

Abstract
Gaucher disease, caused by pathological mutations GBA1, encodes the lysosome-resident enzyme glucocerebrosidase, which cleaves glucosylceramide into glucose and ceramide. In Gaucher disease, glucocerebrosidase deficiency leads to lysosomal accumulation of substrate, primarily in cells of the reticulo-endothelial system. Gaucher disease has broad clinical heterogeneity, and mutations in GBA1 are a risk factor for the development of different synucleinopathies. Insights into the cell biology and biochemistry of glucocerebrosidase have led to new therapeutic approaches for Gaucher disease including small chemical chaperones. Such chaperones facilitate proper enzyme folding and translocation to lysosomes, thereby preventing premature breakdown of the enzyme in the proteasome. This review discusses recent work developing chemical chaperones as a therapy for Gaucher disease, with implications for the treatment of synucleinopathies. It focuses on the development of non-inhibitory glucocerebrosidase chaperones and their therapeutic advantages over inhibitory chaperones, as well as the challenges involved in identifying and validating chemical chaperones.
In 1955, a Belgian biochemist named Christian de Duve described a novel acidic intracellular organelle while he was on a quest to unravel the mechanisms of insulin in the liver. He named this organelle ‘lysosome’, which is Greek for ‘digestive body’ (1, 2). Initially, lysosomes were considered static organelles involved in non-regulated degradation of macromolecules trafficked to lysosomes via different cellular pathways such as autophagy, phagocytosis, and endocytosis (3–8). However, the recent discovery of transcription factor EB (TFEB) has expanded our understanding of lysosomes. Intra- or extra- cellular changes such as starvation or stress can promote translocation of TFEB to the nucleus, where it then regulates expression of the majority of genes involved in lysosomal function and biogenesis. Lysosomes have now emerged as highly regulated dynamic organelles involved in degradation, nutrient sensing, metabolism, and signaling (9–15).
Pathological mutations in lysosome-resident enzymes involved in distinct substrate turnover can cause lysosomal storage disorders (LSDs), which are rare inborn metabolic diseases where lysosomal function is compromised due to accumulation of substrate. Currently, over fifty different LSDs are known, with Gaucher disease (GD) being the most common with an estimated frequency of 1:40,000–60,000 in the general population and a higher prevalence in the Ashkenazi Jewish population (1:850 individuals) (16). GD is an autosomal recessive LSD where pathological mutations in the gene encoding glucocerebrosidase 1 (GBA1) gene lead to deficient activity of the enzyme glucocerebrosidase (GCase), which, in turn, fails to degrade its substrate glucosylceramide (GlcCer) into glucose and ceramide, resulting in subsequent lysosomal accumulation of GlcCer (16). In GD, cells of the reticulo-endothelial system, such as macrophages, are most affected. Aged erythrocytes have GlcCer-rich membranes and are broken down in a phagocytosis-mediated process by macrophages. In patients with GD, macrophages appear engorged due to lysosomal accumulation GlcCer and are referred to as “Gaucher cells” which can infiltrate the spleen, liver, and bone marrow, and cause inflammation and organomegaly (17, 18). Based on the absence or the presence and severity of neurological manifestations, GD has been classified into three different types. The most common form is type 1, non-neuronopathic GD, with clinical symptoms including organomegaly, bone complications, anemia, and thrombocytopenia (16, 19). The rarest and most severe form is acute neuronopathic GD type 2, where rapid neurological deterioration results in death in early infancy (16, 20). Compared to GD type 2, chronic neuronopathic GD type 3 is characterized by later onset and slower progression of neurological symptoms. In addition to visceral and skeletal symptoms, patients exhibit a specific problem with their horizontal eye movements, and other clinical manifestations can include myoclonic epilepsy and ataxia, intellectual deterioration, learning impairments, and developmental delay (18, 20–22). However, due to a broad range of clinical manifestations associated with GD, it is often challenging to diagnose a patient with a specific type of GD (18, 20). The limitations in correlations between the clinical phenotype and molecular genotype, or the amount of accumulated substrate and/or residual GCase enzyme activity adds another layer of complexity to GD (17, 18, 20, 23–25).
GD type1 was long classified as non-neuronopathic, but this has been challenged due to its association with Parkinson disease (PD) and related synucleinopathies. Indeed, longitudinal clinical studies revealed the initial observation of a possible association between GD and the development of Parkinsonism (Tayebi et al., 2001, Bembi et al., 2003, Tayebi et al., 2003). This was followed by reports of an increased incidence of PD in first degree relatives of patients with GD carrying GBA1 mutations, as well as an increased frequency of GBA1 mutations in small cohorts of patients with PD or related synucleinopathies (Goker-Alpan et al., 2004, Lwin et al., 2004, Eblan et al., 2006, Ziegler et al., 2007). Subsequently, large pan-ethnic cohort studies confirmed a strong association between mutations in GBA1 and the development of synucleinopathies such as PD (26–29), dementia with Lewy bodies (DLB) (30), and multiple system atrophy (MSA) (31). Although GBA1 mutations are now the most common genetic risk factor for the development of PD, only a small percentage of GD patients and GBA1 carriers will go on to develop synucleinopathies.
Current FDA-approved treatments for Gaucher disease
Enzyme replacement therapy
For patients with type 1 GD, enzyme replacement therapy (ERT) is the most commonly used and conventional treatment option. In the early 1960s, De Duve was one of the early proponents of ERT, theorizing that the clinical infusion of the deficient enzyme could be an efficient treatment method for lysosomal storage disorders. The idea was based on previous findings that extracellular proteins taken in by the cells are transported to the lysosomes for degradation (32). However, the initial applications of ERT in the clinic proved to be unsuccessful, and it was not until a decade later that the ERT became an effective and well-understood means of medical care (32).
The first successful clinical administration of the enzyme was performed on two patients with GD at the National Institutes of Health (33). The study showed that both patients tolerated the infusion well, and that the intravenously injected glucocerebrosidase primarily localized in the liver (33). Analysis of the patients’ erythrocytes indicated that the infusion of glucocerebrosidase caused a dramatic decrease in glucosylceramide with levels of other lipids being unaffected (34). In 1991, the US Food and Drug Administration approved alglucerase (Ceredase), which was the first human enzyme replacement product for a lysosomal storage disorder. The enzyme was derived from human placenta and was subsequently substituted in 1994 by a human recombinant enzyme, imiglucerase (Cerezyme). Studies have shown that ERT with imigulucerase reversed the organomegaly and anemia, and improved the growth velocity in children and adolescents with GD (35). Over the years, several other recombinant enzymes became available to treat patients with GD including Taliglucerase alfa (Elelyso) and Velaglucrease alfa (36). ERT has not only provided a treatment for patients, but also a greater insight into the molecular mechanism of cellular uptake of exogenous enzyme. Grabowksi and Hopkin found that modified enzymes are endocytosed after binding to cell-surface mannose receptors and localized to lysosomes (37).
ERT has long been the standard medical option for GD type 1 because it alleviates the visceral, hematological and skeletal implications following the disease manifestation. A drawback to ERT is that the recombinant enzymes are unable to cross the blood-brain barrier (BBB), and thus do not alleviate the neurological symptoms seen in neuropathic GD patients (38). Recent efforts by Gramlich and co-workers address this limitation by developing novel GCase recombinant enzymes tagged with peptides that have the potential to carry GCase across the BBB. Enhanced GCase delivery to cultured neurons was observed with the Tat and rabies glycoprotein derived (RDP) peptide tag. Extended treatment of gba−/− mouse neurons with either Tat-GCase or RDP-IgAh-GCase led to significant reduction in lipid substrate glucosylsphigosine (39).
Substrate reduction therapy
While ERT compensates for enzyme deficiency, substrate reduction therapy (SRT) inhibits the synthesis of substrate to be turned over by GCase. The available SRT drugs act as selective inhibitors of glucosylceramide synthase, which is the rate-limiting enzyme in the synthesis of the GCase-specific substrate glucosylceramide. So far, substrate reduction therapy has primarily been used in patients with type 1 GD with moderate symptoms. Despite its discrete approach, SRT is limited in its applicability for type 2 and 3 GD due to pharmacokinetics limitations (40).
Miglustat or NB-DNJ (Zavesca) was approved by the FDA in 2003 as a treatment option for GD type 1 (41). The main limitation for wider use of the glucosylceramide synthase inhibitor miglustat has been the gastrointestinal adverse events. It was shown that out of 37 patients on miglustat therapy for a period ranging from 5 to 8 years, 15 patients had a decrease in absolute platelet count and 17 patients had gastrointestinal side effects (42). A randomized controlled clinical trial revealed that Miglustat treatment did not show significant amelioration of neurological manifestations in GD type 3 patients (43). However, a novel inhibitor of glucosylceramide synthase (Genz-682452) that crosses the BBB holds promise for treatment of neuronopathic GD. Various mouse models representative of neuronopathic GD showed reduced storage of glycolipids in the brain, increased lifespan, and improved neurological manifestations after treatment with Genz-682452 (44).
Eliglustat (Cerdelga), another SRT drug, was FDA-approved in 2014 and is a ceramide analog that selectively reduces endogenous glucosylceramide. Eliglustat showed equivalent alleviation of the clinical symptoms when compared to intravenous imiglucerase (45). Eliglustat showed a similar level of efficacy, indicated by the stable organ volumes and hematological endpoints in patients after 1 year (46). Common adverse effects, which were shown to be present in ≥10% among the patients taking eliglustat, include but are not limited to: fatigue, headache, nausea, diarrhea, back pain, and upper abdominal pain (47). Although SRT has been established as an alternative to ERT, data on comprehensive, long-term observational studies done on patients taking SRT is still insufficient. ERT remains the currently predominant medical care for treatment of GD (48, 49).
Gene Therapy for GD
Recently, progress was made in utilizing gene therapy as a potential therapeutic approach for treatment of GD type I. It was shown that ex vivo autologous bone-marrow-derived GD 1 hematopoietic stem cells were genetically corrected by infection with self-inactivating lentiviral vectors expressing WT gba1 induced by different cellular promotors. Increased GCase enzyme activity, reduced infiltration of Gaucher cells, and reversed splenomegaly were observed post-transplantation (50).
Development of small chemical chaperones for treatment of GD
The development of small chemical chaperones has been one of the most recent treatment approaches for diseases caused by improperly folded proteins (51). Chemical chaperones are small molecules designed to selectively bind to a specific target protein. In case of enzymes, binding of small chemical chaperones can increase enzyme stability, catalytic activity, and increased lysosomal translocation (52). Lysosome-resident enzymes are folded in the endoplasmic reticulum (ER) with the assistance of endogenous cellular chaperones, followed by translocation to lysosomes (Figure 1A). Genetic mutations can lead to misfolding of enzyme and subsequent premature endoplasmic reticulum-associated degradation (ERAD) where mutant misfolded enzyme is broken down in the proteasome and never reaches the lysosome (53) (Figure 1B). Small chemical chaperone therapy aids in protein folding and stabilization as well as lysosomal translocation (Figure 1C). Since small chemical chaperones physically bind to target proteins, the presence of enzyme is required. Fortunately, many of the GBA1 mutations are missense mutations. Based on data from the International Collaborative Gaucher Group Registry Program, 29% of Gaucher patients are homozygous for c.1226A>G (N370S), 48% are heterozygous for c.1226A>G (N370S), and 6% are homozygous for c.1448T>C (L444P) (54). However, these numbers should be interpreted carefully since mutation distribution varies within different ethnic groups. The common c.1226A>G (N370S) mutation represents about 70% of the mutant alleles in the Ashkenazi Jewish population while rarely seen in Japanese or Chinese cohorts (24). Small chemical chaperones are an attractive therapeutic alternative because of their potential for crossing the BBB. This was demonstrated in the lysosomal storage disorder GM1 gangliosidosis where treatment of a mouse model with N-octyl-4-epi-β-valienamine (NOEV) or 6S-NBI-DGJ, both inhibitory β-galactosidase chaperones, showed efficacy in the brain (55–57). For Gaucher disease, pharmacokinetics studies on mice with ambroxol, a mixed-inhibitor of GCase, and NCGC001099758, a non-inhibitory chaperone of GCase, showed significant exposure in the brain (58–60).
Figure 1.
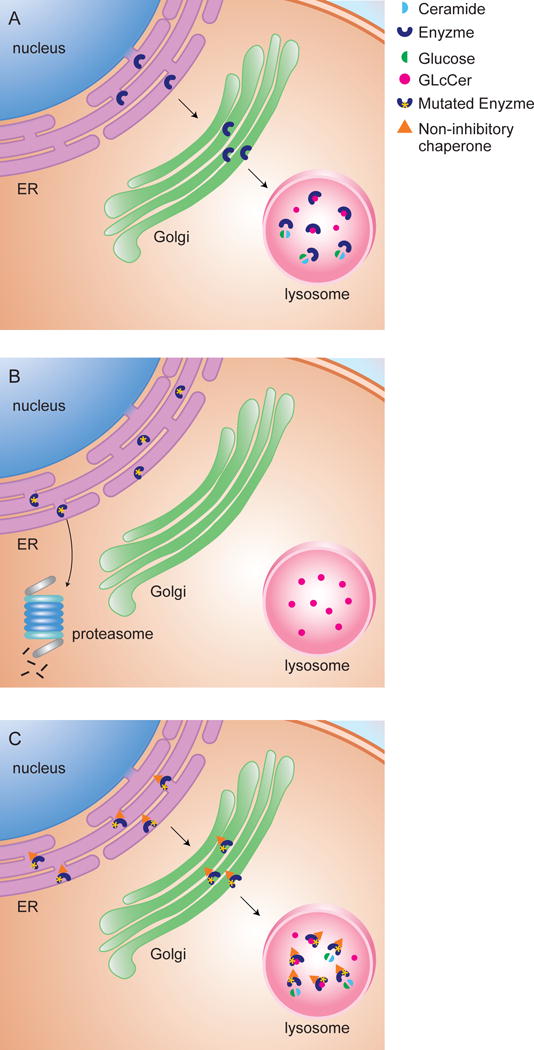
Non-inhibitory chaperones for enhancement of GCase. (A) Wild-type GCase is folded in the ER and translocated to lysosomes where it turns over its substrate. (B) Mutant GCase is misfolded in the ER and undergoes premature degradation in the proteasome with subsequent lysosomal accumulation. (C) Non-inhibitory chaperones facilitate folding and stabilization of mutant GCase in the ER as well as translocation to lysosomes where the residual activity of mutant enzyme is able to turn over substrate.
Potential chaperoning activity of SRT agents
Miglustat, an inhibitor of glucosylceramide synthase and SRT agent for GD type I, has been shown to possess chaperoning activity for mutant GCase (41). Miglustat treatment of COS7 cells transfected with mutant GBA1 cDNA, induced a significant increase in GCase enzyme activity in the S364R, WT, N370S, V15M, and M123T GCase mutant cell lines but no significant differences were observed in the L444P, L336P, and S465del GCase mutants. Increased translocation of GCase to lysosomes was not shown (41). It should be noted that treatment of patient fibroblasts with miglustat did not enhance enzyme activity (61).
Inhibitory chaperones
The majority of pharmacologic chaperones being developed for therapeutics are inhibitors of the target enzyme. These inhibitors bind to the active site of the misfolded enzyme and facilitate proper folding and translocation to lysosomes. Once the mutant enzyme and its bound inhibitor reach lysosomes, the inhibitory chaperone will be out-competed by the substrate. In an ideal situation, the residual enzymatic activity of the mutant enzyme is sufficient to turn over accumulated substrate in lysosomes. It is important to keep in mind that inhibitors bind to the active site of the enzyme. Hence, treatment with inhibitory chaperones can only be done on patients with a genotype that does not impact the integrity of the active site.
In 2002, Sawkar and co-workers observed that the treatment of patient derived fibroblasts homozygous for c.1226A>G (N370S) with sub-inhibitory concentrations of the iminosugar N-nonyl-1-deoxynojirimycin (NN-DNJ) increased mutant GCase activity up to 2-fold (62). Additionally, NN-DNJ simultaneously inhibited glucosylceramide synthase activity in a dose-dependent manner, indicating that NN-DNJ might not be a target-specific drug. Today, many of the GCase inhibitors are iminosugar-based and since they have a high affinity for glycosidases, they can be associated with poor selectivity (63, 64). Additionally, while iminosugar-based molecules act as chaperones at sub-inhibitory concentrations, they function as inhibitors at higher dosages. Indeed, inhibitory chemical chaperones have the capacity to remain inhibitory once the mutant enzyme-chaperone complex becomes lysosome-resident which makes optimization of drug dosing and clinical application difficult (65). An example of this is the iminosugar isofagomine, a competitive inhibitor of GCase. Treatment of cell and mouse models with isofagomine resulted in increased GCase protein levels and enzyme activity, partial rescue of macrophage function, reduction in substrate levels, a delay in the development of neurological manifestations, and increased life span. However, a phase 2 clinical trial failed to improve clinical symptoms in GD patients treated with isofagomine (66–70). Modification of iminosugars to sp2-iminosugars showed increased selectivity for β-glucosidases. However, enzyme activity assays and confocal microscopy-based imaging studies on patient fibroblasts and a neuronal mouse cell line treated with sp2-iminosugars revealed a high inhibitor to chaperone balance (71, 72).
Recent efforts in the development of non-iminosugar inhibitory chaperones identified quinazoline analogues with chaperone activity, high selectivity for GCase, and increased ER to lysosome translocation. Further evaluation will have to determine if these inhibitors have therapeutic potential (73). Another exciting breakthrough was the identification of ambroxol, widely used as cough medicine, as a pH-dependent mixed inhibitor of GCase by high throughput screening (HTS) of an FDA-approved drug library (59). The efficacy of ambroxol as a potent chaperone and translocator of mutant GCase to lysosomes was demonstrated in various independent studies in cells and mice (59, 60, 65, 74). A proof of concept clinical study for ambroxol treatment on twelve GD type 1patients with measurable disease parameters and not receiving ERT showed promise with none of the patients experiencing clinical deterioration (75). This pilot study indicates that ambroxol could be a safe treatment option for GD patients and calls for further evaluation in larger clinical trials.
Non-inhibitory chaperones
As discussed in previous section, inhibitory chaperones must balance inhibitory capacity and chaperoning capacity, which makes clinical development challenging. A non-inhibitory chaperone aids in the folding of mutant enzyme in the ER and translocation to lysosomes by binding to a site that is different from the active site. The non-inhibitory chaperone can then also directly induce the residual activity of mutant enzymes that are newly translocated or already in lysosomes. An ideal non-inhibitory chaperone restores lysosomal enzymatic function through chaperone and enzyme stimulatory effects (76). This makes non-inhibitory chaperones attractive candidates for therapeutic development. However, creating and implementing a practical and accurate methodology for screening of GCase-specific non-inhibitory modulators has been challenging.
In previous HTS using wild type recombinant enzyme, only inhibitory chaperones were identified with a chaperone activities 100 to 1000-fold weaker than their inhibitory activity (77). Such observed differences were attributed to different assay formats utilized between the preliminary HTS, which was done with wild type recombinant GCase and subsequent patient cell-based assays. To improve the methodology of finding non-inhibitory chaperones, a novel screening assay was optimized using mutant GCase in the presence of saposin C, its native activator, and other potential cofactors. For this HTS, protein extracts from spleen tissue derived from splenectomies of GD patients with genotype N370S/N370S were used as the source of mutant enzyme. The spleen-based enzyme assay was then employed to screen a library of 250,000 compounds, identifying novel modulating molecules of mutant GCase. Among those compounds, there were 14 new lead inhibitors and 30 lead non-inhibitory compounds, of which potent chaperone activities were confirmed in subsequent cell-based assays using patient-derived fibroblasts (78). HTS on extracts of spleen as the source of mutant GCase has been proven to discriminate between non-inhibitory chaperones and inhibitors as well as identify GCase-specific, physiologically relevant non-inhibitory chaperones. Since then, analogues of non-inhibitory chaperones, a particular a class of pyrazolpyrimidines, showed specific biochemical activation of GCase and potent chaperone activity in patient fibroblasts (79, 80).
Fibroblast-based models are usually used to study cellular mechanisms of GD and perform follow-up studies for potential candidate drugs, but the fibroblasts lack the hallmark characteristics of the disease which is the glycolipid accumulation in the affected lysosomes (81). In GD patients, macrophages display abnormal lysosomal substrate storage. To overcome this hurdle, Aflaki and co-workers developed primary macrophages (hMacs) that were differentiated from monocytes of patients with GD as well as induced pluripotent stem cells (iPSCs) from dermal fibroblasts that were then differentiated into macrophages (iMacs). The two macrophage types, hMacs and iMacs, were evaluated with the non-inhibitory chaperone molecule NCGC001099758 to determine whether the molecule reversed the disease phenotype.
The study showed that the non-inhibitory chaperone increased GCase activity and lysosomal translocation and significantly reduced substrate storage in hMacs and iMacs with different GD genotypes. Furthermore, the authors showed restoration of chemotaxis in GD hMacs and iMacs after treatment with the non-inhibitory compound. The work henceforth achieved two objectives – first to develop a relevant cell-based model that displays the disease phenotype and second to utilize the said model to effectively evaluate non-inhibitory chaperones.
However, one of the difficulties in accurately identifying potential non-inhibitory chaperones is the fact that non-inhibitory chaperones bind to non-active/enzymatic sites other than the active site, making it difficult to establish a definitive SAR to further develop and design potent non-inhibitory chaperones (76). To go forward with developing effective non-inhibitory chaperones for further clinical applications, novel molecular probes are needed for better evaluation of potential candidates during HTS.
Although not GCase-specific, another promising therapeutic approach for GD is the development of pharmacological agents against key players involved in protein misfolding and proteasome-mediated degradation of mutant GCase, such as heat shock protein 90 (HSP90) and Hsp27. Inhibition of deacetylation of HSP90 and expression of Hsp27 led to upregulation of GCase enzyme activity and amount in GD patient fibroblasts, which makes them promising pharmacological targets (82, 83).
Need for live substrates for evaluation of non-inhibitory chaperones
Currently, there is no efficient and precise method of evaluating non-inhibitory chaperones for GD or other LSDs. When assessing the potency and the efficacy of the non-inhibitory chaperones, it is imperative that the chaperone of interest stabilizes the conformation of mutant GCase, promotes translocation of GCase from the endoplasmic reticulum to the lysosome, and promotes turnover of substrate by mutant GCase within lysosomes. To best assess chaperone efficacy and to distinguish between inhibitory and non-inhibitory chaperones in HTS assays, there is an acute need for GCase-specific fluorescent-based substrates representing GCase activity in lysosomes of live cells.
However, visualizing the activity of endogenous levels of any glycoside hydrolases, including GCase, has been proven to be problematic within live mammalian cells. In 2010, Witte and co-workers designed two elegant fluorescent activity-based inhibitory BODIPY probes for specific in vitro and in vivo labeling of active GCase (84). However, in this application the fluorescent ‘inhibody’ signal represent the amount of active GCase in a biological sample and not the amount of turned over substrate in lysosomes.
Currently, the existing literature on the development of GCase-specific fluorescent substrates is sparse. Yadav and co-workers recently proposed a GCase-specific fluorescence-quenched substrate that favors lysosomal uptake with a quencher group attached to the long aliphatic chain of the ceramide moiety of GlcCer and a fluorophore group on the glucose part. The outcome of the close proximity of the quencher-fluorophore pair is efficient quenching (85). A live cell confocal fluorescence microscopy assay on wild type fibroblasts revealed a time-dependent increase in fluorescence signal in lysosomes due to substrate turnover and subsequent loss of quenching. Treatment of wild type fibroblasts with the GCase-specific suicide inhibitor CBE drastically reduced lysosomal fluorescence signal. On wild type fibroblasts, the difference between untreated and chaperone treated cells is not sufficient for HTS purposes. Unfortunately, treatment of fibroblasts derived from GD patients was missing from this study. Additionally, laser scanning confocal microscopy assays are not feasible for HTS purposes and more suitable read-out platforms such as a fluorescence plate reader platform were not included in this study (85).
In another recent study, a ratiometric two-photon fluorescent substrate was developed for evaluation of β-galactosidase activity in live cells during cell senescence (86). The data showed that treatment of live cells with the pure product indicated a bleed-through signal of the product into the substrate channel in the two-photon microscopy assay (86). Such ratiometric probes are not the most ideal tools in assay development and evaluation of non-inhibitory chaperones because it will not give a definitive evaluation of the activity of chaperone-activated mutant enzyme. Not only would ratiometric substrates be difficult to quantify, most current HTS technologies do not utilize two-photon microscopy to identify lead compounds. It is unfortunate that despite the need for fluorescent substrates suitable for live cell assays in HTS, this particular area of research has not been well established.
Expert review & five-year view
As previously discussed in this review, mutations in GBA1 are a risk factor for the development of synucleinopathies such as PD, DLB, and MSA. The pathological hallmark of all three synucleinopathies is the presence of Lewy bodies and neurite inclusions positive for α-syn aggregates in different parts of the brain (26, 30, 31). In neurons, α-synuclein (α-syn) homeostasis is maintained by a delicate balance between novel α-syn protein synthesis and α-syn turnover by the autophagy-lysosomal pathway (87–89) and the proteasome (90). Increasing evidence suggests that lysosomal impairment plays a role in α-syn aggregation and PD neuropathology (91, 92). Recent experimental evidence favors a reciprocal relationship between GCase activity and α-syn protein levels in which reduced GCase activity can increase α-syn accumulation aggregation and α-syn accumulation can inhibit trafficking of GCase to the lysosome but the exact molecular mechanisms remains elusive (93).
Multiple independent studies in various cell and animal models as well as patient samples with and without GBA1 mutations support the reciprocal relationship. Indeed, diminished GBA1 expression or GCase activity, exogenous introduction of GBA1 mutations or GC substrate enhance accumulation of α-syn while increased levels of α-syn decrease GCase protein and activity levels (93–104). Therefore, therapies that augment GCase activity or decrease GlcCer accumulation could potentially have an impact on α-syn accumulation and aggregation and have a beneficial effect for patients with synucleinopathies. This hypothesis was supported by a proof-of-concept study by Sardi and co-workers where a neuronopathic GD mouse model with GBA1 mutations and a transgenic mouse model with wild type GBA1 and over-expressing A53T α-syn showed significant reduction in α-syn accumulation using virus-mediated infection with wild type GBA1 in the CNS (100).
As mentioned previously, small chemical chaperones have the potential to cross the BBB (80) and could therefore modulate GCase activity and protein levels in the brain, which would make them excellent candidates for treatment of synulceinopathies. To date, there are no reports on the efficacy of non-inhibitory chaperones for treatment of synucleinopathies. Indeed, the recently identified non-inhibitory chaperone that shows promise for the treatment of Gaucher disease has not been evaluated for reduction of α-syn levels in relevant neuronal cell or animal models (58, 78, 105). However, initial studies on cell and mouse models with inhibitory chaperones show promise. Treatment of an α-syn over-expressing neuroblastoma cell line with ambroxol showed reduction of α-syn accumulation (106). Oral administration of isofagomine in an α-syn over-expressing mouse model showed increased GCase activity in brain tissue, improvement motor function, decreased α-syn accumulation in nigral dopaminergic neurons, and decreased inflammation in the brain (107). On the other hand, in a PC12 cell model transfected with WT or mutant GBA1 and over-expressing α-syn, treatment of isofagomine did not significantly reduce α-syn accumulation (95). Treatment of a mouse model representative of neuronopathic GD suggested that isofagomine treatment might restore altered expression levels of miRNA and associated mRNA in processes such as inflammation, axonal guidance pathways, and mitochondrial dysfuntion which are all implicated in PD (108). In another study, patient fibroblasts homozygous for L444P treated with the sp2-iminosugars based inhibitory chaperone NAdBT-AIJ revealed amelioration of mitochondrial dysfunction (109).
Development of therapeutics for a rare disease such as GD has always been of interest to the LSD community. Since the establishment of the link between GBA1 and synucleinopathies, this interest has broadened to common diseases such as PD. Therefore, we anticipate that research and development of small chemical chaperones will continue vigorously in the next five years. Especially non-inhibitory chaperones will be of interest since these compounds will likely not present dosage issues in clinical trials. Advances in the development and evaluation of relevant cell-based assays such as macrophages and neurons will continue and gene-editing technologies such as TALEN and CRISPR will make it possible to turn any wild type cell into a diseased state. Current efforts on the development of relevant live-substrates for measurement of specific lysosomal activity of GCase are premature and not suitable for HTS. We speculate that development of potent live substrates, which will make robust SAR studies possible, will happen over the next five years.
In conclusion, we have discussed the most recent advances in small chemical chaperone therapy development for Gaucher disease. The recently proposed reciprocal relationship between GCase and α-syn has opened new avenues for the application of therapeutics for GD in treatment of synucleinopathies. In this endeavor, GCase-specific non-inhibitory chaperones will be of great interest since they cross the BBB and are not subjected to careful dosing since they do not inhibit GCase in the lysosomes.
Figure 2.
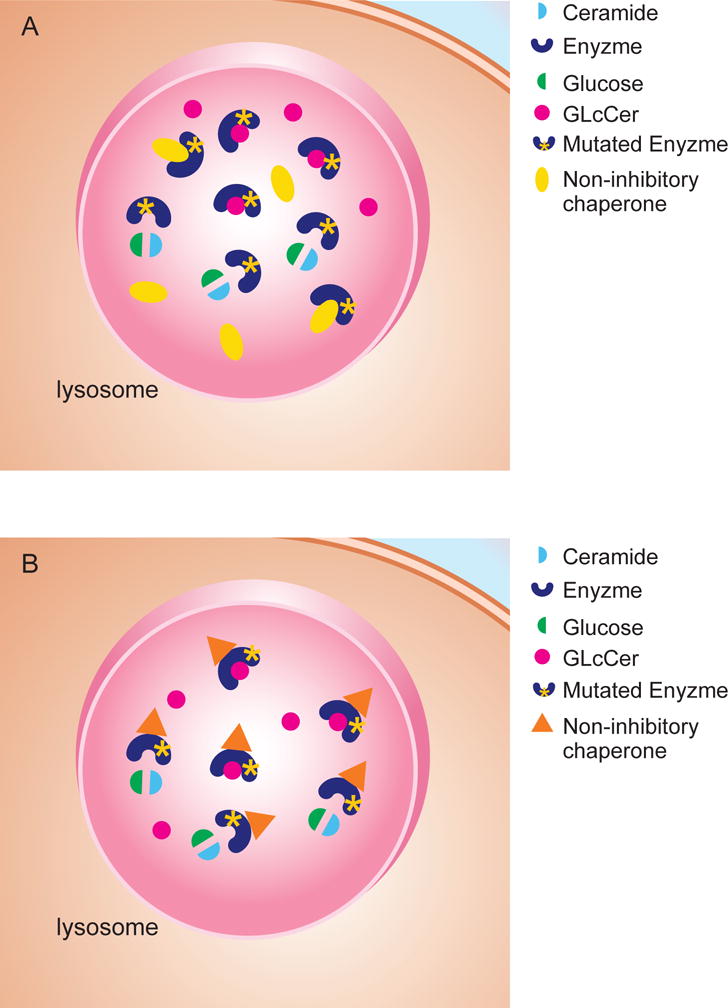
(A) Inhibitory chaperones bind to the active site of mutant GCase. Once the enzyme-inhibitor complex reaches lysosomes, the inhibitor should be out-competed by accumulating substrate. (B) Non-inhibitory chaperones do not bind to the active site of mutant GCase. Substrate binding in the active site of the enzyme can happen without competition.
Key issues.
Gaucher disease (GD) is a rare recessive lysosomal storage disorder in which the mutations in the glucocerebrosidase gene lead to structural instability of the enzyme.
GD can be divided into three common clinical subtype: type 1 is the most common form, type 2 is the most severe, while type 3 is characterized by progressive but milder neurologic symptoms to that of type 2.
In recent years, the non-neuronopathic categorization of GD type1 has been challenged due to its association with PD and related synucleinopathies.
For GD type 1 patients, ERT is the most commonly used and conventional medical care, but the recombinant enzymes fail to cross the BBB.
Substrate reduction therapy has also become an alternative treatment option in recent years; however, SRT is also limited in its potency due to several pharmacokinetics limitations and clinically observed side effects.
There has been a growing movement in utilizing small chemical chaperones as potential therapeutic agents since binding of small chemical chaperones can increase enzyme stability, catalytic activity, and increased lysosomal translocation.
The majority of pharmacologic chaperones being developed for therapeutics are competitive inhibitors of the target enzyme, but the in vivo use of inhibitor chemical chaperones remains challenging since chemical chaperones have the capacity to remain functional for some time once the mutant enzyme-chaperone complex becomes lysosome-resident.
Non-inhibitory chaperons do not interfere with the residual activity of mutant enzymes that are newly translocated or already in lysosomes.
Since non-inhibitory chaperones bind to enzymatic sites other than the active site, which makes it difficult to establish a definitive SAR, it is imperative to develop live substrates to evaluate such non-inhibitory chaperones.
Development of novel therapeutics for GD can have implications for the treatment of synucleinopathies, as treatment with non-inhibitory chemical chaperones can increase GCase protein levels and activity in lysosomes and therefore decrease α-syn protein levels.
Acknowledgments
The authors would like to thank Julia Fekecs for assistance with drafting of the figures. This work was supported by the Intramural Research Programs of the National Human Genome Research Institute, National Center for Advancing Translational Sciences, and National Institutes of Health.
Footnotes
Financial Disclosures
All the authors are employed by the National Institutes of Health and have no other financial disclosures.
References
- 1.De Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. 1955 Aug;60(4):604–17. doi: 10.1042/bj0600604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Duve C. The lysosome turns fifty. Nat Cell Biol. 2005 Sep;7(9):847–9. doi: 10.1038/ncb0905-847. [DOI] [PubMed] [Google Scholar]
- 3.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends in cell biology. 2012 Aug;22(8):407–17. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luzio JP, Parkinson MD, Gray SR, Bright NA. The delivery of endocytosed cargo to lysosomes. Biochemical Society transactions. 2009 Oct;37(Pt 5):1019–21. doi: 10.1042/BST0371019. [DOI] [PubMed] [Google Scholar]
- 5.Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy. 2011 Jul;7(7):673–82. doi: 10.4161/auto.7.7.14733. [DOI] [PubMed] [Google Scholar]
- 6.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008 Feb 28;451(7182):1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saftig P. Lysosomes. Georgetown, Tex. New York, N.Y: LandesBioscience/Eurekah.com; Springer Science+Business Media; 2005. [Google Scholar]
- 8.Settembre C, Ballabio A. Lysosomal adaptation: how the lysosome responds to external cues. Cold Spring Harbor perspectives in biology. 2014 Jun;6(6) doi: 10.1101/cshperspect.a016907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009 Jul 24;325(5939):473–7. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 10.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011 Jun 17;332(6036):1429–33. doi: 10.1126/science.1204592. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulze H, Kolter T, Sandhoff K. Principles of lysosomal membrane degradation: Cellular topology and biochemistry of lysosomal lipid degradation. Biochimica et biophysica acta. 2009 Apr;1793(4):674–83. doi: 10.1016/j.bbamcr.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 12.Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Science signaling. 2012 Jun 12;5(228):ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. The EMBO journal. 2012 Mar 7;31(5):1095–108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012 Jun;8(6):903–14. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013 Jun;15(6):647–58. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beutler E, Grabowski GA. Gaucher disease. In: Scriver CBA, Beaudet AL, Sly WS, Valle D, editors. The metabolic & molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3635–68. [Google Scholar]
- 17.Lachmann RH, Grant IR, Halsall D, Cox TM. Twin pairs showing discordance of phenotype in adult Gaucher’s disease. Qjm. 2004 Apr;97(4):199–204. doi: 10.1093/qjmed/hch036. Research Support, Non-U.S. Gov’t Twin Study. [DOI] [PubMed] [Google Scholar]
- 18.Sidransky E. Gaucher disease: insights from a rare Mendelian disorder. Discov Med. 2012 Oct;14(77):273–81. Research Support, N.I.H., Intramural Review. [PMC free article] [PubMed] [Google Scholar]
- 19.Pastores GM, Patel MJ, Firooznia H. Bone and joint complications related to Gaucher disease. Curr Rheumatol Rep. 2000 Apr;2(2):175–80. doi: 10.1007/s11926-000-0059-x. Review. [DOI] [PubMed] [Google Scholar]
- 20.Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004 Sep-Oct;83(1–2):6–15. doi: 10.1016/j.ymgme.2004.08.015. Review. [DOI] [PubMed] [Google Scholar]
- 21.Gupta N, Oppenheim IM, Kauvar EF, Tayebi N, Sidransky E. Type 2 Gaucher disease: phenotypic variation and genotypic heterogeneity. Blood Cells Mol Dis. 2011 Jan 15;46(1):75–84. doi: 10.1016/j.bcmd.2010.08.012. Case Reports Research Support, N.I.H., Intramural Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012 Nov;11(11):986–98. doi: 10.1016/S1474-4422(12)70190-4. Research Support, N.I.H., Extramural Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goker-Alpan O, Hruska KS, Orvisky E, Kishnani PS, Stubblefield BK, Schiffmann R, et al. Divergent phenotypes in Gaucher disease implicate the role of modifiers. J Med Genet. 2005 Jun;42(6):e37. doi: 10.1136/jmg.2004.028019. Letter Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA) Hum Mutat. 2008 May;29(5):567–83. doi: 10.1002/humu.20676. Review. [DOI] [PubMed] [Google Scholar]
- 25.Biegstraaten M, van Schaik IN, Aerts JM, Langeveld M, Mannens MM, Bour LJ, et al. A monozygotic twin pair with highly discordant Gaucher phenotypes. Blood Cells Mol Dis. 2011 Jan 15;46(1):39–41. doi: 10.1016/j.bcmd.2010.10.007. Case Reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009 Oct 22;361(17):1651–61. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siebert M, Sidransky E, Westbroek W. Glucocerebrosidase is shaking up the synucleinopathies. Brain. 2014 May;137(Pt 5):1304–22. doi: 10.1093/brain/awu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malec-Litwinowicz M, Rudzinska M, Szubiga M, Michalski M, Tomaszewski T, Szczudlik A. Cognitive impairment in carriers of glucocerebrosidase gene mutation in Parkinson disease patients. Neurol Neurochir Pol. 2014;48(4):258–61. doi: 10.1016/j.pjnns.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Pulkes T, Choubtum L, Chitphuk S, Thakkinstian A, Pongpakdee S, Kulkantrakorn K, et al. Glucocerebrosidase mutations in Thai patients with Parkinson’s disease. Parkinsonism Relat Disord. 2014 Sep;20(9):986–91. doi: 10.1016/j.parkreldis.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013 Jun;70(6):727–35. doi: 10.1001/jamaneurol.2013.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitsui J, Matsukawa T, Sasaki H, Yabe I, Matsushima M, Durr A, et al. Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neurol. 2015 Apr;2(4):417–26. doi: 10.1002/acn3.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Duve C. Lysosomal Storage Disorders. Springer US; 2007. From Lysosomes to Storage Diseases and Back: A Personal Reminiscence; pp. 1–5. [Google Scholar]
- 33.Brady RO, Pentchev PG, Gal AE, Hibbert SR, Dekaban AS. Replacement Therapy for Inherited Enzyme Deficiency. New England Journal of Medicine. 1974;291(19):989–93. doi: 10.1056/NEJM197411072911901. [DOI] [PubMed] [Google Scholar]
- 34.Barton NW, Furbish FS, Murray GJ, Garfield M, Brady RO. Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proceedings of the National Academy of Sciences. 1990 Mar 1;87(5):1913–6. doi: 10.1073/pnas.87.5.1913. 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doneda D, Netto CB, Moulin CC, Schwartz IVD. Effects of imiglucerase on the growth and metabolism of Gaucher disease type I patients: a systematic review. Nutrition & Metabolism. 2013;10:34. doi: 10.1186/1743-7075-10-34. 04/09 12/13/received 03/22/accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris JL. Velaglucerase Alfa for the Management of Type 1 Gaucher Disease. Clinical Therapeutics. 2012;34(2):259–71. doi: 10.1016/j.clinthera.2011.12.017. 2// [DOI] [PubMed] [Google Scholar]
- 37.G GA, Hopkin RJ. ENZYME THERAPY FOR LYSOSOMAL STORAGE DISEASE: Principles, Practice, and Prospects. Annual Review of Genomics and Human Genetics. 2003;4(1):403–36. doi: 10.1146/annurev.genom.4.070802.110415. [DOI] [PubMed] [Google Scholar]
- 38.Valayannopoulos V. Chapter 190 - Enzyme replacement therapy and substrate reduction therapy in lysosomal storage disorders with neurological expression. In: Olivier Dulac ML, Harvey BS, editors. Handbook of Clinical Neurology: Elsevier. 2013. pp. 1851–7. [DOI] [PubMed] [Google Scholar]
- 39.Gramlich PA, Westbroek W, Feldman RA, Awad O, Mello N, Remington MP, et al. A peptide-linked recombinant glucocerebrosidase for targeted neuronal delivery: Design, production, and assessment. Journal of Biotechnology. 2016;221:1–12. doi: 10.1016/j.jbiotec.2016.01.015. 3/10/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henley WE, Anderson LJ, Wyatt KM, Nikolaou V, Anderson R, Logan S. The NCS-LSD cohort study: a description of the methods and analyses used to assess the long-term effectiveness of enzyme replacement therapy and substrate reduction therapy in patients with lysosomal storage disorders. J Inherit Metab Dis. 2014;37(6):939–44. doi: 10.1007/s10545-014-9679-6. 2014/11/01. [DOI] [PubMed] [Google Scholar]
- 41.Alfonso P, Pampín S, Estrada J, Rodríguez-Rey JC, Giraldo P, Sancho J, et al. Miglustat (NB-DNJ) works as a chaperone for mutated acid β-glucosidase in cells transfected with several Gaucher disease mutations. Blood Cells, Molecules, and Diseases. 2005;35(2):268–76. doi: 10.1016/j.bcmd.2005.05.007. 9// [DOI] [PubMed] [Google Scholar]
- 42.Andrade MM, Medrano B, Alfonso P, Irún P, Atutxa K, Fernandez-Galan A, et al. Substrate Reduction Therapy With Miglustat In Type 1 Gaucher Disease In Spain. Nine Years Outcomes Update On ZAGAL Study. Blood. 2013;122(21):4713. 2013-11-15 00:00:00. [Google Scholar]
- 43.Schiffmann R, Fitzgibbon EJ, Harris C, DeVile C, Davies EH, Abel L, et al. Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol. 2008 Nov;64(5):514–22. doi: 10.1002/ana.21491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marshall J, Sun Y, Bangari DS, Budman E, Park H, Nietupski JB, et al. CNS-Accessible Inhibitor of Glucosylceramide Synthase for Substrate Reduction Therapy of Neuronopathic Gaucher Disease. Mol Ther. 2016 Mar;:7. doi: 10.1038/mt.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scott L. Eliglustat: A Review in Gaucher Disease Type 1. Drugs. 2015;75(14):1669–78. doi: 10.1007/s40265-015-0468-9. 2015/09/01. [DOI] [PubMed] [Google Scholar]
- 46.Balwani M, Burrow TA, Charrow J, Goker-Alpan O, Kaplan P, Kishnani PS, et al. Recommendations for the use of eliglustat in the treatment of adults with Gaucher disease type 1 in the United States. Mol Genet Metab. doi: 10.1016/j.ymgme.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 47.Bennett LL, Turcotte K. Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease. Drug Design, Development and Therapy. 2015;9:4639–47. doi: 10.2147/DDDT.S77760. 08/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.GM P, DA H, RA P, MP A, HH A, et al. e. Gaucher Disease. GeneReviews®. 2000 Review. [Google Scholar]
- 49.Ficicioglu C. Review of miglustat for clinical management in Gaucher disease type 1. Therapeutics and Clinical Risk Management. 2008;4(2):425–31. doi: 10.2147/tcrm.s6865. 04/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dahl M, Doyle A, Olsson K, Mansson JE, Marques AR, Mirzaian M, et al. Lentiviral gene therapy using cellular promoters cures type 1 Gaucher disease in mice. Mol Ther. 2015 May;23(5):835–44. doi: 10.1038/mt.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Compain P, Martin OR, Boucheron C, Godin G, Yu L, Ikeda K, et al. Design and Synthesis of Highly Potent and Selective Pharmacological Chaperones for the Treatment of Gaucher’s disease. ChemBioChem. 2006;7(9):1356–9. doi: 10.1002/cbic.200600217. [DOI] [PubMed] [Google Scholar]
- 52.Lieberman RL, Wustman BA, Huertas P, Powe AC, Jr, Pine CW, Khanna R, et al. Structure of acid beta-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat Chem Biol. 2007 Feb;3(2):101–7. doi: 10.1038/nchembio850. [DOI] [PubMed] [Google Scholar]
- 53.Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nature cell biology. 2005 Aug;7(8):766–72. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 54.Pastores GM, Hughes DA. Gaucher Disease. GeneReviews®. 2000:2000. [Google Scholar]
- 55.Suzuki Y, Ichinomiya S, Kurosawa M, Ohkubo M, Watanabe H, Iwasaki H, et al. Chemical chaperone therapy: clinical effect in murine G(M1)-gangliosidosis. Ann Neurol. 2007 Dec;62(6):671–5. doi: 10.1002/ana.21284. [DOI] [PubMed] [Google Scholar]
- 56.Takamura A, Higaki K, Ninomiya H, Takai T, Matsuda J, Iida M, et al. Lysosomal accumulation of Trk protein in brain of GM(1) -gangliosidosis mouse and its restoration by chemical chaperone. J Neurochem. 2011 Aug;118(3):399–406. doi: 10.1111/j.1471-4159.2011.07310.x. [DOI] [PubMed] [Google Scholar]
- 57.Takamura A, Higaki K, Kajimaki K, Otsuka S, Ninomiya H, Matsuda J, et al. Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochem Biophys Res Commun. 2008 Mar 14;367(3):616–22. doi: 10.1016/j.bbrc.2007.12.187. [DOI] [PubMed] [Google Scholar]
- 58.Patnaik S, Zheng W, Choi JH, Motabar O, Southall N, Westbroek W, et al. Discovery, structure-activity relationship, and biological evaluation of noninhibitory small molecule chaperones of glucocerebrosidase. Journal of medicinal chemistry. 2012 Jun 28;55(12):5734–48. doi: 10.1021/jm300063b. Research Support, N.I.H., Extramural Research Support, N.I.H., Intramural. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maegawa GH, Tropak MB, Buttner JD, Rigat BA, Fuller M, Pandit D, et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J Biol Chem. 2009 Aug 28;284(35):23502–16. doi: 10.1074/jbc.M109.012393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luan Z, Li L, Higaki K, Nanba E, Suzuki Y, Ohno K. The chaperone activity and toxicity of ambroxol on Gaucher cells and normal mice. Brain Dev. 2013 Apr;35(4):317–22. doi: 10.1016/j.braindev.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 61.Sánchez-Ollé G, Duque J, Egido-Gabás M, Casas J, Lluch M, Chabás A, et al. Promising results of the chaperone effect caused by iminosugars and aminocyclitol derivatives on mutant glucocerebrosidases causing Gaucher disease. Blood Cells, Molecules, and Diseases. 2009;42(2):159–66. doi: 10.1016/j.bcmd.2008.11.002. 3// [DOI] [PubMed] [Google Scholar]
- 62.Sawkar AR, Cheng W-C, Beutler E, Wong C-H, Balch WE, Kelly JW. Chemical chaperones increase the cellular activity of N370S β-glucosidase: A therapeutic strategy for Gaucher disease. Proceedings of the National Academy of Sciences. 2002 Nov 26;99(24):15428–33. doi: 10.1073/pnas.192582899. 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gloster TM, Davies GJ. Glycosidase inhibition: assessing mimicry of the transition state. Org Biomol Chem. 2010 Jan 21;8(2):305–20. doi: 10.1039/b915870g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Butters TD, Dwek RA, Platt FM. Imino sugar inhibitors for treating the lysosomal glycosphingolipidoses. Glycobiology. 2005 Oct;15(10):43R–52R. doi: 10.1093/glycob/cwi076. [DOI] [PubMed] [Google Scholar]
- 65.Babajani G, Tropak MB, Mahuran DJ, Kermode AR. Pharmacological chaperones facilitate the post-ER transport of recombinant N370S mutant β-glucocerebrosidase in plant cells: Evidence that N370S is a folding mutant. Mol Genet Metab. 2012;106(3):323–9. doi: 10.1016/j.ymgme.2012.04.018. 04/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khanna R, Benjamin ER, Pellegrino L, Schilling A, Rigat BA, Soska R, et al. The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of beta-glucosidase. Febs J. 2010 Apr;277(7):1618–38. doi: 10.1111/j.1742-4658.2010.07588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun Y, Liou B, Xu YH, Quinn B, Zhang W, Hamler R, et al. Ex vivo and in vivo effects of isofagomine on acid beta-glucosidase variants and substrate levels in Gaucher disease. J Biol Chem. 2012 Feb 3;287(6):4275–87. doi: 10.1074/jbc.M111.280016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun Y, Ran H, Liou B, Quinn B, Zamzow M, Zhang W, et al. Isofagomine in vivo effects in a neuronopathic Gaucher disease mouse. PLoS ONE. 2011;6(4):e19037. doi: 10.1371/journal.pone.0019037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Panicker LM, Miller D, Park TS, Patel B, Azevedo JL, Awad O, et al. Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc Natl Acad Sci U S A. 2012 Oct 30;109(44):18054–9. doi: 10.1073/pnas.1207889109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zimran A. How I treat Gaucher disease. Blood. 2011 Aug 11;118(6):1463–71. doi: 10.1182/blood-2011-04-308890. [DOI] [PubMed] [Google Scholar]
- 71.Mena-Barragán T, García-Moreno MI, Nanba E, Higaki K, Concia AL, Clapés P, et al. Inhibitor versus chaperone behaviour of d-fagomine, DAB and LAB sp2-iminosugar conjugates against glycosidases: A structure–activity relationship study in Gaucher fibroblasts. European Journal of Medicinal Chemistry. doi: 10.1016/j.ejmech.2015.08.038. [DOI] [PubMed] [Google Scholar]
- 72.Luan Z, Higaki K, Aguilar-Moncayo M, Li L, Ninomiya H, Nanba E, et al. A Fluorescent sp2-Iminosugar With Pharmacological Chaperone Activity for Gaucher Disease: Synthesis and Intracellular Distribution Studies. ChemBioChem. 2010;11(17):2453–64. doi: 10.1002/cbic.201000323. [DOI] [PubMed] [Google Scholar]
- 73.Marugan JJ, Zheng W, Motabar O, Southall N, Goldin E, Westbroek W, et al. Evaluation of Quinazoline analogues as Glucocerebrosidase Inhibitors with Chaperone activity. Journal of medicinal chemistry. 2011;54(4):1033–58. doi: 10.1021/jm1008902. 01/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bendikov-Bar I, Maor G, Filocamo M, Horowitz M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol Dis. 2013 Feb;50(2):141–5. doi: 10.1016/j.bcmd.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zimran A, Altarescu G, Elstein D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells, Molecules, and Diseases. 2013;50(2):134–7. doi: 10.1016/j.bcmd.2012.09.006. 2// [DOI] [PubMed] [Google Scholar]
- 76.Lee B, Scaglia F. Inborn Errors of Metabolism: From Neonatal Screening to Metabolic Pathways. Oxford University Press; 2014. [Google Scholar]
- 77.Zheng W, Padia J, Urban DJ, Jadhav A, Goker-Alpan O, Simeonov A, et al. Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease. Proc Natl Acad Sci U S A. 2007 Aug 7;104(32):13192–7. doi: 10.1073/pnas.0705637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goldin E, Zheng W, Motabar O, Southall N, Choi JH, Marugan J, et al. High throughput screening for small molecule therapy for Gaucher disease using patient tissue as the source of mutant glucocerebrosidase. PloS one. 2012;7(1):e29861. doi: 10.1371/journal.pone.0029861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rogers S, Patnaik S, Schoenen F, Zheng W, Choi J, Motabar O, et al. Discovery, SAR, and Biological Evaluation of Non-inhibitory Chaperones of Glucocerebrosidase. 2010 [PubMed] [Google Scholar]
- 80.Patnaik S, Zheng W, Choi JH, Motabar O, Southall N, Westbroek W, et al. Discovery, Structure–Activity Relationship, and Biological Evaluation of Noninhibitory Small Molecule Chaperones of Glucocerebrosidase. Journal of Medicinal Chemistry. 2012;55(12):5734–48. doi: 10.1021/jm300063b. 2012/06/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee RE. The fine structure of the cerebroside occurring in Gaucher’s disease. Proceedings of the National Academy of Sciences of the United States of America. 1968;61(2):484–9. doi: 10.1073/pnas.61.2.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang C, Rahimpour S, Lu J, Pacak K, Ikejiri B, Brady RO, et al. Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc Natl Acad Sci U S A. 2013 Jan 15;110(3):966–71. doi: 10.1073/pnas.1221046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang C, Wang H, Zhu D, Hong CS, Dmitriev P, Zhang C, et al. Mutant glucocerebrosidase in Gaucher disease recruits Hsp27 to the Hsp90 chaperone complex for proteasomal degradation. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(4):1137–42. doi: 10.1073/pnas.1424288112. 01/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Witte MD, Kallemeijn WW, Aten J, Li KY, Strijland A, Donker-Koopman WE, et al. Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat Chem Biol. 2010 Dec;6(12):907–13. doi: 10.1038/nchembio.466. [DOI] [PubMed] [Google Scholar]
- 85.Yadav AK, Shen DL, Shan X, He X, Kermode AR, Vocadlo DJ. Fluorescence-Quenched Substrates for Live Cell Imaging of Human Glucocerebrosidase Activity. Journal of the American Chemical Society. 2015;137(3):1181–9. doi: 10.1021/ja5106738. 2015/01/28. [DOI] [PubMed] [Google Scholar]
- 86.Lee HW, Heo CH, Sen D, Byun H-O, Kwak IH, Yoon G, et al. Ratiometric Two-Photon Fluorescent Probe for Quantitative Detection of β-Galactosidase Activity in Senescent Cells. Analytical Chemistry. 2014;86(20):10001–5. doi: 10.1021/ac5031013. 2014/10/21. [DOI] [PubMed] [Google Scholar]
- 87.Cook C, Stetler C, Petrucelli L. Disruption of protein quality control in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012 May;2(5):a009423. doi: 10.1101/cshperspect.a009423. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011 Apr;23(2):184–9. doi: 10.1016/j.ceb.2010.10.009. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010 Sep;12(9):814–22. doi: 10.1038/ncb0910-814. Historical Article Research Support, N.I.H., Extramural Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu M, Sun XL, Qiao C, Liu Y, Ding JH, Hu G. Uncoupling protein 2 deficiency aggravates astrocytic endoplasmic reticulum stress and nod-like receptor protein 3 inflammasome activation. Neurobiol Aging. 2013 Sep;:13. doi: 10.1016/j.neurobiolaging.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 91.Tofaris GK. Lysosome-dependent pathways as a unifying theme in Parkinson’s disease. Mov Disord. 2012 Sep 15;27(11):1364–9. doi: 10.1002/mds.25136. Research Support, Non-U.S. Gov’t Review. [DOI] [PubMed] [Google Scholar]
- 92.Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Human molecular genetics. 2013 May 15;22(10):2067–82. doi: 10.1093/hmg/ddt057. Research Support, N.I.H., Extramural Research Support, N.I.H., Intramural. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011 Jul 8;146(1):37–52. doi: 10.1016/j.cell.2011.06.001. Research Support, N.I.H., Extramural Research Support, N.I.H., Intramural. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cleeter MW, Chau KY, Gluck C, Mehta A, Hughes DA, Duchen M, et al. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem Int. 2013 Jan;62(1):1–7. doi: 10.1016/j.neuint.2012.10.010. Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann Neurol. 2011 Jun;69(6):940–53. doi: 10.1002/ana.22400. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 96.Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, et al. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol. 2012 Sep;72(3):455–63. doi: 10.1002/ana.23614. Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Manning-Bog AB, Schule B, Langston JW. Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology. 2009 Nov;30(6):1127–32. doi: 10.1016/j.neuro.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 98.Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S, et al. Mitochondria and quality control defects in a mouse model of Gaucher disease–links to Parkinson’s disease. Cell Metab. 2013 Jun 4;17(6):941–53. doi: 10.1016/j.cmet.2013.04.014. Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proceedings of the National Academy of Sciences of the United States of America. 2011 Jul 19;108(29):12101–6. doi: 10.1073/pnas.1108197108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sardi SP, Clarke J, Viel C, Chan M, Tamsett TJ, Treleaven CM, et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proceedings of the National Academy of Sciences of the United States of America. 2013 Feb 26;110(9):3537–42. doi: 10.1073/pnas.1220464110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Murphy KE, Halliday GM. Glucocerebrosidase deficits in sporadic Parkinson disease. Autophagy. 2014 Jul;10(7):1350–1. doi: 10.4161/auto.29074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chiasserini D, Paciotti S, Eusebi P, Persichetti E, Tasegian A, Kurzawa-Akanbi M, et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol Neurodegener. 2015;10(1):15. doi: 10.1186/s13024-015-0010-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Du TT, Wang L, Duan CL, Lu LL, Zhang JL, Gao G, et al. GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy. 2015 Oct 3;11(10):1803–20. doi: 10.1080/15548627.2015.1086055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rocha EM, Smith GA, Park E, Cao H, Graham AR, Brown E, et al. Sustained Systemic Glucocerebrosidase Inhibition Induces Brain alpha-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid Redox Signal. 2015 Aug 20;23(6):550–64. doi: 10.1089/ars.2015.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Aflaki E, Stubblefield BK, Maniwang E, Lopez G, Moaven N, Goldin E, et al. Macrophage Models of Gaucher Disease for Evaluating Disease Pathogenesis and Candidate Drugs. Science translational medicine. 2014;6(240):240ra73–ra73. doi: 10.1126/scitranslmed.3008659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. 2014 May;137(Pt 5):1481–95. doi: 10.1093/brain/awu020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Richter F, Fleming SM, Watson M, Lemesre V, Pellegrino L, Ranes B, et al. A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics. 2014 Oct;11(4):840–56. doi: 10.1007/s13311-014-0294-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dasgupta N, Xu Y-h, Li R, Peng Y, Pandey MK, Tinch SL, et al. Neuronopathic Gaucher disease: dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Human Molecular Genetics. 2015 Dec 15;24(24):7031–48. doi: 10.1093/hmg/ddv404. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.de la Mata M, Cotán D, Oropesa-Ávila M, Garrido-Maraver J, Cordero MD, Villanueva Paz M, et al. Pharmacological Chaperones and Coenzyme Q(10) Treatment Improves Mutant β-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Scientific Reports. 2015;5:10903. doi: 10.1038/srep10903. 06/05 01/20/received 04/27/accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]