

Abstract
The TCR repertoire of regulatory T cells (Tregs) is highly diverse. The relevance of this diversity to maintain self-tolerance remains unknown. We established a model where the TCR repertoire of normal polyclonal Tregs was limited by serial transfers into IL-2Rβ−/− mice, which lack functional Tregs. After a primary transfer, the donor Treg TCR repertoire was substantially narrowed, yet the recipients remained autoimmune-free. Importantly, upon purification and transfer of donor-derived Tregs from an individual primary recipient into neonatal IL-2Rβ−/− mice, these secondary recipients developed autoimmunity. Here the Treg TCRβ repertoire was reshaped and further narrowed. In contrast, secondary IL-2Rβ recipients showed less symptoms of autoimmunity when they received donor Tregs that were pre-mixed from several primary recipients to increase their TCRβ repertoire diversity. About 8–11% of the Treg TCRβ repertoire was estimated to be the minimum required to establish and maintain tolerance in primary IL-2Rβ−/− recipients. Collectively, these data quantify where limitations imposed on Treg TCRβ repertoire results in a population of Tregs that cannot fully suppress polyclonal autoreactive T cells. Our data favor a model where the high diversity of the Treg TCR provides a mechanism for Tregs to actively adapt and effectively suppress autoreactive T cells, which are not fixed, but are evolving as they encounter self-antigens.
Introduction
Foxp3+ regulatory T cells (Tregs) importantly maintain tolerance by suppressing autoreactive T cells but also regulate ongoing immune responses to infections agents, tumors, and transplanted tissues and cells. To mediate these activities, Tregs express a TCR repertoire that is highly diverse, with only a partial and somewhat limited overlap with the repertoire found on conventional Foxp3− T cells (1–3). Tregs show heterogeneity in their TCR repertoire based on the anatomical location of draining secondary lymphoid tissues (4) or within distinct non-lymphoid tissue sites (5). These findings are consistent with localized responses by Tregs to tissue-specific antigens.
An important unanswered question is what fraction of this diverse Treg TCR repertoire is required to maintain peripheral tolerance and homeostasis of the immune system as this represents an indispensable function of Tregs. Several studies have shown that control of autoimmunity is impaired under conditions where the TCR repertoire has been substantially limited (6–9). These studies, however, do not indicate the extent to which the Treg repertoire can be limited and still control autoimmunity. In this regard, neonatal IL-2Rβ−/− mice were protected from autoimmunity when they received wild-type (WT) Tregs (10–12), but not Tregs from TCRβ transgenic mice, where the inherently limited diversity of their TCR repertoire is further constrained (6).
IL-2Rβ−/− mice contain only immature non-functional Foxp3l° CD25− cells due to a block during an essential step of thymic Treg development (13–15). A recent study showed that conditional knockout of IL-2Rβ in Tregs led to a more rapid disease onset (16), similar to Scurfy or Foxp3-deficient mice, indicating that the autoimmunity associated with germline IL-2Rβ-deficiency is a combination of impaired Tregs and impaired autoreactive T cells, the latter which slow the tempo of autoimmunity. Thus, the lethal autoimmunity associated with IL-2Rβ−/− mice is due to defective suppression of polyclonal autoreactive T cells that escape thymic negative selection. In the present study, WT Tregs were serially transferred into IL-2Rβ−/− recipients, where the diversity of their Treg TCR repertoire is actively limited. Our assessment of disease parameters and the Treg TCR repertoire in these mice allowed us to approximate the physiologic limits on Treg TCR diversity that are necessary to effectively suppress polyclonal autoreactive T cells that circulate in the periphery.
Material and Methods
Mice
Foxp3/RFP-reporter (17), IL-2Rβ−/− (18), and CD45.1-congenic C57BL/6 mice were bred within the specific pathogen-free animal facility at the University of Miami. Spleen cells (1 × 107) or the indicated purified T cells (2 × 105) from Foxp3/RFP or CD45.1 mice were adoptively transferred i.v. into 1–3 day old neonatal IL-2Rβ−/− mice. Male and female recipients were typically analyzed at 8–16 weeks of age. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Miami.
Flow cytometry and cell purification
Monoclonal antibodies to the indicated molecules are: CD4 (GK1.5 or RM4-5), CD8 (53–6.7), CD62L (MEL-14) Foxp3 (FKJ-16s), IFNγ (XMG1.2), CD45.1 (A20), and CD44 (Pgp-1). Samples were analyzed on a LSR-Fortessa-HTS. Typically, more than 100,000 total events were collected per sample. For adoptive transfers, CD4+ Tregs were purified by enrichment of CD4+ CD25hi T cells as previously described (12). CD8+ T cells were isolated by anti-CD8 magnetic MicroBeads (Miltenyi Biotec) and then CD8+ CD122+ cells were isolated by sorting using a BD FACSAria. For the TCRβ repertoire analysis, CD4+ T cells were enriched from the spleen with anti-CD4 magnetic beads and the CD4+ RFP+ Tregs were sorted using a BD FACSAria. All sorted cells were >95% pure.
Assessments of autoimmunity
Determination of the hematocrit and pathological analysis were performed as previously described (12). In brief, major organs were collected at necropsy, fixed in 10% neutral buffered formalin, sectioned and stained with hematoxylin-eosin. The tissues were coded and examined by a board certified veterinary pathologist. Inflammation was scored as: mild (<10%), moderate (10–40%), and severe (>40%) leukocyte inflammation of the indicated tissues. Intracellular IFNγ production was determined by culturing spleen cells with PMA and ionomycin for 4 hrs in the presence of brefeldin A as previously described (19).
TCRβ repertoire analysis
Spectratype analysis using reverse-transcribed RNA from purified Tregs and calculations of diversity (D)-scores were performed as previously described (6, 20). Deep sequencing of TCRβ V-regions was performed using DNA from purified Tregs (19) at Adaptive Biotechnologies (Seattle, WA) using the immunoSEQ platform and their bioinformatics software for CDR3 region analysis (21, 22). The resulting sequences are of sufficient length to assign the V subgroups and J regions associated with each CDR3. The resulting sequences were further analyzed in part by using ImmunoSEQ Analyzer 2.0 software. This software calculates repertoire overlap between two samples as the sum of the read counts of each unique rearrangement in sample 1 and sample 2 divided by the sum of total read counts in sample 1 and sample 2 (23) and is expressed as a percentage. This software also assesses the clonality of the repertoire as 1 minus the Shannon Entropy of all productive clones in a sample (24), where 1 equals monoclonal and 0 as the most diverse.
Statistical analysis
Data were analyzed by one way ANOVA applying Tukey’s multiple comparison test using Graph Pad Prism 6.0 software. In all Figures, data are reported as the mean ± SD where significant differences are indicated as *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Results
Model to define Treg TCR repertoire limits in suppressing autoimmunity
C57BL/6 IL-2Rβ-deficient mice die from lethal systemic autoimmunity, usually at 8–12 weeks of age (12, 18). Over the years, we have routinely maintained an autoimmune-free breeding colony of IL-2Rβ-deficient mice by the transfer of purified Tregs, purified CD4+ T cells, or more recently by the transfer of 10–20 × 106 spleen cells into 1–3 day old neonatal mice. Donor Tregs from such transfers represent essentially the entire Treg population in recipient IL-2Rβ-deficient mice and are approximately 10% of peripheral CD4+ T cells by 7–10 days of life (25). These donor Tregs persist at this proportion for at least 9–12 months of age. Autoimmunity associated with IL-2Rβ-deficient mice is effectively controlled by the transfer of 1–2 × 105 WT Tregs during neonatal life (10–12). After transfer, the donor Treg repertoire is narrowed and reshaped (6).
To define the limits where donor Tregs cannot effectively control autoimmunity due to too few TCR specificities, we performed serial transfers of donor Tregs from a primary (1°) “cured” IL-2Rβ−/− recipient to a new neonatal IL-2Rβ−/− mouse to serve as a secondary (2°) recipient, and so forth, as necessary (Fig. 1A). Upon each serial transfer, the Treg repertoire was expected to show progressive narrowing. Through monitoring the health of the recipient mice in conjunction with spectratyping and sequencing the Vβ-region of donor Treg TCRs, we expected to find a point where limitations on Treg TCR repertoire were associated with failed control of autoimmunity.
Figure 1.
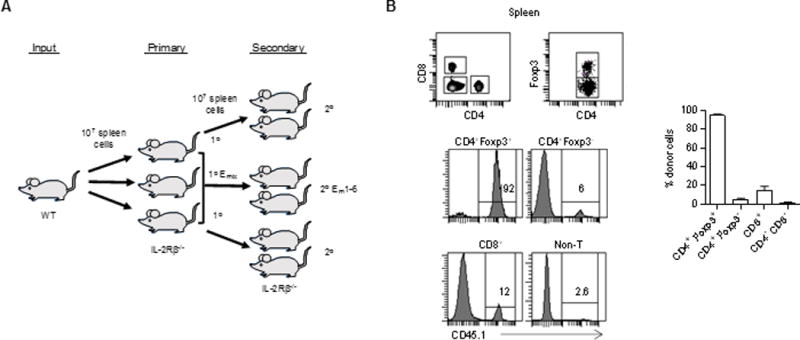
Serial transfer model to follow Treg repertoire in control of autoimmunity. (A) Experimental scheme. (B) Neonatal IL-2Rβ−/− mice were adoptively transferred with CD45.1-unfractionated spleens. The gated regions in top panels were assessed for donor cell engraftment (CD45.1+) in the spleen 16 weeks post-transfer (n=4).
To maintain the limit on the donor Treg TCRs imposed after transfer into 1° recipients, the experimental design required that 2° IL-2Rβ recipients receive donor Tregs derived from a single 1° IL-2Rβ−/− mouse (Fig. 1A). Another important consideration was that lowering of the TCR repertoire could not be achieved simply by lowering the number of donor Tregs. In this case, failure to control autoimmunity might be due to an inadequate number of Tregs that could not initially keep up with the autoreactive T cells rather than too few TCR specificities. A technical consideration was that the 1° recipient needed to provide donor-derived Tregs for adoptive transfer into 2° recipients and RNA and/or DNA for TCR repertoire analysis. To satisfy these requirements, donor Tregs were transferred i.v. using unfractionated spleen cells (10 × 106). As donor Tregs comprise approximately 3% (3.1± 0.9%, n=6) of the spleen of normal mice and 2% (2.1% ± 0.4, n=12) from a 1° recipient, several 2° recipients could receive an optimal number of Tregs (~2 × 105) from a single 1° IL-2Rβ−/− recipient, while sufficient spleen cells remained for sorting the RFP+ Tregs to prepare RNA or DNA for TCR analysis.
The distribution of donor spleen cells (CD45.1+) was assessed for several IL-2Rβ−/− recipients (CD45.2+) (Fig. 1B). As expected, donor cells accounted for nearly all CD4+ Foxp3+ T cells (>90%) whereas they constituted only a minority of all other lymphoid cells in these recipients. The second most prominent donor-derived population was CD8+ T cells (approximately 15%); this result was also expected (12) and likely due to favorable competition of the WT CD8+ T cells for IL-15, which depends on IL-2Rβ signaling. Very few conventional CD4+ T cells and non-T lymphocytes, the latter as assessed after gating on the CD4− and CD8− cells, were donor derived. These results indicate that Tregs are the primary lymphoid subset that engraft and persist after transfer of spleen cells into neonatal IL-2Rβ-deficient mice.
Autoimmunity is not suppressed in 2° IL-2Rβ−/− recipients
IL-2Rβ−/− recipients were examined for several parameters associated with their autoimmune disease, especially the appearance of activated conventional CD4+ and CD8+ T cells, as this is an early sign predictive of autoimmunity (11, 12). As expected, 1° IL-2Rβ−/− recipients showed a normal complement of donor Tregs the majority of which expressed CD25 (Fig. 2A), lacked hemolytic anemia as measured by the hematocrit (Fig. 2B), contained few activated CD44hi CD62Ll° conventional CD4+ and CD8+ T cells (Fig. 2C), and had a relatively low fraction of IFNγ-producing CD4+ T cells (Fig. 2D). Most of these indicators were abnormal in 2° IL-2Rβ−/− recipients and are characteristic of autoimmunity in untreated IL-2Rβ−/− mice. Although on average the proportion of Tregs in the CD4+ T cell compartment of 2° recipients was similar to 1° recipients, several mice had a lower proportion of Tregs than the average, consistent with an inability to keep up with autoreactive T cells. Most 2° recipients exhibited hemolytic anemia, contained a high fraction of activated CD44hi CD62Ll° recipient-derived CD4+ and CD8+ T cells, and increased proportion of IFNγ-producing CD4+, but not CD8+, T cells.
Figure 2.
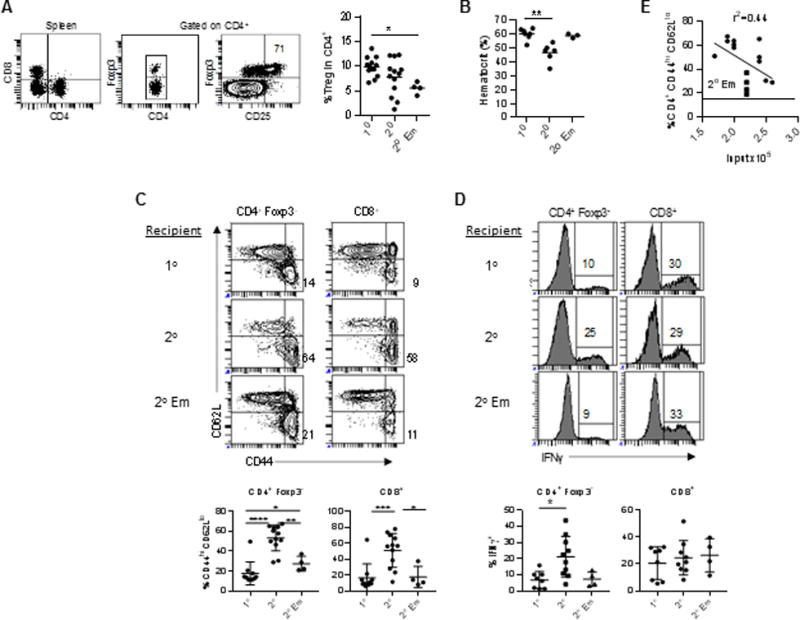
Autoimmune profile of 1° and 2° IL-2Rβ−/− recipients. The x-axis in the graphs represent the type of recipients (see Fig. 1A) where 2° EM represents the results from 2° recipients that received a mixture of 1° donor Tregs from 4 mice (A) Donor Treg representation in the spleen of 1° recipients, (B) the hematocrit, (C) the proportion of activated CD44hi CD62Ll° T cells in the spleen, and (D) IFNγ production by CD4+ and CD8+ T cells in the spleen. (E) The relationship between donor Treg numbers that were used for the 2° transfer into IL-2Rβ−/− recipients vs. autoimmune symptoms of the 2° recipients, as measured by the proportion of activated CD4+ Foxp3− CD44hi CD62Ll° T cells in the spleen. Solid circles represent the results from 2° recipients that received 1° donor Tregs from a single mouse; these data were used for linear regression analysis. The solid squares (n=4) represents the results from 2° recipients that received a mixture of 1° donor Tregs from 4 mice (2° EM). The horizontal line in the graph represents the average proportion (n=9) of activated CD4+ T cells in autoimmune-free 1° recipients, where the one 1° recipient with autoimmune symptoms was excluded.
The pathological changes in 2° recipients consisted primarily of perivascular accumulations of lymphocytes, plasma cells, and monocytes (Fig. 3A). There was extension into the surrounding parenchyma. The lesions were scored as mild, moderate, or severe. With the exception of one 1° recipient with a high proportion (50–60%) of activated CD4+ and CD8+ T cells and moderate hyperplasia of the lymph nodes, the pathological analysis of multiple tissues of the remaining 1° recipients was largely unremarkable. Mild inflammation was noted for the lung of five 1° recipients, but similar lung pathology was also found in some normal C57BL/6 mice in our colony (not shown). Thus, this indicator is not associated with autoimmunity. In marked contrast, moderate to severe inflammatory infiltrates were detected in multiple organs, typically the salivary gland, lung, pancreas, and small intestine in almost all 2° IL-2Rβ−/− recipients (Fig. 3B). The lymph nodes of these 2° recipient mice usually showed hyperplasia, consistent with increased lymphocyte proliferation. These data, therefore, establish that the donor Tregs in the 2° recipients are unable to fully suppress autoimmunity.
Figure 3.
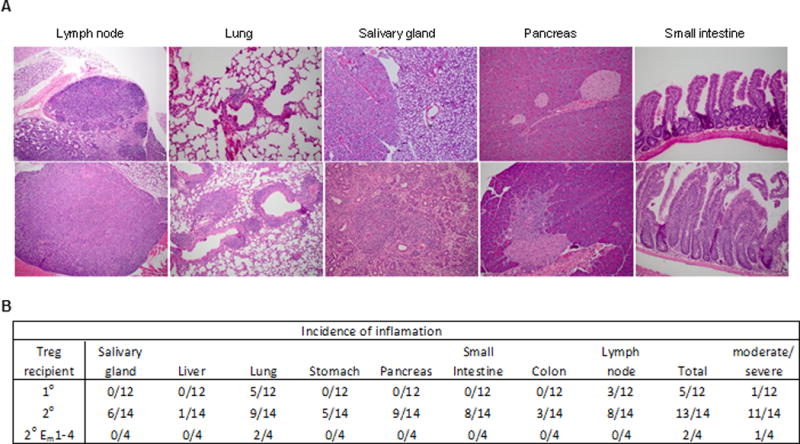
Inflammation associated with 1° and 2° IL-2Rβ−/− recipients. (A) The indicated tissues were assessed for histopathological changes after hematoxylin and eosin staining and scoring by a Board Certified Veterinary Pathologist. For the lung from a 1° recipient, a representative is shown that was scored as mild lymphocytic infiltration. (B) Summary of the histopathological analysis, where any scored instance of tissue inflammation or lymph node hyperplasia is enumerated. “Total” refers to the number of mice that showed any pathological abnormalities. Any mouse that showed in one or more tissues moderate/severe inflammation or lymph node hyperplasia was listed in the right column and these mice have clear pathological signs consistent with autoimmunity. 2° EM represents the results from 2° recipients that received a mixture of 1° donor Tregs from 4 mice.
Donor Tregs in 1° IL-2Rβ−/− recipients primarily acquire a phenotype, i.e. CD62Ll° and CD103+, associated with effector Tregs (eTregs) (19, 26). This skewing of the donor Tregs raised the possibility that the autoimmunity in 2° recipients might be due to this or another undefined change in the engrafted Tregs rather than a potential decrease in Treg TCR repertoire diversity. To address this issue, the diversity of the donor Tregs from 1° recipients was intentionally increased by mixing spleen cells from four 1° recipients in a single experiment (designated 1° Emix) prior to transfer into 2° IL-2Rβ−/− neonatal recipients (designated 2° Em). Notably, 2° Em IL-2Rβ−/− recipients showed an intermediate phenotype when compared to 1° and 2° recipients, with a consistent trend for less severe autoimmune symptoms based on immunologic (Fig. 2) and pathological (Fig. 3) assessments. One sign that the donor Tregs in these 2° Em recipients do not fully suppress autoimmunity is a somewhat elevated proportion of activated CD4+ conventional T cells (Fig. 2C). Collectively, these data show that severe autoimmunity develops upon serial passage of WT Tregs into IL-2Rβ−/− mice that is evident in 2° recipients and this may be due to a decrease in expression of effective TCR specificities.
Our experimental design was to transfer 10 × 106 spleen cells from a 1° recipient into 2° neonatal IL-2Rβ recipients. All these recipients received ≥1.7 × 105 donor Tregs, a number that prevents autoimmunity when obtained from normal WT mice. However, by transferring a fixed number of spleen cells, individual 2° recipients received different numbers (1.7–2.6 × 105) of Tregs. Although all these 2° mice showed symptoms of autoimmunity, the severity of disease was somewhat different based on the proportion of activated CD4+ CD44hi CD62Ll° conventional T cells in the spleen (Fig. 2E). A moderate relationship was noted where fewer activated T cells was associated with mice that received a greater numbers of Tregs, where TCR diversity is expected to be greater. This result, therefore, is analogous to the mixing experiment and suggests that a potentially modest increase (<2-fold) in TCR diversity is useful to moderate autoimmunity in this model.
CD8+ T cells do not contribute to suppression of autoimmunity in IL-2Rβ-deficient mice
Our past work has shown that WT conventional CD4+ T cells do not substantially contribute to suppression of autoimmunity in IL-2Rβ−/− mice (12). However, donor CD8+ T cells readily engraft and persist in IL-2Rβ-deficient recipients (Fig. 1B). We directly assessed if autoimmunity in IL-2Rβ−/− mice might be down-regulated by CD8+ T cells, including the CD122+ subset that contains regulatory activity in some settings (27). This analysis focused on the proportion of activated conventional CD4+ T cells and a decrease in the hematocrit in these IL-2Rβ−/− recipients, as these represent the initial and a progressive state of autoimmunity, respectively, in untreated IL-2Rβ-deficient mice (6, 12). Neonatal IL-2Rβ−/− mice that received unfractionated CD8+ or CD122+ CD8+ T cells, but not CD4+ Tregs, contained a high proportion of recipient CD62Ll° activated CD4+ and CD8+ T cells (Fig. 4A) and exhibited a low hematocrit (Fig.4B), consistent with severe autoimmunity and hemolytic anemia associated with untreated IL-2Rβ−/− mice. In addition, without the transfer of CD4+ Tregs, a majority of the CD8+ T cell compartment was accounted for by donor CD8+ T cells (Fig. 4C), with <0.3% of the CD4+ compartment derived from a few contaminating Tregs (not shown). This result contrasts with IL-2Rβ-deficient recipients that received purified Tregs, where a few contaminating CD8+ T cells expand and persist [Fig 4C and (12)], or unfractionated spleen cells, where donor-derived CD8+ T cells are readily detected (Fig. 1B). Thus, these data indicate that WT CD8+ T cells on their own do not contribute to suppression of autoimmunity in IL-2Rβ−/− mice and that Tregs regulate the expansion of CD8+ T cells in this model. From these results and the distribution of donor cells in recipient IL-2Rβ−/− mice (Fig. 1B), we conclude that control of autoimmunity after transfer of spleen cells is primarily attributed to donor CD4+ Tregs.
Figure 4.
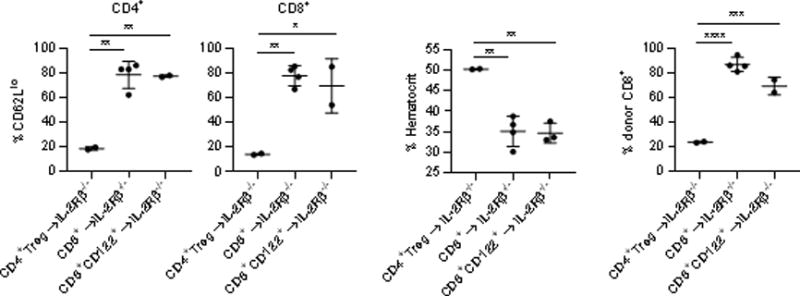
Donor CD8+ T cells do not suppress autoimmunity after transfer into IL-2Rβ−/− mice. The indicated cell populations (2 × 105) were transferred into neonatal IL-2Rβ−/− recipients and mice were assessed at 4-8 weeks post-transfer. (A) The proportion of activated CD62Ll° CD4+ and CD8+ recipient-derived splenic T cells, (B) the hematocrit, and (C) the proportion of the splenic CD8+ T cells compartment that was donor cell derived. Data were derived from two independent experiments.
Spectratype analysis reveals TCR repertoire narrowing in 10 and 2° IL-2Rβ−/− recipients
Spectratype analysis was performed for 8 Vβ subgroups after serial transfer of Tregs (Fig. 5A). Plots of CDR3 fragment lengths showed a typical polyclonal pattern for the input purified Tregs that were used for the initial transfers into IL-2Rβ−/− neonates. However, the plots for the CDR3s from Tregs purified after the 1° and 2° transfers showed skewing, consistent with repertoire narrowing. D-scores were calculated based on differences in individual peak heights when compared to a polyclonal reference sample, where a larger D-score represents a less diverse TCR repertoire. In comparison to low D-scores associated with input Tregs with a largely Gaussian distribution of CDR3s, a progressive increase in D-scores was noted for Vβ subgroups, either individually (Fig. 5B) or in aggregate (Fig. 5C), for the donor Tregs between the 1° and 2° recipients. Thus, although most 1° IL-2Rβ−/− recipients did not show any signs of autoimmunity, there was a substantial reduction in the diversity of their TCR repertoire. However, further narrowing of the TCRβ repertoire led to autoimmunity, as evidenced by spectratype analysis of the 2° recipients. Notably, the increase in the D-scores for donor Tregs between the 1° and 2° recipients was relatively modest when compared to that between input and 1° Tregs, suggesting that a relatively small decrease in the TCRβ repertoire diversity observed in Tregs from 2° recipients is associated with a failure to suppress polyclonal autoreactive T cells.
Figure 5.
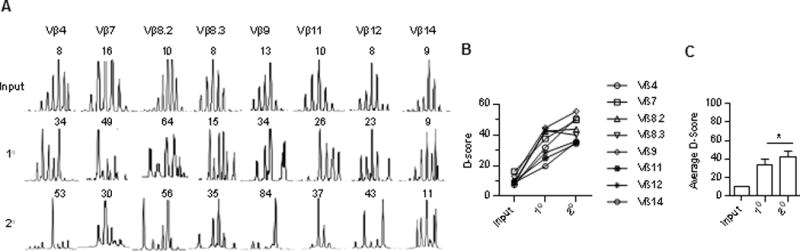
Spectratype analysis of selected TCRβ subgroups of donor Tregs from serial transferred IL-2Rβ−/− mice. 10 × 106 spleen cells were transferred into each recipient. (A) Representative plots of CDR3 lengths, where the number in each plot is the D-score. (B) D-scores for reference normal (Input) Tregs or donor Foxp3/RFP+ Tregs from 1° (n=4) and 2° (n=11) IL-2Rβ−/− mice. (C) Averaged D-scores for each sample calculated as: Average D-score = D-score for: Vβ4+Vβ 7+Vβ8.3+Vβ9+Vβ11+Vβ12+Vβ14/8. Data are from 2 independent experiments where the input spleen cells were pooled from 3 mice and used for the 1° recipients. Spleen cells from 5 different 1° donors were used for the 11 2° recipients.
Direct measure of the Treg TCRβ repertoire by deep sequencing
Selected samples of purified Tregs from two independent experiments were subjected to deep sequencing to evaluate the TCRβ diversity of the fresh input Tregs and those Tregs that engrafted in the 1° and 2° IL-2Rβ−/− recipients. The pedigree of the samples is shown in Fig. 6A. On average, 590,000 ± 90,000 (SEM) purified Tregs were assessed, which resulted in 490,000 ± 23,000 (SEM) productive in-frame sequences/sample (Fig. 6B, top left). Sequence coverage, i.e. the average number of reads that aligned to known reference bases, ranged from 7.5–42 and averaged 16.6 ± 2.8 (SEM). The percentage (Fig. 6B, top right) and number (Fig. 6B, bottom left) of unique productive sequences decreased as the Tregs underwent serial transfers into IL-2Rβ−/− mice. An assessment of repertoire clonality (Fig. 6B, lower right), where 1 is monoclonal, supports a reduction in the TCRβ repertoire as the Tregs undergo serial transfers. In addition, a plot of the most dominant clonotypes, as assessed by the V-region nucleotide sequences with the most reads, is consistent with increased clonal expansion and less repertoire diversity with increased serial transfer of Tregs (Fig. 6C).
Figure 6.
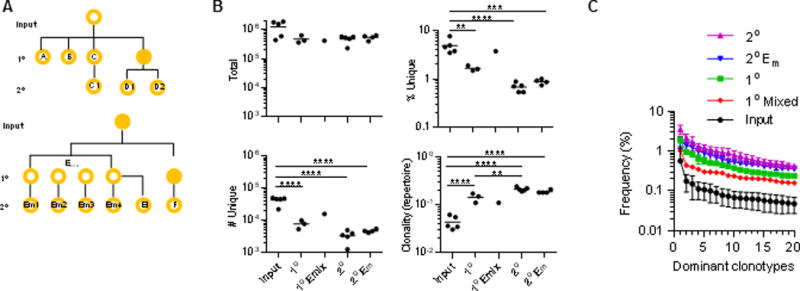
Deep sequencing of the Vβ TCR repertoire of donor Tregs from serial transferred IL-2Rβ−/− mice. (A) Pedigree of sample relationship, where a solid circle indicates an unsequenced sample. Em1-4 represent 4 individual 2° recipients that received 1° Tregs from 4 donor spleens that were premixed before transfer. E1 represents a distinct 2° recipient that received Tregs from a single 1° donor that was also used to prepare Emix. C1, D1, D2 and F represent individual 2° recipients whose TCRs were sequenced, where only the TCRs of the 1° donor for C1 was sequenced. (B) Repertoire characteristics of each sample. (C) The clone size, represented as % reads, of the 20 most dominant TCRβ VDJ nucleotide sequences for each group, as indicated.
The average number ± SEM of unique productive clonotypes was: 1° recipients, 7608 ± 1321; 2° recipients; 3309 ± 604; and 2° Em1–4 recipients, 4673 ± 279. These modest changes in TCRβ repertoire diversity, nevertheless, are biologically relevant because the health of the mice was clearly impacted (Fig. 2 and 3). Due to the narrowing of the TCRβ repertoires after transferring Tregs, sequencing depth increases such that the numbers of observed TCRβ clonotypes may approach the actual number. Thus, a population of Tregs with approximately 7608 ± 1321 distinct TCRβ clonotypes represents an estimate for the minimal TCRβ repertoire diversity required to suppress autoreactive T cells that escape thymic selection. This number of TCRβ clonotypes represents 18.0 ± 3.1% (SEM) or approximately 15–21% of the total TCRβ repertoire as on average 42,225 TCRβ clonotypes were identified after deep sequencing five independent samples of Tregs isolated from normal mice (Fig. 6B, top right).
A predictor of autoimmunity in the IL-2Rβ-deficient model is the proportion of activated CD44hi CD62Ll° conventional T cells in the spleen (11, 12). We plotted the number of donor Treg TCRβ clonotypes for individual 1° and 2° recipients, including Em 1–4, to the percentage of CD4+ Foxp3− CD44hi CD62Ll° T cells in the spleen (Fig 7A). This analysis revealed a moderate relationship between recipients with a more diverse TCRβ repertoire and a low proportion of activated CD4+ conventional T cells. In contrast, plots of the proportion of Tregs in the spleen (Fig. 7B) or within total splenic CD4+ T cells (Fig. 7C) showed a lower correlation with the levels of activated CD4+ T cells. These data provide further support that Treg TCRβ diversity, and not Treg proportions per se, is a more reliable predictor of autoimmunity in this model system.
Figure 7.
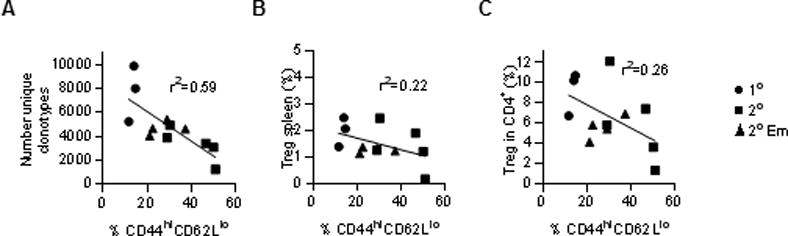
Comparison of donor Treg TCRβ diversity to symptoms of autoimmunity in IL-2Rβ−/− mice. For all 1° and 2° recipients, (A) the number of donor Treg TCRβ clonotypes, (B) the percentage of donor Tregs in the spleen or (C) within the CD4+ splenic T cells compartment were plotted against the percentage of activated CD4+ Foxp3− CD44hi CD62L+ T cells from the same recipient mouse. Linear regression analysis was performed for these data. Symbols in the graphs represent the origin of the data with respect to the types of recipients.
TCRβ repertoire relationships after serial transfer of Tregs to IL-2Rβ−/− recipients
Pairwise comparisons were made for all samples sequenced from the pedigree shown in Fig. 6A. An approximately 15–20% overlap in TCRβ repertoires was noted for comparisons between input Tregs and Tregs from 1° and 2° recipients or between Tregs from any 1° and 2° recipients from unrelated pedigrees (Fig. 8A). In contrast, a 50–70% overlap in TCRβ repertoires was noted between Tregs from 1° and 2° recipients of a related pedigree or among Tregs from the 2° Em1–4 recipients that received 1° Emix donor Tregs, i.e. 1° C vs 2° C1; 2° D1 vs. 2° D2; 1° Emix vs. 2° Em1–4, and 1° Emix vs. 2° Ei. These data suggest that useful TCRβs were selected in the autoimmune-free primary recipients and were retained by the 2° recipients, but autoimmunity develops perhaps due to further repertoire narrowing.
Figure 8.
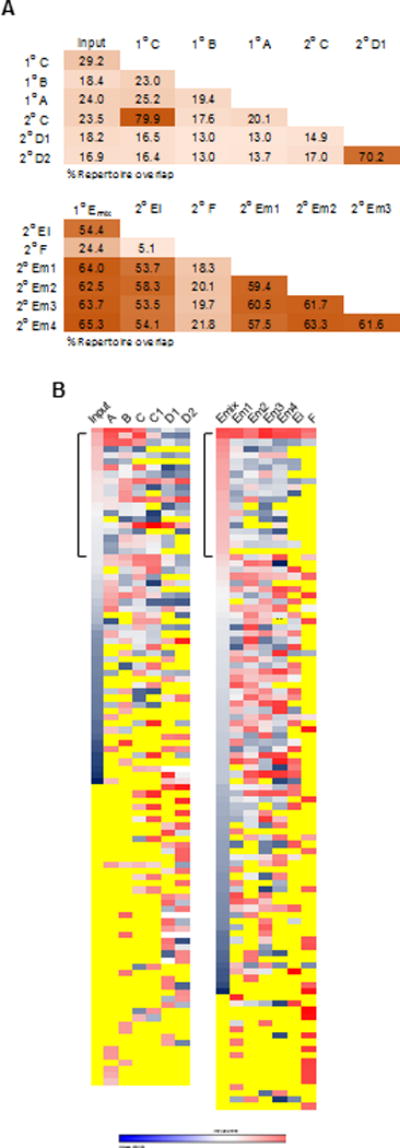
Characteristics of the Vβ TCR repertoire of donor Tregs from serial transferred IL-2Rβ−/− mice. (A) Repertoire overlap based on the total number of shared productive VDJ nucleotide sequence reads for two samples calculated vs. the total number of productive reads for those samples. (B) The relative frequency of the 20 most dominant clonotypes (nucleotide VDJ) was determined for each sample from both experiments. Heat maps were constructed that indicate the relative level of the each clonotype in all samples from low (blue) to high (red) frequency. Yellow indicates the absence of a clonotype. The bracket designates the top 20 clonotypes for the Input (left) and 1° Emix (right). Ei represents donor Tregs from an individual 2° recipients that received spleen cells from only one of the mice that were used to prepare Emix.
The top 20 TCRβ clonotypes were identified for each Treg sample based on the frequency of sequence reads. As each analysis contained 7 samples, 140 individual clonotypes were identified. However, because some of the sequences from one sample were also detected as a top 20 clone for a second sample, only 103 (Fig. 8B, left) and 127 (Fig. 8B, right) unique clonotypes were identified and compared. The distribution of these TCRβ clonotypes, i.e. their relative presence (blue-red heat map) or absence (yellow), was quantified for the entire repertoire of each sample (Fig. 8B). For Input and Emix, they are shown sequentially from highest to lowest frequency. However, this type of distribution is not possible in this comparison for the remaining samples; for example, some clonotypes in the Input were not detected in the repertoire of some of the other recipients while other clonotypes were more abundant in some recipients than found in the Input sample.
The top 20 TCRβ clonotypes from fresh input Tregs were readily found in most 1° and 2° recipients, sometimes even at a higher frequency after serial transfers (Fig. 8B, left). This strongly supports the notion that these clonotypes represent useful specificities and are selected after adoptive transfer into IL-2Rβ−/− mice. For the dominant clonotypes associated with the 1° and 2° recipients, a majority were found in the input TCRβ repertoire while 46% were not identified. This directly shows that some TCRβ clonotypes present at a low frequency in the input Tregs were selected and expanded upon transfer to IL-2Rβ−/− mice. The failure to detect 46% of the dominant clonotypes for 1° and 2° recipients in the input Treg TCRβ sequences suggests that the deep sequencing missed nearly half of the TCRβ clonotypes associated with the input sample. Thus, the 7608 ± 1321 donor Treg TCRβ clonotypes required to effectively suppress autoimmunity in 1° recipients more likely represents 8.1–11.3% of the total repertoire, not the 15–21% derived above by simply calculating the averge number of TCRβ clonotypes divided by the number of unique clonotypes observed for the input samples.
In the one experiments where we compared the dominant TCRβ clonotypes from 1° Emix to the repertoires of individual 2° Em1–4, clear evidence was provided for repertoire reshaping (Fig. 8B, right). For the top 20 clonotypes from 1° Emix, most were found in 2° Em1–4. However, these clonotypes were usually at a lower frequency in 2° Em1–4. Dominant clonotypes of 1° Emix were less frequently found in Tregs from the Ei 2° recipient, which received Tregs from only 1 of the 4 mice that comprised the 1° Emix. This result is expected because some of the dominant clonotypes in 1° Emix were probably derived from the other 3 donor mice. Furthermore, many of the dominant TCRβ clonotypes of Em1–4 were readily shared among each other, but were at a low frequency in the 1° Emix Tregs. In contrast, the dominant clones in the Treg TCRβ repertoire from the 2° F recipient showed much less repertoire sharing with the unrelated Treg repertoires of Emix or Em 1–4. Overall, these data and those in Fig. 6 indicate that considerable TCRβ repertoire narrowing and reshaping occurs in 1° recipients, but after this process occurs, the TCRβ repertoire is more moderately altered in 2° IL-2Rβ−/− recipients.
Discussion
The Treg TCR repertoire exhibits diversity that is equivalent to that of conventional T cells (1–3). The extent that this diversity is required to maintain peripheral T cell tolerance versus regulation of other immune responses remains unanswered. Past studies have demonstrated that when severe constraints were placed on the Treg TCR repertoire, e.g. use of Tregs that express a single TCR β-chain, autoimmunity was ineffectively controlled (6–9). In this study we addressed how much of the Treg TCRβ TCR repertoire is required to maintain peripheral T cell tolerance under conditions where unconstrained polyclonal autoreactive T cells lead to rapid systemic autoimmunity. IL-2Rβ−/− mice were used as a model because these mice contain only immature non-functional Foxp3l° Tregs that lead to rapid fatal autoimmunity (15, 18). When compared to scurfy and Foxp3-deficient mice, germline IL-2Rβ−/− mice succumb to autoimmunity later in life (typically 8–12 weeks of age). However, the tempo of this disease is accelerated when IL-2Rβ signaling is conditionally ablated only in Treg cells (16), probably because in these latter mice, the autoreactive T cells normally respond to IL-2 and IL-15 and contain more aggressive self-reactive T cells. Adoptively transferred WT Tregs effectively control this autoimmunity, represent the only Tregs detected in these recipient, and persist long-term (10–12), providing a model system to assess the Treg repertoire in maintaining peripheral self-tolerance.
By following the fate of the Treg TCRβ repertoire in serial transferred IL-2Rβ−/− recipients, we established a point where Treg TCRβ diversity of the transferred cells was insufficient and associated with several characteristic features of autoimmunity in untreated IL-2Rβ−/− mice. Thus, after a 1° transfer of Tregs into IL-2Rβ−/− mice, almost all recipients were autoimmune-free whereas after a 2° transfer essentially all recipients exhibited clear signs of autoimmune disease. Since highly purified splenic Tregs, which are largely thymic-derived (4), persist long-term and prevent autoimmunity when transferred to IL-2Rβ−/− mice (10–12), we assume in this study that thymic-derived Tregs represent the main protective component. However, since total spleen cells were transferred, we cannot rule out that some of the persistent donor Tregs are peripherally induced.
This study using normal polyclonal Tregs has defined that ~8–11% of the Treg TCRβ repertoire is sufficient to suppress autoimmunity in 1° recipients in this model system. This value is based on TCRβ sequencing of input donor Tregs from the spleen and then the engrafted purified donor splenic Tregs in 1° recipients. As we focused on the spleen, our measure of the TCRβ repertoire does not take into consideration that unique Treg TCRβ specificities may be uniquely expressed within lymphoid tissues draining distinct anatomical sites (4). The 8–11% value is derived based on a direct calculation of the number of unique TCRβ clonotypes associated with Tregs that engraft and persist in 1° recipients in relationship to the number of unique clonotypes measured for the input cells prior to adoptive transfer. This value was arrived at after correction based on the observation that almost 50% of the dominant TCRβ clonotypes from the Tregs in the 1° recipients were not found in the input cells. The reasons for this difference likely reflect that the pool of Tregs used for the input cannot represent the entire repertoire as another aliquot of these cells was used for the transfer into IL-2Rβ−/− neonates and almost certainly contained specificities not included in the aliquot sequenced. In addition, the deep sequencing of the input cells may not have been sufficient to detect all TCRβ clonotypes. In any case, from these data, ~8–11% of the TCRβ repertoire represents an estimate for the amount of TCRβ Treg diversity required to maintain self-tolerance in control of polyclonal autoreactive T cells. However, this calculation does not fully account for some uncertainty concerning the TCR repertoire diversity of the initial input Tregs and potential donor TCRβ specificities that are unique to draining lymphoid tissues that are not represented in the spleen.
An interesting feature of this study is a rather small decrease in the TCRβ clonotypes (approximately 2.3-fold) was noted in the persistent donor Tregs obtained from 1° vs. 2° IL-2Rβ−/− recipients, yet this decrease in the Treg TCRβ repertoire was associated with autoimmunity. This notion is supported by both the spectratype analysis and deep sequencing. Analysis of these data correlated TCRβ clonotype number as the best predictor of autoimmunity when using a sensitive measure of autoimmunity, i.e. the presence of activated CD4+ T cells. In addition, in our model system modest differences in the number of donor 1° Tregs transferred into 2° recipients also showed some association with the extent of autoimmunity, i.e. fewer activated CD4+ T cells in mice that received more Tregs, which by necessity should increase TCR diversity. Thus, we conclude that this modest TCRβ repertoire narrowing accounts for the failure to effectively suppress autoimmunity in 2° recipients and that the diversity of the donor Treg TCRβ repertoire in the 1° recipients is near the limits required to effectively control autoimmunity.
There remains, nevertheless, several caveats to this conclusion. First, substantial selection and reshaping of the Treg TCR repertoire occurs after transfer of Tregs into a 1° recipients. Thus, the failure of Tregs from a 1° transfer to control autoimmunity in a 2° recipient might reflect that the selected Treg TCRβ repertoire of 1° recipients have important holes in their repertoire and that an unselected Treg TCRβ repertoire of similar restricted diversity might suppress autoimmunity. Second, Tregs from 1° recipients acquired a more activated phenotype (19) and this shift toward eTregs might also contribute to failed control of autoimmunity in the 2° recipients. Third, donor CD8+ T cells along with Tregs are selected in 1° recipients and are transferred into the 2° recipients. Although our data rule out that CD8+ T cells substantially suppress autoimmunity in IL-2Rβ−/− mice, these CD8+ T cells might promote autoimmunity in 2° recipients.
Although we cannot entirely rule out these possibilities, the ability to more effectively suppress autoimmunity in 2° recipients that received donor Tregs that were premixed from several 1° recipients provides some evidence that argues against these concerns. Premixing spleen cells from the 1° recipients inherently increased the diversity of the TCRβ repertoire while retaining its enrichment of cells with an eTreg phenotype. Indeed, the TCRβ repertoire of the persistent Tregs from these 2° recipients, which received mixed donor cells and showed mild symptoms of autoimmunity, contained an intermediate number of TCRβ clonotypes when compared to 1° and secondary 2° recipients that received Tregs from an individual donor. These premixed transfers also contained donor CD8+ T cells, but these cells did not lead to severe autoimmunity. Furthermore, when considering the frequency of activated CD4+ CD44hi CD62Ll° conventional T cells, where an increased level is an early sign of autoimmunity, the number of Treg TCRβ clonotypes is a better predictor of self-tolerance than the proportion of Tregs found in IL-2Rβ−/− recipient mice. However, these data from a single mixing experiment with one pool of donor splenocytes does not provide firm data to refine the estimate for Treg TCRβ diversity needed to suppress autoimmunity and does not take into account the potential for increased repertoire diversity outside of the spleen related to unique specificities in tissues and lymph nodes (4) that might contribute to self-tolerance.
Treg exhaustion is also unlikely involved to account for autoimmunity in 2° IL-2Rβ−/− recipients. Our experience is that a single transfer of Tregs into neonatal IL-2Rβ−/− mice is sufficient for life-long persistence of the donor Tregs cells at normal proportions when compared to total CD4+ T cells (10–12). Notably, a normal level of donor Tregs was also observed for most 2° recipients. Thus, approximately 8–11% of the TCRβ clonotypes expressed by donor Tregs in the spleen is required to maintain self-tolerance in IL-2Rβ−/− recipients and represents an estimate of the TCRβ repertoire diversity required to effectively maintain self-tolerance in an environment that contains polyclonal autoreactive T cells with distinctive specificities due to random generation of their TCRs. This estimate assumes that this relationship holds for the unique TCRβ chains found in lymph nodes and tissues sites before and after transfer into 1° IL-2Rβ−/− recipients but were not assayed in this study.
This process, which we have identified for IL-2Rβ-deficient recipients, likely also occurs under normal physiological conditions. Our data support a model where high diversity of the Treg TCR repertoire provides an effective means over a lifetime to adapt and change in a way to select TCR specificities that are optimized to suppress T cells directed toward self-antigens and commensal micro-organisms. The specificities expressed by auto-reactive T cells are not fixed, but quantitatively and qualitatively evolve as the immune system responds to foreign and self-antigens. Even though only a fraction of the Treg TCRβ repertoire is very effective in maintaining tolerance, the high diversity of the Treg TCR repertoire provides a mechanism with excess spare capacity geared to ensure tolerance. An additional benefit of this diversity is that it also provides specificities for Tregs to participate in regulation of other immune responses, including those to foreign antigens.
Acknowledgments
We thank Kevin Toomer for critically reading this manuscript and the flow cytometry cores of the Sylvester Comprehensive Cancer Center and the Diabetes Research Institute at the University of Miami for help with FACS analysis and sorting.
This work was supported by National Institutes of Health Grants R01 AI055815
Abbreviations used in this article
- D
diversity
- eTreg
effector regulatory T cell
- Treg
regulatory T cell
- WT
wild-type
- 1°
primary
- 2°
secondary
Footnotes
Disclosures
The authors declare no competing financial interest.
References
- 1.Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006;7:401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- 2.Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity. 2006;25:249–259. doi: 10.1016/j.immuni.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 3.Wong J, Mathis D, Benoist C. TCR-based lineage tracing: no evidence for conversion of conventional into regulatory T cells in response to a natural self-antigen in pancreatic islets. J Exp Med. 2007;204:2039–2045. doi: 10.1084/jem.20070822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lathrop SK, Santacruz NA, Pham D, Luo J, Hsieh CS. Antigen-specific peripheral shaping of the natural regulatory T cell population. J Exp Med. 2008;205:3105–3117. doi: 10.1084/jem.20081359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, Hsieh CS. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478:250–254. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adeegbe D, Matsutani T, Yang J, Altman NH, Malek TR. CD4+ CD25+ Foxp3+ T regulatory cells with limited TCR diversity in control of autoimmunity. J Immunol. 2010;184:56–66. doi: 10.4049/jimmunol.0902379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fohse L, Suffner J, Suhre K, Wahl B, Lindner C, Lee CW, Schmitz S, Haas JD, Lamprecht S, Koenecke C, Bleich A, Hammerling GJ, Malissen B, Suerbaum S, Forster R, Prinz I. High TCR diversity ensures optimal function and homeostasis of Foxp3+ regulatory T cells. Eur J Immunol. 2011;41:3101–3113. doi: 10.1002/eji.201141986. [DOI] [PubMed] [Google Scholar]
- 8.Hori S, Haury M, Coutinho A, Demengeot J. Specificity requirements for selection and effector functions of CD25+4+ regulatory T cells in anti-myelin basic protein T cell receptor transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8213–8218. doi: 10.1073/pnas.122224799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishio J, Baba M, Atarashi K, Tanoue T, Negishi H, Yanai H, Habu S, Hori S, Honda K, Taniguchi T. Requirement of full TCR repertoire for regulatory T cells to maintain intestinal homeostasis. Proc Natl Acad Sci U S A. 2015;112:12770–12775. doi: 10.1073/pnas.1516617112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adeegbe D, Bayer AL, Levy RB, Malek TR. Cutting edge: allogeneic CD4+CD25+Foxp3+ T regulatory cells suppress autoimmunity while establishing transplantation tolerance. J Immunol. 2006;176:7149–7153. doi: 10.4049/jimmunol.176.12.7149. [DOI] [PubMed] [Google Scholar]
- 11.Adeegbe D, Levy RB, Malek TR. Allogeneic T regulatory cell-mediated transplantation tolerance in adoptive therapy depends on dominant peripheral suppression and central tolerance. Blood. 2010;115:1932–1940. doi: 10.1182/blood-2009-08-238584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rβ-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 2002;17:167–178. doi: 10.1016/s1074-7613(02)00367-9. [DOI] [PubMed] [Google Scholar]
- 13.Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28:100–111. doi: 10.1016/j.immuni.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burchill MA, Yang J, Vang KB, Moon JJ, Chu HH, Lio CW, Vegoe AL, Hsieh CS, Jenkins MK, Farrar MA. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity. 2008;28:112–121. doi: 10.1016/j.immuni.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng G, Yu A, Dee MJ, Malek TR. IL-2R signaling is essential for functional maturation of regulatory T cells during thymic development. J Immunol. 2013;190:1567–1575. doi: 10.4049/jimmunol.1201218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, Gasteiger G, Feng Y, Fontenot JD, Rudensky AY. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. 2016 doi: 10.1038/ni.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci U S A. 2005;102:5126–5131. doi: 10.1073/pnas.0501701102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, Schmits R, Simard JJL, Ohashi PS, Griesser H, Taniguchi T, Paige CJ, Mak TW. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor β. Science. 1995;268:1472–1476. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- 19.Toomer KH, Yuan X, Yang J, Dee MJ, Yu A, Malek TR. Developmental progression and interrelationship of central and effector regulatory T cell subsets. J Immunol. 2016;196:3665–3676. doi: 10.4049/jimmunol.1500595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorochov G, Neumann AU, Kereveur A, Parizot C, Li T, Katlama C, Karmochkine M, Raguin G, Autran B, Debre P. Perturbation of CD4+ and CD8+ T-cell repertoires during progression to AIDS and regulation of the CD4+ repertoire during antiviral therapy. Nat Med. 1998;4:215–221. doi: 10.1038/nm0298-215. [DOI] [PubMed] [Google Scholar]
- 21.Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, Steen MS, LaMadrid-Herrmannsfeldt MA, Williamson DW, Livingston RJ, Wu D, Wood BL, Rieder MJ, Robins H. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun. 2013;4:2680. doi: 10.1038/ncomms3680. [DOI] [PubMed] [Google Scholar]
- 22.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, Carlson CS. Comprehensive assessment of T-cell receptor β-chain diversity in alphabeta T cells. Blood. 2009;114:4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emerson RO, Sherwood AM, Rieder MJ, Guenthoer J, Williamson DW, Carlson CS, Drescher CW, Tewari M, Bielas JH, Robins HS. High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J Path. 2013;231:433–440. doi: 10.1002/path.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stewart JJ, Lee CY, Ibrahim S, Watts P, Shlomchik M, Weigert M, Litwin S. A Shannon entropy analysis of immunoglobulin and T cell receptor. Mol Immunol. 1997;34:1067–1082. doi: 10.1016/s0161-5890(97)00130-2. [DOI] [PubMed] [Google Scholar]
- 25.Bayer AL, Yu A, Adeegbe D, Malek TR. Essential role for interleukin-2 for CD4+CD25+ T regulatory cell development during the neonatal period. J Exp Med. 2005;201:769–777. doi: 10.1084/jem.20041179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan X, Cheng G, Malek TR. The importance of regulatory T-cell heterogeneity in maintaining self-tolerance. Immunol Rev. 2014;259:103–114. doi: 10.1111/imr.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim HJ, Cantor H. Regulation of self-tolerance by Qa-1-restricted CD8+ regulatory T cells. Sem Immunol. 2011;23:446–452. doi: 10.1016/j.smim.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]