seven studies, showed that the variant does not influence these traits [35].
THE ROLE OF GENETICS IN THE STRATIFICATION OF INDIVIDUALS AT RISK AND PREDICTION OF THERAPEUTIC INTERVENTION
The remarkable progress in the understanding of the genetic risk of NAFLD and NASH offers the unique opportunity to translate this information into the clinical practice. Specifically, the design and identification of novel diagnostic tools, as well as promising therapeutic targets, would benefit from thorough knowledge of the individual genetic makeup of affected patients [4].
Several strategies were proposed for the application of genetic variants in the risk prediction, particularly the rs738409 in PNPLA3, for improving the diagnostic accuracy of NASH or predicting non-invasively the response to lifestyle or surgery intervention (a detailed explanation is shown in Table 1).
Table 1.
The role of genetic variation in PNPLA3 (rs738409) in the prediction of NAFLD disease risk and therapeutic response
| Author | Sample size and characterization of NAFLD | Design strategy | Outcome |
|---|---|---|---|
| Noninvasive risk estimates of nonalcoholic steatohepatitis | |||
| Kotronen et al. (2009) [69] | 470 subjects. | Performance of predicting NAFLD by combining routine clinical and laboratory data and the rs738409 genotypes: | Addition of rs738409 to the score improved the accuracy of the prediction by only <1%. |
| Liver fat content measured using proton magnetic resonance spectroscopy. | |||
| Two stage study (estimation and validation) | |||
| Guichelaar et al. (2013) [71] | 144 patients undergoing bariatric surgery; histologic (liver biopsy) assessment | Probability of developing NASH by the combination of four risk factors: rs738409 G allele, CK-18 >145 IU/l, Glucose >100 mg/dL, C-reactive protein (CRP) >0.8 mg/dL | 82% probability in the presence of all four risk factors versus 9% in their absence |
| Zhou et al. (2016) [68] | 318 subjects who underwent a liver biopsy. | NASH ClinLipMet Score: glutamate, isoleucine, glycine, lysophosphatidylcholine 16:0, phosphoethanolamine 40:6, AST, and fasting insulin, along with rs738409 genotypes | Identified patients with NASH with an AUROC of 0.866 (95% confidence interval, 0.820-0.913). |
| Two stage study (estimation and validation) | |||
| Francque et al. (2012) [72] | 542 overweight or obese patients, liver biopsy is a sub-sample. | ALT, fasting levels of C-peptide, and ultrasound | AUROC values of 0.854 in the design cohort and 0.823 in the validation cohort. |
| Two stage study (estimation and validation) | steatosis scores along with rs738409 genotypes | rs738409 correlated with development of NASH but did not add value. | |
| Noninvasive assessment of response to therapeutic intervention | |||
| Shen et al. (2015) [73] | 154 patients randomized into the intervention and control group. | NAFLD patients received a 12-month communitybased randomized controlled trial on a lifestyle modification program | Allele G was associated with greater reduction in IHTG (6%), body weight, waist-to-hip ratio, blood total cholesterol, and LDL levels |
| Liver fat content measured using proton magnetic resonance spectroscopy | |||
| Sevastianova et al. (2011) [74] | 18 patients | To explore whether the rs738409 influences the ability of weight loss, to decrease liver fat or change insulin sensitivity. | Weight loss was not associated to rs738409 G or C allele. |
| Liver fat content measured using proton magnetic resonance spectroscopy | Euglycemic clamp technique and lipolysis were measured before and after the diet. | Liver fat decreased by 45% in the rs738409 GG group (P<0.001) and by 18% in the CC group (P<0.01). | |
| Krawczyk et al. (2016) [75] | 84 obese individuals scheduled for bariatric surgery | To explored improvement of hepatic steatosis after bariatric surgery | Patients carrying G lost more weight (P<0.01) and liver fat (P=0.04) one year after surgery, as compared to individuals having the common genotype. |
| The PNPLA3 genotype and initial grade of steatosis, but not the TM6SF2 or MBOAT7 variants, were independent predictors of NAFLD improvement | |||
AUROC, area under the receiver operating characteristic. NASH, non-alcoholic steatohepatitis; IHTG, intrahepatic tryglicerides; LDL, low density lipoprotein cholesterol.
Although a priori use of genetic testing appears encouraging, the overall experience suggests that the value of utilizing SNPs for improving the efficacy in the diagnosis of NASH is still inconclusive. Some Authors of some studies support the introduction of rs738409 into diagnostic tests, while others do not advocate its use (Table 1). For example, authors of the NASH ClinLipMet Score suggested that the use of plasma metabolites—including glutamate, isoleucine, glycine, lysophosphatidylcholine 16:0, and phosphoethanolamine 40:6—along with routine biochemical test (AST, and fasting insulin), together with rs738409 genotypes [68], predicts NASH with an area under the receiver operating characteristic of 0.866 (95% confidence interval, 0.820-0.913) [68]. Conversely, Kotronen and coworkers evaluated the performance of predicting NAFLD by combining routine clinical and laboratory data and the rs738409 genotypes, and observed a sensitivity of 86% and specificity of 71% in the estimation of increased liver fat content. However, the incorporation of the genetic information into the test score improved the accuracy of the prediction by less than 1% [69].
A more complex, yet poorly explored, scenario is the stratification of patients’ response to medications or lifestyle intervention according to their genetic background. Authors of several extant studies have explored this issue, but further research is urgently needed. In particular, future prospective studies should involve large sample size, a proper design and long-term follow-up of the patients. Future research programs that integrate health records—ideally electronic ones—with patients’ genomic, transcriptomic, proteomic, and metabolomic information will change our ability to control or cure the disease. For example, we recently showed that NASH is associated with a state of betaine insufficiency and a missense variant (rs1805074- p.Ser646Pro) in DMGDH (dimethylglycine dehydrogenase mitochondrial) that modulates the levels of betaine and related metabolites is associated with the disease severity [70]. We hope that the exact and precise genetic knowledge gained with the tremendous advance in omics techniques would allow the medical community to fully benefit from the application of Personalized or Precision Medicine in the context of this relatively new “pandemic” disease.
Abbreviations
- ALT
alanine-aminotransferase
- GCKR
glucokinase gene regulator
- EWAS
exome wide association studies
- GWAS
genome wide association studies
- MetS
metabolic syndrome
- MBOAT7
Membrane Bound O-Acyltransferase Domain Containing 7
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PNPLA3
patatin-like phospholipase domain containing 3
- SNP
single nucleotide polymorphism
- TM6SF2
transmembrane 6 superfamily member 2
Footnotes
Funding Support
This study was supported partially by grants PICT 2014-543, PICT 2014-1816 and PICT 2015-0551 (Agencia Nacional de Promoción Científica y Tecnológica
Conflicts of Interest: The authors have no conflicts to disclose.
REFERENCES
- 1.Satapathy SK, Sanyal AJ. Epidemiology and Natural History of Nonalcoholic Fatty Liver Disease. Semin Liver Dis. 2015;35:221–235. doi: 10.1055/s-0035-1562943. [DOI] [PubMed] [Google Scholar]
- 2.Brunt EM, Wong VW, Nobili V, Day CP, Sookoian S, Maher JJ, et al. Nonalcoholic fatty liver disease. Nat Rev Dis Primers. 2015;1:15080. doi: 10.1038/nrdp.2015.80. [DOI] [PubMed] [Google Scholar]
- 3.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–2273. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- 4.Sookoian S, Pirola CJ. The genetic epidemiology of nonalcoholic fatty liver disease: toward a personalized medicine. Clin Liver Dis. 2012;16:467–485. doi: 10.1016/j.cld.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 5.Ahrens M, Ammerpohl O, von Schönfels W, Kolarova J, Bens S, Itzel T, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013;18:296–302. doi: 10.1016/j.cmet.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, et al. Relationship between methylome and transcriptome in patients with nonalcoholic Fatty liver disease. Gastroenterology. 2013;145:1076–1087. doi: 10.1053/j.gastro.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pirola CJ, Gianotti TF, Burgueño AL, Rey-Funes M, Loidl CF, Mallardi P, et al. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut. 2013;62:1356–1363. doi: 10.1136/gutjnl-2012-302962. [DOI] [PubMed] [Google Scholar]
- 8.Pirola CJ, Fernández Gianotti T, Castaño GO, Mallardi P, San Martino J, Mora Gonzalez Lopez Ledesma M, et al. Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut. 2015;64:800–812. doi: 10.1136/gutjnl-2014-306996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pirola CJ, Scian R, Gianotti TF, Dopazo H, Rohr C, Martino JS, et al. Epigenetic Modifications in the Biology of Nonalcoholic Fatty Liver Disease: The Role of DNA Hydroxymethylation and TET Proteins. Medicine (Baltimore) 2015;94:e1480. doi: 10.1097/MD.0000000000001480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sookoian S, Rosselli MS, Gemma C, Burgueño AL, Fernández Gianotti T, Castaño GO, et al. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology. 2010;52:1992–2000. doi: 10.1002/hep.23927. [DOI] [PubMed] [Google Scholar]
- 11.Zeybel M, Hardy T, Robinson SM, Fox C, Anstee QM, Ness T, et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin Epigenetics. 2015;7:25. doi: 10.1186/s13148-015-0056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Struben VM, Hespenheide EE, Caldwell SH. Nonalcoholic steatohepatitis and cryptogenic cirrhosis within kindreds. Am J Med. 2000;108:9–13. doi: 10.1016/s0002-9343(99)00315-0. [DOI] [PubMed] [Google Scholar]
- 13.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagenknecht LE, Scherzinger AL, Stamm ER, Hanley AJ, Norris JM, Chen YD, et al. Correlates and heritability of nonalcoholic fatty liver disease in a minority cohort. Obesity (Silver Spring) 2009;17:1240–1246. doi: 10.1038/oby.2009.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palmer ND, Musani SK, Yerges-Armstrong LM, Feitosa MF, Bielak LF, Hernaez R, et al. Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology. 2013;58:966–975. doi: 10.1002/hep.26440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwimmer JB, Celedon MA, Lavine JE, Salem R, Campbell N, Schork NJ, et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology. 2009;136:1585–1592. doi: 10.1053/j.gastro.2009.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loomba R, Schork N, Chen CH, Bettencourt R, Bhatt A, Ang B, et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology. 2015;149:1784–1793. doi: 10.1053/j.gastro.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui J, Chen CH, Lo MT, Schork N, Bettencourt R, Gonzalez MP, et al. Shared genetic effects between hepatic steatosis and fibrosis: A prospective twin study. Hepatology. 2016;64:1547–1558. doi: 10.1002/hep.28674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarnoki AD, Tarnoki DL, Bata P, Littvay L, Osztovits J, Jermendy G, et al. Heritability of non-alcoholic fatty liver disease and association with abnormal vascular parameters: a twin study. Liver Int. 2012;32:1287–1293. doi: 10.1111/j.1478-3231.2012.02823.x. [DOI] [PubMed] [Google Scholar]
- 20.Sookoian S, Pirola CJ. Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: a systematic review. J Hepatol. 2008;49:600–607. doi: 10.1016/j.jhep.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 21.Sookoian S, Pirola CJ. Nonalcoholic fatty liver disease and metabolic syndrome: Shared genetic basis of pathogenesis. Hepatology. 2016;64:1417–1420. doi: 10.1002/hep.28746. [DOI] [PubMed] [Google Scholar]
- 22.Chalasani N, Guo X, Loomba R, Goodarzi MO, Haritunians T, Kwon S, et al. Genome-wide association study identifies variants associated with histologic features of nonalcoholic Fatty liver disease. Gastroenterology. 2010;139:1567–1576. doi: 10.1053/j.gastro.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feitosa MF, Wojczynski MK, North KE, Zhang Q, Province MA, Carr JJ, et al. The ERLIN1-CHUK-CWF19L1 gene cluster influences liver fat deposition and hepatic inflammation in the NHLBI Family Heart Study. Atherosclerosis. 2013;228:175–180. doi: 10.1016/j.atherosclerosis.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawaguchi T, Sumida Y, Umemura A, Matsuo K, Takahashi M, Takamura T, et al. Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PLoS One. 2012;7:e38322. doi: 10.1371/journal.pone.0038322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitamoto T, Kitamoto A, Yoneda M, Hyogo H, Ochi H, Nakamura T, et al. Genome-wide scan revealed that polymorphisms in the PNPLA3, SAMM50, and PARVB genes are associated with development and progression of nonalcoholic fatty liver disease in Japan. Hum Genet. 2013;132:783–792. doi: 10.1007/s00439-013-1294-3. [DOI] [PubMed] [Google Scholar]
- 26.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46:352–356. doi: 10.1038/ng.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sookoian S, Castaño GO, Burgueño AL, Gianotti TF, Rosselli MS, Pirola CJ. The nuclear receptor PXR gene variants are associated with liver injury in nonalcoholic fatty liver disease. Pharmacogenet Genomics. 2010;20:1–8. doi: 10.1097/FPC.0b013e328333a1dd. [DOI] [PubMed] [Google Scholar]
- 29.Sookoian S, Castaño G, Gianotti TF, Gemma C, Pirola CJ. Polymorphisms of MRP2 (ABCC2) are associated with susceptibility to nonalcoholic fatty liver disease. J Nutr Biochem. 2009;20:765–770. doi: 10.1016/j.jnutbio.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 30.Sookoian S, Castaño G, Gianotti TF, Gemma C, Rosselli MS, Pirola CJ. Genetic variants in STAT3 are associated with nonalcoholic fatty liver disease. Cytokine. 2008;44:201–206. doi: 10.1016/j.cyto.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Sookoian S, Castaño G, Gemma C, Gianotti TF, Pirola CJ. Common genetic variations in CLOCK transcription factor are associated with nonalcoholic fatty liver disease. World J Gastroenterol. 2007;13:4242–4248. doi: 10.3748/wjg.v13.i31.4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hernaez R. Genetic factors associated with the presence and progression of nonalcoholic fatty liver disease: a narrative review. Gastroenterol Hepatol. 2012;35:32–41. doi: 10.1016/j.gastrohep.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology. 2016;150:1147–1159. doi: 10.1053/j.gastro.2016.01.038. [DOI] [PubMed] [Google Scholar]
- 34.Sookoian S, Pirola CJ. Elafibranor for the treatment of NAFLD: One pill, two molecular targets and multiple effects in a complex phenotype. Ann Hepatol. 2016;15:604–609. [PubMed] [Google Scholar]
- 35.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53:1883–1894. doi: 10.1002/hep.24283. [DOI] [PubMed] [Google Scholar]
- 36.Sookoian S, Castaño GO, Burgueño AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res. 2009;50:2111–2116. doi: 10.1194/jlr.P900013-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sookoian S, Pirola CJ. PNPLA3, the triacylglycerol synthesis/hydrolysis/storage dilemma, and nonalcoholic fatty liver disease. World J Gastroenterol. 2012;18:6018–6026. doi: 10.3748/wjg.v18.i42.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rae-Whitcombe SM, Kennedy D, Voyles M, Thompson MP. Regulation of the promoter region of the human adiponutrin/PNPLA3 gene by glucose and insulin. Biochem Biophys Res Commun. 2010;402:767–772. doi: 10.1016/j.bbrc.2010.10.106. [DOI] [PubMed] [Google Scholar]
- 39.He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285:6706–6715. doi: 10.1074/jbc.M109.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61:108–118. doi: 10.1002/hep.27242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J Biol Chem. 2016;291:10659–10676. doi: 10.1074/jbc.M116.719955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min HK, Sookoian SC, Pirola CJ, Cheng J, Mirshahi F, Sanyal AJ. Metabolic profiling reveals that PNPLA3 induces widespread effects on metabolism beyond triacylglycerol remodeling in Huh-7 hepatoma cells. Am J Physiol Gastrointest Liver Physiol. 2014;307:G66–76. doi: 10.1152/ajpgi.00335.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DiStefano JK, Kingsley C, Craig WG, Chu X, Argyropoulos G, Still CD, et al. Genome-wide analysis of hepatic lipid content in extreme obesity. Acta Diabetol. 2015;52:373–382. doi: 10.1007/s00592-014-0654-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, van der Harst P, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43:1131–1138. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan X, Waterworth D, Perry JR, Lim N, Song K, Chambers JC, et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet. 2008;83:520–528. doi: 10.1016/j.ajhg.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology. 2015;61:506–514. doi: 10.1002/hep.27490. [DOI] [PubMed] [Google Scholar]
- 47.Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JB, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. 2014;5:4309. doi: 10.1038/ncomms5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pirola CJ, Sookoian S. The dual and opposite role of the TM6SF2-rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: A meta-analysis. Hepatology. 2015;62:1742–1756. doi: 10.1002/hep.28142. [DOI] [PubMed] [Google Scholar]
- 49.Sookoian S, Castaño GO, Scian R, Mallardi P, Fernández Gianotti T, Burgueño AL, et al. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology. 2015;61:515–525. doi: 10.1002/hep.27556. [DOI] [PubMed] [Google Scholar]
- 50.Wang X, Liu Z, Peng Z, Liu W. The TM6SF2 rs58542926 T Allele Is Significantly Associated with Nonalcoholic Fatty Liver Disease in Chinese. J Hepatol. 2015;62:1438–1439. doi: 10.1016/j.jhep.2015.01.040. [DOI] [PubMed] [Google Scholar]
- 51.Wong VW, Wong GL, Tse CH, Chan HL. Prevalence of the TM6SF2 variant and non-alcoholic fatty liver disease in Chinese. J Hepatol. 2014;61:708–709. doi: 10.1016/j.jhep.2014.04.047. [DOI] [PubMed] [Google Scholar]
- 52.Mahdessian H, Taxiarchis A, Popov S, Silveira A, Franco-Cereceda A, Hamsten A, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A. 2014;111:8913–8918. doi: 10.1073/pnas.1323785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holmen OL, Zhang H, Fan Y, Hovelson DH, Schmidt EM, Zhou W, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet. 2014;46:345–351. doi: 10.1038/ng.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sookoian S, Pirola CJ. Meta-analysis of the influence of TM6SF2 E167K variant on Plasma Concentration of Aminotransferases across different Populations and Diverse Liver Phenotypes. Sci Rep. 2016;6:27718. doi: 10.1038/srep27718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zain SM, Mohamed Z, Mohamed R. Common variant in the glucokinase regulatory gene rs780094 and risk of nonalcoholic fatty liver disease: a meta-analysis. J Gastroenterol Hepatol. 2015;30:21–27. doi: 10.1111/jgh.12714. [DOI] [PubMed] [Google Scholar]
- 56.Dimas AS, Lagou V, Barker A, Knowles JW, Magi R, Hivert MF, et al. Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes. 2014;63:2158–2171. doi: 10.2337/db13-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology. 2016;150:1219–1230. doi: 10.1053/j.gastro.2016.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sookoian S, Flichman D, Garaycoechea M, Gazzi C, Castano G, Pirola CJ. Genetic Basis for Nonalcoholic Fatty Liver Disease: The role of MBOAT7-p.Gly17Glu-missense variant. Hepatology. 2016;64(Suppl 1):1114. [Google Scholar]
- 59.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sookoian S, Flichman D, Scian R, Rohr C, Dopazo H, Gianotti TF, et al. Mitochondrial genome architecture in non-alcoholic fatty liver disease. J Pathol. 2016;240:437–449. doi: 10.1002/path.4803. [DOI] [PubMed] [Google Scholar]
- 61.Reinberg MH, Lipson M. The association of Laennec’s cirrhosis with diabetes mellitus. Ann Intern Med. 1950;33:1195–1202. doi: 10.7326/0003-4819-33-5-1195. [DOI] [PubMed] [Google Scholar]
- 62.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 63.Sookoian S, Gianotti TF, Rosselli MS, Burgueño AL, Castaño GO, Pirola CJ. Liver transcriptional profile of atherosclerosis-related genes in human nonalcoholic fatty liver disease. Atherosclerosis. 2011;218:378–385. doi: 10.1016/j.atherosclerosis.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 64.Leung JC, Loong TC, Wei JL, Wong GL, Chan AW, Choi PC, et al. Histological severity and clinical outcomes of nonalcoholic fatty liver disease in nonobese patients. Hepatology. 2017;65:54–64. doi: 10.1002/hep.28697. [DOI] [PubMed] [Google Scholar]
- 65.Wei JL, Leung JC, Loong TC, Wong GL, Yeung DK, Chan RS, et al. Prevalence and Severity of Nonalcoholic Fatty Liver Disease in Non-Obese Patients: A Population Study Using Proton-Magnetic Resonance Spectroscopy. Am J Gastroenterol. 2015;110:1306–1314. doi: 10.1038/ajg.2015.235. [DOI] [PubMed] [Google Scholar]
- 66.Cho HC. Prevalence and Factors Associated with Nonalcoholic Fatty Liver Disease in a Nonobese Korean Population. Gut Liver. 2016;10:117–125. doi: 10.5009/gnl14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feldman A, Eder SK, Felder TK, Kedenko L, Paulweber B, Stadlmayr A, et al. Clinical and Metabolic Characterization of Lean Caucasian Subjects With Non-alcoholic Fatty Liver. Am J Gastroenterol. 2017;112:102–110. doi: 10.1038/ajg.2016.318. [DOI] [PubMed] [Google Scholar]
- 68.Zhou Y, Orešič M, Leivonen M, Gopalacharyulu P, Hyysalo J, Arola J, et al. Noninvasive Detection of Nonalcoholic Steatohepatitis Using Clinical Markers and Circulating Levels of Lipids and Metabolites. Clin Gastroenterol Hepatol. 2016;14:1463–1472. doi: 10.1016/j.cgh.2016.05.046. [DOI] [PubMed] [Google Scholar]
- 69.Kotronen A, Peltonen M, Hakkarainen A, Sevastianova K, Bergholm R, Johansson LM, et al. Prediction of non-alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology. 2009;137:865–872. doi: 10.1053/j.gastro.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 70.Sookoian S, Puri P, Castaño GO, Scian R, Mirshahi F, Sanyal AJ, et al. Nonalcoholic steatohepatitis is associated with a state of betaine insufficiency. Liver Int. 2016 doi: 10.1111/liv.13249. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 71.Gawrieh S, Gawrieh S, Olivier M, Viker K, Krishnan A, Sanderson S, et al. Interactions of allelic variance of PNPLA3 with nongenetic factors in predicting nonalcoholic steatohepatitis and nonhepatic complications of severe obesity. Obesity (Silver Spring) 2013;21:1935–1941. doi: 10.1002/oby.20327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Francque SM, Verrijken A, Mertens I, Hubens G, Van Marck E, Pelckmans P, et al. Noninvasive assessment of nonalcoholic fatty liver disease in obese or overweight patients. Clin Gastroenterol Hepatol. 2012;10:1162–1168. doi: 10.1016/j.cgh.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 73.Shen J, Wong GL, Chan HL, Chan RS, Chan HY, Chu WC, et al. PNPLA3 gene polymorphism and response to lifestyle modification in patients with nonalcoholic fatty liver disease. J Gastroenterol Hepatol. 2015;30:139–146. doi: 10.1111/jgh.12656. [DOI] [PubMed] [Google Scholar]
- 74.Sevastianova K, Kotronen A, Gastaldelli A, Perttilä J, Hakkarainen A, Lundbom J, et al. Genetic variation in PNPLA3 (adiponutrin) confers sensitivity to weight loss-induced decrease in liver fat in humans. Am J Clin Nutr. 2011;94:104–111. doi: 10.3945/ajcn.111.012369. [DOI] [PubMed] [Google Scholar]
- 75.Krawczyk M, Jiménez-Agüero R, Alustiza JM, Emparanza JI, Bujanda L, et al. PNPLA3 p.I148M variant is associated with greater reduction of liver fat content after bariatric surgery. Surg Obes Relat Dis. 2016;12:1838–1846. doi: 10.1016/j.soard.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 76.Sookoian S, Pirola CJ. PNPLA3, the history of an orphan gene of the potato tuber protein family that found an organ: the liver. Hepatology. 2014;59:2068–2071. doi: 10.1002/hep.26895. [DOI] [PubMed] [Google Scholar]