

Abstract
Peptidoglycan (PG) protects bacteria from osmotic lysis, and its biogenesis is a key antibiotic target. A central step in PG biosynthesis is to flip the lipid-linked PG precursor lipid II across the cytoplasmic membrane for subsequent incorporation into peptidoglycan. MurJ, part of the multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) transporter superfamily, was recently shown to carry out this process. However, our understanding of how MurJ flips lipid II and how MOP transporters operate in general remains limited by a lack of structural information. Here we present a crystal structure of MurJ from Thermosipho africanus in an inward-facing conformation at 2.0 Å resolution. A hydrophobic groove is formed by two C-terminal transmembrane helices, which leads into a large central cavity that is mostly cationic. Our studies not only provide the first structural glimpse of MurJ but also suggest that alternating access is important for MurJ function, which may be applicable to other MOP superfamily transporters.
INTRODUCTION
The building block of peptidoglycan, GlcNAc-MurNAc-pentapeptide, is synthesized in the cytosol and attached to the inner leaflet of the cytoplasmic membrane as the lipid-linked precursor undecaprenyl diphosphate-MurNAc-pentapeptide-GlcNAc, known as lipid II1,2. Subsequently, lipid II is flipped from the cytoplasmic side to the periplasmic side of the membrane for polymerization into peptidoglycan3,4. While the precursor synthesis and polymerization steps of peptidoglycan biogenesis are well studied, lipid II flipping is poorly understood and flippase candidates MurJ, Amj, and FtsW have been identified only recently5–9.
Recent studies strongly suggest that MurJ is the long-sought-after lipid II flippase7,9,10. Depletion of MurJ (formerly known as MviN) results in an accumulation of peptidoglycan precursors, a decrease in peptidoglycan biosynthesis, and ultimately cell lysis in Escherichia coli and Burkholderia cenocepacia7,9,11. Notably, MurJ-dependent flipping of radiolabeled lipid II was detected in E. coli cells and spheroplasts10. Furthermore, the FtsW paralog RodA was recently characterized to be a peptidoglycan glycosyltransferase12.
MurJ belongs to the mouse virulence factor (MVF) family of multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) superfamily transporters, which also includes the multidrug and toxic compound extrusion (MATE) transporter and eukaryotic oligosaccharidyl-lipid flippase (OLF) families13. In addition to the canonical MOP transporter core consisting of 12 transmembrane helices (TMs), MurJ has two additional C-terminal TMs (13 and 14) of unknown function. The outward-facing conformation of E. coli MurJ has been probed extensively by homology modeling, in vivo substituted cysteine-accessibility method and mutagenesis studies, revealing a solvent-exposed cavity that is essential for function14,15. However, our understanding of how MurJ can flip such a large (~1900 Da) and negatively charged lipid substrate remains elusive due to a lack of crystal structures. Another intriguing question is whether MurJ-dependent lipid II flipping occurs via an alternating-access mechanism; recent studies have suggested that alternating access is not employed for lipid-linked oligosaccharide flipping by the ABC transporter PglK16. Furthermore, among the MOP transporter superfamily, only MATE family transporters have been crystallized to date, all in outward-facing conformations17–21. As such, the molecular mechanism of alternating-access transport by MOP transporters is still poorly understood.
To understand the structural basis of lipid II flipping by MurJ, we carried out structural and mechanistic studies of MurJ. We report the crystal structure of MurJ. Our structural analyses, combined with mutagenesis studies, have allowed us to propose a model of lipid II flipping by MurJ. Our studies provide foundational data for an atomic-level understanding of MurJ function.
RESULTS
Overall architecture of MurJ
We solved the structure of MurJ from the hyperthermophilic bacterium Thermosipho africanus (MurJTA) to 2.0 Å resolution from crystals grown in lipidic cubic phase. Experimental phases to 3.5 Å resolution were determined by single wavelength anomalous dispersion from selenomethionine-substituted crystals (Supplementary Fig. 1). The electron density map allowed residues 4 to 470 (out of 475 residues) to be built unambiguously, and the final model was refined to good geometry and a free R factor of 21.4% (Table 1). The locations and identities of ions in the structure were deduced from anomalous scattering experiments (Supplementary Fig. 2 and Supplementary Table 1).
Table 1.
Data collection and refinement statistics
| Native (PDB 5T77)* | SeMet† | |
|---|---|---|
| Data collection | ||
| Space group | C2 | C2 |
| Cell dimensions | ||
| a, b, c (Å) | 94.16, 99.57, 74.66 | 93.86, 99.74, 75.67 |
| α, β, γ (°) | 90, 112.91, 90 | 90, 101.84, 90 |
|
Peak
|
||
| Wavelength (Å) | 0.9791 | 0.9791 |
| Resolution (Å) | 100–2.00 (2.07–2.00)a | 70–3.15 (3.26–3.15)a |
| Rpim (%) | 6.5 (55.0) | 4.2 (25.0) |
| I/σ(I) | 9.47 (1.06) | 30.8 (4.10) |
| CC1/2 | 0.99 (0.50) | 1.00 (0.87) |
| Completeness (%) | 98.0 (94.0) | 99.5 (95.0) |
| Redundancy | 4.2 (2.2) | 50.4 (30.5) |
| Refinement | ||
| Resolution (Å) | 34.2–2.00 (2.07–2.00) | |
| No. reflections | 41992 (4018) | |
| Rwork / Rfree (%) | 18.9 / 21.4 | |
| No. atoms | 4404 | |
| Protein | 3743 | |
| Monoolein/PEG/ | 453/48/5/1/3/2 | |
| Cl−/Zn2+/Ca2+/Na+ | ||
| Water | 149 | |
| B factors | ||
| Protein | 38.2 | |
| Monoolein/PEG/ | 68.5 | |
| Cl−/Zn2+/Ca2+/Na+ | ||
| Water | 43.2 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.003 | |
| Bond angles (°) | 0.55 |
Merged from 3 native crystals grown in LCP.
Merged from 12 SeMet crystals grown in LCP.
Values in parentheses are for highest-resolution shell.
Our structure of MurJTA is strikingly different from existing MATE structures. The structure of MurJTA has the N-lobe (TMs 1–6) and C-lobe (TMs 7–14) arranged in an inward-facing N-shape conformation (Fig. 1a–b) rather than the outward-facing V-shape conformation observed in all existing MATE transporter structures17–21. Cytoplasmic orientation of the termini and the positive-inside surface electrostatics (Supplementary Fig. 1e) support the inward-facing topology. Although strong pseudo two-fold rotational symmetry is observed between the two lobes in the MATE structures17,19,20, the symmetry between the N-lobe and the core C-lobe (TMs 7–12) of MurJTA is significantly distorted (Fig. 1c). In particular, the conformations of TM1 and TM2 are very different from those of the symmetry-related TM7 and TM8 (Fig. 1c). TM1 extends out from the transporter core instead of interacting with the C-lobe as in other MATE structures, while TM2 is broken at the Gly(Ala) 56–Glu 57–Gly 58–Ala 59 motif that is conserved in Gram-negative bacteria (Supplementary Note 1), allowing this region to protrude into the central cavity. In addition, both lobes of the core transport domain of MurJTA are substantially wider and shorter than those in MATE transporters (Fig. 1c).
Figure 1. Architecture of MurJTA. a.
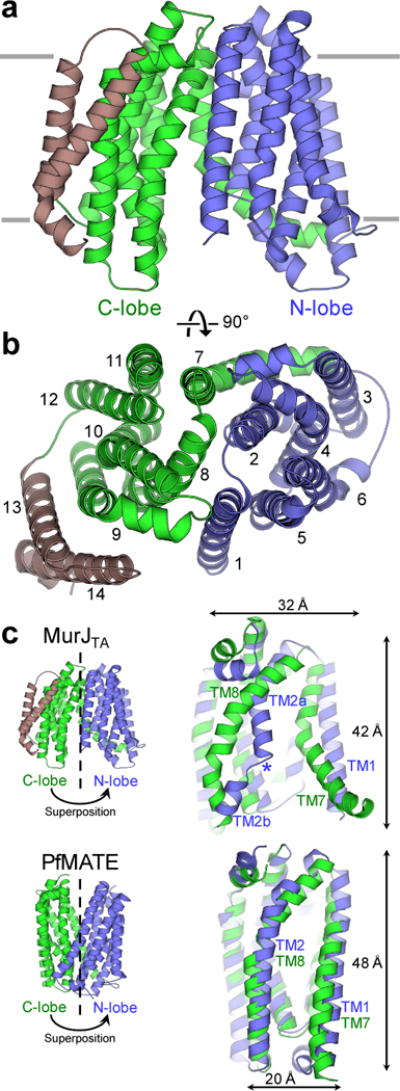
View from within the membrane. The N-lobe (TMs 1–6) and C-lobe (TMs 7–12) of the core transport domain are denoted in blue and green respectively, while the additional TMs 13 and 14 are shown in brown. b, Extracellular view of MurJTA, showing the arrangement of the N-lobe (TMs 1–6, blue) and C-lobe (TMs 7–12, green). c, Structural superposition between the N-lobe (blue) and C-lobe (green) of MurJTA and of PfMATE (PDB ID: 3VVN), showing the broken symmetry in MurJTA, in particular between TMs 1 and 7 as well as between TMs 2 and 8. The core transport domain of MurJTA is also shorter and wider than that of PfMATE. The helix-breaking Gly56–Glu57–Gly58–Ala59 motif in TM2 is denoted by the blue asterisk.
The hydrophobic groove and central cavity of MurJTA
C-terminal TMs 13 and 14 extend out from the transporter core and are oriented beside TM9. These two additional TMs, absent in MATE proteins, form a curved groove (Fig. 2a) that is ~20 Å long. This groove is lined mostly with hydrophobic residues from TMs 1, 8, 9, and 14. The hydrophobic groove penetrates into the central transport cavity via a membrane portal located between TMs 1 and 8 (Fig. 2b). Most of the residues lining the portal have small side chains with a notable exception of Arg 18, which is essential for E. coli MurJ function15. Beyond the portal, the groove opens up into a large inward-facing central cavity ~20 Å wide and penetrating up to ~20 Å into the membrane (Fig. 2c). This cavity can be subdivided into a proximal site (directly connected to the hydrophobic groove) and a distal site (far from the groove). The proximal site is highly positively charged (Fig. 2d), mainly due to the presence of Arg 24, Arg 52 and Arg 255, counterparts of which were found to be important for the function of E. coli MurJ15. In our structure, a chloride ion, identified by anomalous signal from a bromide-soaked crystal, is located inside the proximal site (Fig. 2c and Supplementary Fig. 2b). Whether this chloride ion is physiologically relevant remains unknown: the chloride might be nonspecifically bound to MurJ due to the high concentration of chloride in the crystallization drop (containing 400 mM CaCl2).
Figure 2. The hydrophobic groove of MurJTA is linked to the central cavity. a.
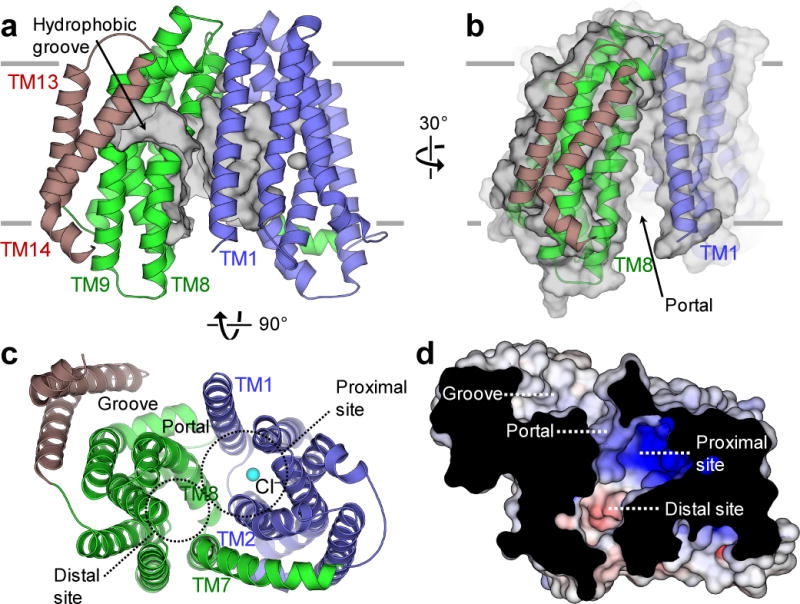
TMs 13 and 14 (brown) form a hydrophobic groove (surface shown in gray). b, The hydrophobic groove leads into the central cavity through a portal between TMs 1 and 8. c, The central cavity is formed mainly by TMs 1, 2, 7, and 8. This large cavity can be subdivided into proximal (near groove) and distal (away from groove) sites. A chloride ion is found in the proximal site (cyan sphere). d, The proximal site is strongly cationic, whereas the distal site is mildly anionic.
Model of lipid II binding to MurJTA
Although we observed unassigned electron density peaks at both the membrane portal (Supplementary Fig. 3a) and the distal site of the central cavity (Supplementary Fig. 3b), the quality of the density peaks was not sufficient to assign them unambiguously. We therefore used a ligand docking approach to test whether the binding of lipid II to our MurJTA structure was feasible. In the docking result with the highest score, lipid II was docked into the groove and cavity with good geometry and feasible electrostatic interactions (Fig. 3a). In this model, the undecaprenyl tail of lipid II associates with the hydrophobic groove, the diphosphate and sugar moieties are placed in the proximal site, and the pentapeptide moiety is located in the distal site of the cavity. In particular, the unassigned electron density at the distal site was where the pentapeptide moiety was located in our docking result. Based on this observation, we could build the pentapeptide moiety partially into the electron density (Supplementary Fig. 3c), but it was excluded from refinement due to the uncertainty of our assignment.
Figure 3. Model of lipid II binding to MurJTA. a.
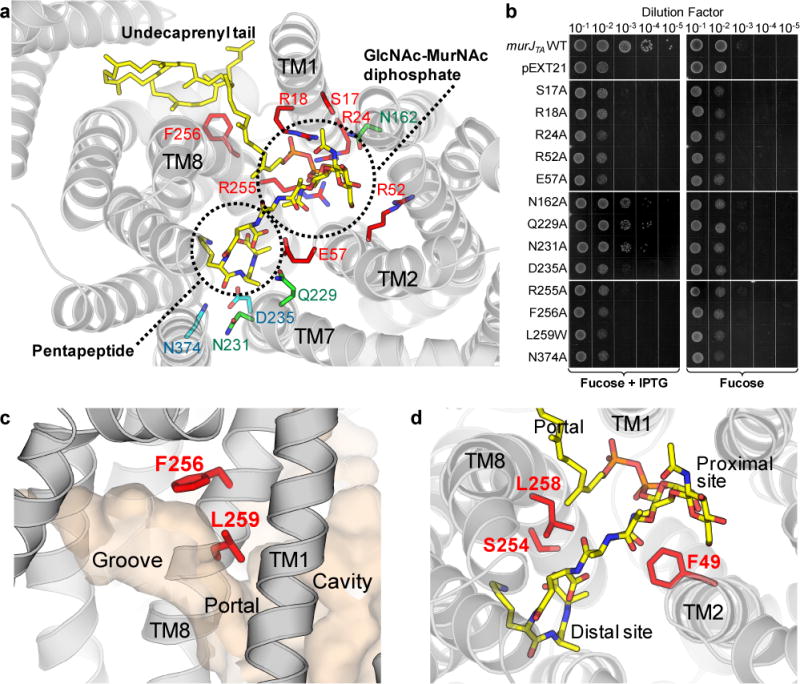
A model of lipid II (yellow sticks) docked into the structure of MurJTA. Each chemical moiety of lipid II is indicated. Residues that were mutated to alanine and subjected to the complementation assay in panel b are shown as sticks. Residues essential for the function of MurJTA are colored red, non-essential residues are colored green, while residues of which the alanine mutants failed to express are shown in cyan. b, Functional complementation data for MurJTA wild-type and mutants in E. coli strain NR1154. Plasmid pEXT21 encoding either MurJTA (wild-type or mutant) or without an insert, were transformed into NR1154 cells. Cells were subsequently depleted of endogenous MurJ by serial dilution and plating with the anti-inducer fucose in the absence of arabinose, and MurJTA was expressed by the addition of IPTG (representative of n = 3, technical replicates). c, Occluding the portal (by substituting Leu 259 with a bulky tryptophan residue) or perturbing the groove surface (by mutating Phe 256 to alanine) results in a protein that is unable to complement MurJ activity. d, Mapping of MTSES-sensitive positions from E. coli MurJ, where cell lysis or cell shape defects were observed upon MTSES treatment of cysteine mutants10, to the structure of MurJTA. These positions (49, 254, 258; red sticks) do not appear to interact with the diphosphate or sugar groups and are not strongly conserved (Supplementary Note 1).
Guided by our docking result, we next tested conserved residues in the portal and central cavity for their importance to lipid II binding by performing functional complementation assays as previously described (Fig. 3b)7,11. We utilized E. coli strain NR1154 in which the expression of endogenous MurJ (murJEC) is under the control of arabinose7. Introduction of a plasmid containing murJTA under the control of an IPTG-inducible promoter allowed growth of this strain in the absence of arabinose, provided that IPTG was present (Fig. 3b). Mutation of conserved residues (Ser 17, Arg 18, Arg 24, Arg 52, and Arg 255) at the proximal site failed to complement MurJEC function, consistent with the idea that these residues are important for recognizing the diphosphate and/or sugar moieties of lipid II (Fig. 3a). Occluding the membrane portal (by mutating Leu 259 to tryptophan) or changing the shape of the groove (by mutating Phe 256 to alanine) also led to loss of complementation, suggesting disruption of lipid II binding (Fig. 3c). As failure to complement could alternatively be due to loss of protein expression, we analyzed total membrane fractions by Western blot. All mutant MurJTA proteins expressed except D235A and N374A, which we removed from our analysis (Supplementary Fig. 4).
We next analyzed previous in vivo chemical genetics data in the context of our structure to gain further insight into lipid II binding. Ruiz and coworkers found that while a single cysteine mutant of E. coli MurJ (MurJEC A29C) was functional, addition of the membrane impermeant cysteine-reactive agent 2-sulfonatoethyl methanethiosulfonate (MTSES) inhibited flippase activity and led to cell lysis10. They identified a total of five positions that exhibited a similar MTSES-dependent phenotype in MurJEC10,14. Because we showed that MurJTA can complement MurJEC and that the essential residues are conserved between the two orthologs, we mapped these residues to the structure of MurJTA. Three of the residues (Phe 49, Ser 254, and Leu 258) are located at the junction between the proximal and distal sites in the central cavity (Fig. 3d) but are not strongly conserved (Supplementary Note 1), suggesting that they do not directly participate in lipid II binding. Consistent with our model, mutation to cysteine at these positions would not affect lipid II binding but adding a bulky adduct from MTSES would likely obstruct lipid II binding (Fig. 3d). However, we caution against excessive interpretation of our docking and complementation results, as these experiments are not direct measurements of lipid II binding. We performed these experiments to refine our hypothesis of where the different chemical moieties of lipid II would likely be located, not to propose a definitive structural model of lipid II bound to MurJ.
Importance of alternating access for lipid II flipping by MurJ
Because our MurJTA structure is the first structure of a MOP transporter captured in an inward-facing conformation, it offers an opportunity to study the possibility of an alternating-access transport mechanism. We observed from our inward-facing structure that TM7 is bent by ~45° about Ser 228 as opposed to running straight in the outward-facing MATE structures. As most MurJ/MATE sequences have either a serine or a threonine residue around this region (Supplementary Note 1), we hypothesized that bending of TM7 about this residue might play a role in the transition to the outward-facing state. We generated an outward-facing model of MurJTA (Supplementary Data Set 1) by splitting the model at the middle of TM7 (Ser 228) and aligning the N-terminal fragment (TM1–TM7a) and the C-terminal fragment (TM7b–TM12) as rigid bodies to the structure of PfMATE (PDB ID 3VVN), which was chosen as it is the highest resolution MATE structure published20. The resulting model aligned better at the C-lobe than at the N-lobe. Because of the substantial structural difference between MurJ and MATE transporters, our outward-facing model generated based on the MATE structure should be interpreted with caution.
To test the validity of our outward-facing model, we mapped those five positions that are functionally sensitive to MTSES10 to the inward-facing structure and outward-facing model. All five positions are accessible from the periplasm in the outward-facing model but not in the inward-facing structure, showing that our model is consistent with the MTSES data (Fig. 4a,b). Notably, mapping two MTSES-sensitive positions located at the periplasmic ends of TM1 (A29) or TM8 (S248) suggests that alternating access is important for lipid II flipping. In our inward-facing MurJ structure, these positions are located at the interface between the N- and C-lobes, thereby playing important roles in occluding access to the periplasmic side. MTSES adduct formation at these sites likely disrupted the inward-facing state of MurJ, thereby trapping MurJ in the outward-facing state, resulting in disruption of lipid II flipping and cell lysis (Fig. 4c,d). An alternate hypothesis where the adducts occlude the outward-facing cavity is ruled out because mutations of these sites to large polar or charged amino acids (but smaller than the adduct) other than cysteine also led to cell lysis10. These data suggest both the inward-facing and outward-facing states exist for MurJ, and that alternating access might be important for lipid II flipping.
Figure 4. Putative mechanism of lipid II flipping by MurJ.
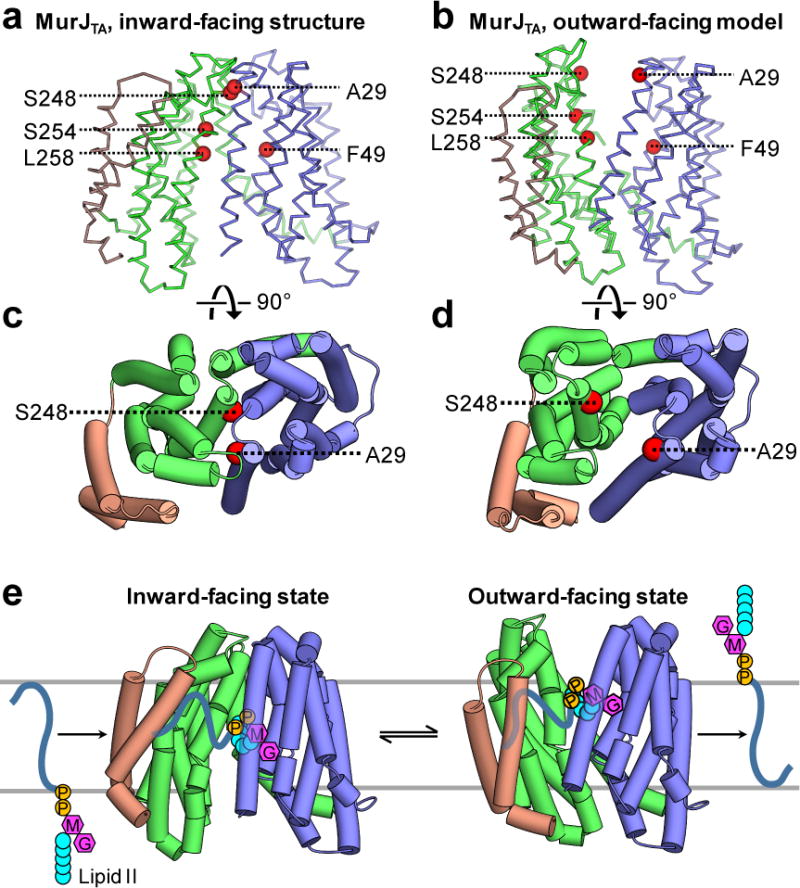
Previously reported MTSES-sensitive positions (red spheres) that resulted in loss of MurJ function upon MTSES treatment10 were mapped to the structure of MurJTA. a, These residues are not accessible to the membrane-impermeable agent MTSES in the inward-facing state. b, In the outward-facing model (Supplementary Data Set 1), all of these residues are exposed to the periplasm, explaining the MTSES-sensitivity of cysteine substitutions at these positions. Both Ala 29 and Ser 248 are located at the interface between the N-lobe and the C-lobe in the inward-facing structure of MurJTA (c), suggesting that adducts at these positions would trap the protein in the outward-facing state (d) by sterically blocking transition to the inward-facing state. e, Proposed rocker-switch model of lipid II flipping by MurJ. The substrate is captured in the central cavity between the N-lobe and C-lobe. Rocker-switch motion of both lobes translocates lipid II across the membrane, which is then released. The hydrophobic groove formed by TMs 13 and 14 of MurJ facilitates capture of the undecaprenyl tail, which remains associated with the groove during transition to the outward-facing state.
DISCUSSION
While MATE proteins have been characterized to catalyze either Na+ or H+-coupled antiport transport of substrates17–20, it is unclear whether MurJ requires any counter-ion for lipid II flipping. Unfortunately, our structure does not provide an answer to this question. Most of the ions observed in the structure are unlikely to function as the counter-transporting ions for lipid II flipping, as they were located outside the central cavity and are coordinated mostly by water molecules (Supplementary Fig. 2). This does not rule out the involvement of Na+ or H+ as they are not only difficult to be identified, but also likely to be bound only in the outward-facing state. However, the chloride ion in the central cavity and a zinc ion at the beginning of TM7 might be functionally important. The chloride ion (Cl− 1, Supplementary Fig. 2b) is located in the proximal site near Arg 24 and Arg 255, which were critical for the function of both MurJTA and E. coli MurJ14,15. The putative zinc ion is coordinated by two histidine residues and two chloride ions (Supplementary Fig. 2c,d,e), which is surprising as at no point was zinc added during the protein purification or crystallization process. Due to its strategic location at the start of the hinge helix TM7, it might influence gating between the two alternate states. The importance of the chloride or zinc ions in lipid II flipping remains to be tested.
Our studies show that the proximal site and membrane portal are critical for MurJTA function. Importantly, this region of the protein is formed by TMs 1, 2, and 8 where the structural difference is most substantial compared to the MATE structures (Supplementary Fig. 5a). It is possible that the observed structural difference in these three TMs is to accommodate the large lipid II head group. The first two TMs in MVF and OLF flippase families are amphipathic13, consistent with the idea that the role of these TMs is to recognize the diphosphate and sugar moieties of the substrates. Furthermore, because of the arginine residues located in these three TMs, the cavity of MurJTA is mainly positively charged unlike typical MATE transporters, consistent with the idea that MurJTA has evolved to bind the large and negatively charged lipid II molecule (Supplementary Fig. 5b). Alternatively, this structural difference could be due to the differences between MurJ and MATEs in their conformational states or potential ion coupling mechanisms.
Based on the structure of MurJTA and the MTSES functional data on E. coli MurJ10, we propose a rocker-switch model of lipid II flipping by MurJ (Fig. 4e). The substrate is captured in the central cavity between the N-lobe and C-lobe. This rocker-switch motion is generated about a hinge on TM7, translocating lipid II across the membrane, which is then released. The hydrophobic groove formed by TMs 13 and 14 of MurJ facilitates capture of the undecaprenyl tail, which remains associated with the groove during transition to the outward-facing state. Our model of lipid II flipping is strikingly different from that proposed for the lipid-linked oligosaccharide flippase PglK, where only the outward-facing state is necessary for flipping16, indicating diverse mechanisms of lipid flipping. Because it is thought that all MOP transporters utilize a shared transport mechanism13, this rocker-switch motion is possibly utilized by all MOP transporters for alternating-access transport.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Protein expression and purification
The gene of full-length MurJ from Thermosipho africanus was synthesized (Bio Basic Inc.) and cloned into a modified pET26 expression vector encoding maltose binding protein (MBP) and polyhistidine tags flanked by PreScission protease cleavage sites (PPX), such that the final construct is His10-MBP-PPX-MurJ-PPX-His10. A plate of transformed C41(DE3) cells was used to inoculate 4 L of terrific broth. Isopropyl β-D-thiogalactopyranoside was added to 0.4 mM upon reaching an optical density (OD600) of 0.8. Protein was overexpressed for 3 hours at 37°C with shaking. Cell pellets from each liter of culture were resuspended in 10 mL lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl) containing the following additives: 10 mM β-mercaptoethanol, 1 μg/mL leupeptin, 1 μg/mL pepstatin, 5 mU/mL aprotinin, 20 μg/mL deoxyribonuclease I, and 2.5 mM phenylmethylsulfonyl fluoride. Cells were disrupted by a Microfluidizer and another 2.5 mM phenylmethylsulfonyl fluoride was added. Membrane proteins were extracted by stirring with 30 mM n-dodecyl-β-D-maltopyranoside (DDM, Anatrace) for 1 hour at 4°C. Upon centrifugation at 20,000 g for 40 min, the supernatant was rotated with 1.5 mL of TALON Cobalt resin (Clontech) for 1 hour at 4°C in the presence of 5 mM imidazole. Resin was recovered by centrifugation at 800 g for 5 minutes and resuspended in 7.5 mL of buffer A (50 mM Tris-HCl pH 8.0, 500 mM NaCl, 15 mM imidazole, 1 mM DDM). The slurry was poured into a gravity column, and subsequently washed twice with 7.5 mL buffer B (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 15 mM imidazole, 1 mM DDM). Protein was eluted in 6 mL of buffer B with imidazole added to 200 mM, and incubated overnight at 4°C with 40 μg/mL PreScission protease and 5 mM β-mercaptoethanol. PreScission protease-treated MurJ was purified by gel filtration on a Superdex 200 10/300 column in 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM dithiothreitol, and 0.3 mM decyl maltose neopentyl glycol (DMNG, Anatrace). For preparation of selenomethionine (SeMet)-labeled protein, the same plasmid was transformed into B834(DE3) cells, protein was overexpressed by autoinduction in the presence of 125 mg/L L-selenomethionine (ACROS Organics) according to established protocol22, and purified as above.
Crystallization
MurJTA was crystallized in lipidic cubic phase (LCP) according to established protocol23 but with modifications. Briefly, purified MurJTA was concentrated to 15 mg/mL with a 50 kD MW cutoff centrifugal filter (EMD Millipore). Tris(2-carboxyethyl)phosphine (ThermoFisher) was added to 2 mM, and protein was centrifuged at 16,000 g for 30 minutes before mixing with molten monoolein (Sigma) in a 2:3 (w/w) protein:lipid ratio using a twin-syringe setup. LCP was dispensed on 96-well glass sandwich plates as 150 nL drops by a Gryphon LCP robot (Art Robbins Instruments), and overlaid with 1 μl of precipitant solution. Both native and SeMet MurJTA crystallized at room temperature in solutions containing 30–45% (v/v) PEG400, 200–400 mM CaCl2, pH 6.5–9.0. Crystals grew to full size in 4 weeks and were flash-frozen in liquid nitrogen without additional cryo-protectant.
Addition of lipid II improved diffraction quality and was performed for native crystals as follows: the L-lysine form of lipid II (BaCWAN, University of Warwick) was added to monoolein powder in a glass vial to a final concentration of 1.5 mM (in molten monoolein). The monoolein powder was dissolved in 1 mL of chloroform and mixed thoroughly. This mixture was dried under argon stream and washed with 0.5 mL pentane. Finally, organic solvent was removed by drying completely under argon stream before melting the lipids in a 42°C water bath. LCP trays were set up as described above.
For the bromide soak experiment, a window was cut from the coverslip of selected wells and the precipitant solution was wicked away. Fresh precipitant solution (of the same composition except with CaCl2 replaced by CaBr2) was injected and the well was resealed with tape. Soaking was performed for 2 hours, after which crystals were immediately harvested and frozen.
Data collection and structure determination
The structure of MurJ was determined by single wavelength anomalous dispersion (SAD). Data was collected at the NECAT 24-ID-C and 24-ID-E beamlines (Advanced Photon Source, Argonne National Laboratory) with a wavelength of 0.9791 Å and processed by HKL2000 (HKL Research). Due to the high isomorphism between crystals, selected frames from 12 SeMet crystals were merged together in HKL2000 to generate a dataset with over 50-fold average redundancy and signal extending to 3.2 Å resolution. Heavy atom sites were located by SHELXD24, and phasing was performed by PHENIX AutoSol25 initially to a high resolution cutoff of 5.0 Å, producing a figure of merit of 0.50. Phases were gradually extended to 3.5 Å using MR-SAD by placing idealized helical fragments into the density and density modification (Supplementary Fig. 1). The 8 observed Se sites, corresponding to all the methionine residues in the protein except the initiator, were sufficient for unambiguous assignment of topology and register (Supplementary Fig. 1). For the high-resolution data, selected frames from 3 native crystals crystallized in the presence of lipid II were merged together to generate a dataset with signal extending to 2.0 Å resolution. Phases were obtained from the model built from the SeMet data by PHASER26, and improved by cycles of model building in COOT and refinement in PHENIX.refine to a final Rwork/Rfree of 18.9/21.4 (Table 1), good geometry (98% Ramachandran favored, 0% Ramachandran outliers) and minimal clashes (clashscore of 1). MurJTA crystallized in space group C2 with a single MurJ molecule in the asymmetric unit. The electron density map (Supplementary Fig. 1d) was of sufficient quality to unambiguously build residues 4–470 (out of a total of 475 residues), as well as place a shell of ordered monoolein and water molecules around the protein. For the bromide soak experiment, data was collected at beamline 24-ID-C with a wavelength of 0.9197 Å. Phases were obtained from the high resolution model by PHASER, and the anomalous difference Fourier map was calculated by PHENIX.maps.
Docking of lipid II and structure analysis
Docking lipid II to the structure of MurJTA was performed by Autodock Vina27. The L-lysine form of lipid II was chosen for docking instead of the meso-diaminopimelate (mDAP) form, as the related bacterium Thermotoga maritima was shown to incorporate both L- and D-lysine into peptidoglycan instead of mDAP28,29. Initial lipid II coordinates were generated from 2D geometry in PHENIX.eLBOW, and stereochemistry was manually corrected in PHENIX.REEL, with reference to the NMR structure of lipid II in complex with nisin (PDB ID: 1WCO)30. In particular, effort was made to ensure that all the 16 chiral centers have the correct R/S configuration, the peptide and polyprenyl C=C bonds have the right cis/trans configuration and planarity, and the sugars have the correct pucker and glycosidic linkage. The remaining 68 torsions were freely rotatable during docking. In addition, the side chains of Arg 18 and Glu 57 of MurJTA were treated as flexible for docking as they appeared to be flexible in our structure (high B factors as compared to side chains of adjacent residues). Docking was performed over a search space of 30 × 30 × 36 Å covering the central cavity, portal, and hydrophobic groove. Surface electrostatics were calculated by the APBS31 PyMOL plugin. Molecular graphics were created in PyMOL32 and CueMol (http://www.cuemol.org/). Sequence alignment was performed with structure restraints by PROMALS3D33, and structure superposition was done in UCSF Chimera34.
MurJ complementation assay
MurJTA was cloned into pEXT21 with a C-terminal FLAG tag and was subjected to site-directed mutagenesis using standard recombinant DNA methods. Expression of MurJTA is controlled by the tac promoter in pEXT2111. The pEXT21 plasmid bearing wild-type MurJTA or a mutant thereof was transformed into the conditional MurJ-depletion E. coli strain NR11547, which was a gift from Dr. Natividad Ruiz (The Ohio State University). The endogenous MurJ in NR1154 is placed under the control of the PBAD promoter; therefore, NR1154 growth depends on the presence of L-arabinose. D-fucose is an anti-inducer of endogenous E. coli MurJ expression in NR1154. Transformants were selected with 80 μg mL−1 spectinomycin on LB/agar plates also supplemented with 0.2% arabinose. Overnight cultures were diluted to an OD600 of 0.2 and grown to an OD600 of 0.6 in the presence of 80 μg mL−1 spectinomycin and 0.2% arabinose. IPTG was then added to the duplicate of each culture at a final concentration of 0.1 mM. Cultures were further incubated with shaking for 3 hours at 37°C before cells were harvested by centrifugation. For the complementation assay, cells were washed three times with PBS and normalized to an OD600 of 0.5. Each culture was then diluted to 0.02, followed by 10-fold serial dilutions and spotted (5 μL) onto LB/agar plates supplemented with either 0.05% fucose or 0.05% fucose with 0.1 mM IPTG.
For the Western blot to check for protein expression, cells were lysed by microfluidizer and total membrane was isolated by ultracentrifugation. Samples were prepared by resuspending the membrane in 50 mM Tris-HCl pH 8, 150 mM NaCl, mixing with a loading buffer (final concentrations of 3 M urea, 2.5% SDS, 60 mM Tris-HCl pH 6.8, 0.01% bromophenol blue, 1 mM β-mercaptoethanol), and subjecting to 3 min sonication prior to loading onto a 10% SDS-PAGE gel. Bands were transferred to a PVDF membrane, which was probed with mouse anti-FLAG primary antibody (1:1200 dilution, Sigma F3165) followed by donkey anti-mouse secondary antibody (1:10,000 dilution, LI-COR 926-32212). Results were visualized on a LI-COR Odyssey CLx imaging system.
Data availability
Atomic coordinates and structure factors for the reported crystal structure are deposited in the Protein Data Bank under accession code 5T77. Atomic coordinates for the outward-facing model is included as Supplementary Data Set 1. Any other data pertaining to this paper is available upon request.
Supplementary Material
Acknowledgments
We thank N. Ruiz (The Ohio State University) for sharing E. coli strains NR1154 and NR1157. We thank Z. Guan for mass spectrometry analysis of MurJ and lipid II. Data for this study were collected at beamlines NECAT 24-ID-C and 24-ID-E at the Advanced Photon Source. This work was supported by Duke startup funds (S.-Y.L.). Beamlines 24-ID-C and 24-ID-E are funded by P41GM103403 and S10 RR029205.
Footnotes
AUTHOR CONTRIBUTIONS
A.K. and S.-Y.L. conceived the project. A.K. expressed, purified, crystallized, and solved the structure of MurJTA under the guidance of S.-Y.L. E.M carried out the functional complementation assays under the guidance of S.-Y.L. A.K. and S.-Y.L. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
ACCESSION CODES
Atomic coordinates and structure factors for the reported crystal structure are deposited in the Protein Data Bank under accession code 5T77. Atomic coordinates for the outward-facing model is included as Supplementary Data Set 1. Any other data pertaining to this paper is available upon request.
Note: Any Supplementary Information files are available in the online version of the paper.
References
- 1.Bugg TD, Braddick D, Dowson CG, Roper DI. Bacterial cell wall assembly: still an attractive antibacterial target. Trends Biotechnol. 2011;29:167–73. doi: 10.1016/j.tibtech.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 2.de Kruijff B, van Dam V, Breukink E. Lipid II: a central component in bacterial cell wall synthesis and a target for antibiotics. Prostaglandins Leukot Essent Fatty Acids. 2008;79:117–21. doi: 10.1016/j.plefa.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 3.Ruiz N. Lipid Flippases for Bacterial Peptidoglycan Biosynthesis. Lipid Insights. 2015;8:21–31. doi: 10.4137/LPI.S31783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruiz N. Filling holes in peptidoglycan biogenesis of Escherichia coli. Curr Opin Microbiol. 2016;34:1–6. doi: 10.1016/j.mib.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohammadi T, et al. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 2011;30:1425–32. doi: 10.1038/emboj.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohammadi T, et al. Specificity of the transport of Lipid II by FtsW in Escherichia coli. J Biol Chem. 2014 doi: 10.1074/jbc.M114.557371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruiz N. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc Natl Acad Sci U S A. 2008;105:15553–7. doi: 10.1073/pnas.0808352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meeske AJ, et al. MurJ and a novel lipid II flippase are required for cell wall biogenesis in Bacillus subtilis. Proc Natl Acad Sci U S A. 2015;112:6437–42. doi: 10.1073/pnas.1504967112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inoue A, et al. Involvement of an essential gene, mviN, in murein synthesis in Escherichia coli. J Bacteriol. 2008;190:7298–301. doi: 10.1128/JB.00551-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sham LT, et al. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science. 2014;345:220–222. doi: 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohamed YF, Valvano MA. A Burkholderia cenocepacia MurJ (MviN) homolog is essential for cell wall peptidoglycan synthesis and bacterial viability. Glycobiology. 2014;24:564–76. doi: 10.1093/glycob/cwu025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meeske AJ, et al. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature. 2016 doi: 10.1038/nature19331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hvorup RN, et al. The multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily. Eur J Biochem. 2003;270:799–813. doi: 10.1046/j.1432-1033.2003.03418.x. [DOI] [PubMed] [Google Scholar]
- 14.Butler EK, Davis RM, Bari V, Nicholson PA, Ruiz N. Structure-function analysis of MurJ reveals a solvent-exposed cavity containing residues essential for peptidoglycan biogenesis in Escherichia coli. J Bacteriol. 2013;195:4639–49. doi: 10.1128/JB.00731-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler EK, Tan WB, Joseph H, Ruiz N. Charge requirements of lipid II flippase activity in Escherichia coli. J Bacteriol. 2014;196:4111–9. doi: 10.1128/JB.02172-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez C, et al. Structure and mechanism of an active lipid-linked oligosaccharide flippase. Nature. 2015;524:433–8. doi: 10.1038/nature14953. [DOI] [PubMed] [Google Scholar]
- 17.He X, et al. Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature. 2010;467:991–4. doi: 10.1038/nature09408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu M, Radchenko M, Symersky J, Nie R, Guo Y. Structural insights into H+-coupled multidrug extrusion by a MATE transporter. Nat Struct Mol Biol. 2013;20:1310–7. doi: 10.1038/nsmb.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu M, et al. Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc Natl Acad Sci U S A. 2013;110:2099–104. doi: 10.1073/pnas.1219901110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka Y, et al. Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature. 2013;496:247–51. doi: 10.1038/nature12014. [DOI] [PubMed] [Google Scholar]
- 21.Mousa JJ, et al. MATE transport of the E. coli-derived genotoxin colibactin. Nature Microbiology. 2016;1:15009. doi: 10.1038/nmicrobiol.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fox BG, Blommel PG. Autoinduction of protein expression. Curr Protoc Protein Sci. 2009 doi: 10.1002/0471140864.ps0523s56. Chapter 5, Unit 5.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–31. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–22. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 25.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huber R, et al. Thermotoga maritima sp. nov. represents a new genus of unique extremely thermophilic eubacteria growing up to 90°C. Archives of Microbiology. 1986;144:324–333. [Google Scholar]
- 29.Boniface A, Bouhss A, Mengin-Lecreulx D, Blanot D. The MurE synthetase from Thermotoga maritima is endowed with an unusual D-lysine adding activity. J Biol Chem. 2006;281:15680–6. doi: 10.1074/jbc.M506311200. [DOI] [PubMed] [Google Scholar]
- 30.Hsu ST, et al. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat Struct Mol Biol. 2004;11:963–7. doi: 10.1038/nsmb830. [DOI] [PubMed] [Google Scholar]
- 31.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–41. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delano WL. The PyMOL Molecular Graphics System. 2002 [Google Scholar]
- 33.Pei J, Kim BH, Grishin NV. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008;36:2295–300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structure are deposited in the Protein Data Bank under accession code 5T77. Atomic coordinates for the outward-facing model is included as Supplementary Data Set 1. Any other data pertaining to this paper is available upon request.