

Abstract
The NF-κB pathway, a critical regulator of apoptosis, plays a key role in many normal cellular functions. Genetic alterations and other mechanisms leading to constitutive activation of the NF-κB pathway contribute to cancer development, progression and therapy resistance by activation of downstream anti-apoptotic pathways, unfavorable microenvironment interactions, and gene dysregulation. Not surprisingly, given its importance to normal and cancer cell function, the NF-κB pathway has emerged as a target for therapy. In the review, we present the physiologic role of the NF-κB pathway and recent advances in better understanding of the pathologic roles of the NF-κB pathway in major types of lymphoid neoplasms. We also provide an update of clinical trials that use NF-κB pathway inhibitors. These trials are exploring the clinical efficiency of combining NF-κB pathway inhibitors with various agents that target diverse mechanisms of action with the goal being to optimize novel therapeutic opportunities for targeting oncogenic pathways to eradicate cancer cells.
Keywords: NF-κB pathway, B-cell lymphoma, Apoptosis, miRNA, Tumor microenvironment, EBV
1. Introduction
B-cell lymphomas are a heterogeneous group of neoplasms that originate from B cells that normally reside in lymphoid structures and extra-nodal tissues. Molecular analysis of lymphomas has shown that lymphoma cells co-opt normal cellular pathways to support their own growth and dissemination. In addition, the tumor microenvironment is important to the survival of lymphoma cells via tumor cell-microenviroment cellular interactions [1-3]. One of the most important normal cellular pathways is the nuclear factor-kappaB (NF-κB) pathway. NF-κB transcription factor family members are involved in many physiologic cellular functions including inflammation, apoptosis, cell survival, proliferation, angiogenesis, and innate and acquired immunity.
Constitutive activation of the NF-κB pathway is a feature of most types of B-cell lymphoma. NF-κB can be activated by acquired genetic lesions of different NF-κB members or key signaling molecules including cancer-related chromosomal translocations, deletion or mutations. These signaling molecules include mucosa-associated lymphoid tissue 1 (MALT1), BCL-10, caspase recruitment domain-containing protein 11 (CARD11), tumor necrosis factor receptor-associated factors 3 (TNFAIP3, A20), NF-κB2, and infection by viruses that produce oncoproteins [4-9]. Constitutive activation of the NF-κB pathway inhibits cell differentiation andapoptosis, promotes cell proliferation, and increases angiogenesis, cancer-related inflammation and metastatic potential [3, 10]. Consequently, activated NF-κB is one of the prime therapeutic targets in lymphoma cells.
In this review, we summarize the essential functions of the NF-κB pathway and its signaling components in normal and lymphoma cells with a particular focus on novel molecular, clinical and preclinical studies. NF-κB functions as a crucial modulator of the extrinsic B-cell TME and intrinsic survival signaling pathways. Recent advances have greatly enhanced our understanding of NF-κB expression and have led to new insights into mechanisms involved in dysregulated gene expression in various subtypes of lymphoma. The new knowledge has yielded cellular targets of mechanism-mediated drug resistance and pointed to new therapeutic approaches for the treatment of patients with lymphoma.
2. Two NF-κB pathways
The NF-κB transcription factor family is composed of five subunits: RelA or p65, RelB, c-Rel or Rel, and p50 and p52 (with their precursors p105 and p100, respectively and known as NF-κB 1 and NF-κB2). The Rel homology region, a ∼300 residue long homologous element shared by all subunits, is responsible for dimerization, inhibitor binding, nuclear localization, and DNA binding. Homodimeric and heterodimeric combinations formed by NF-κB proteins allow for functional NF-κB activation, with the exception of RelB that does not form a stable, detectable homodimer, but forms heterodimers selectively with p50 and p52 [11]. In quiescent cells, NF-κB transcription factors are located in the cytoplasm, whereas IκBα releases NF-κB factors allowing their translocation into the nucleus as stimulated by intracellular cues through a variety of surface receptors [12].
NF-κB signaling is categorized into canonical and non-canonical pathways that represent two independent, yet interlinked, pathways. The canonical pathway preferentially involves certain receptors, such as the B cell receptor (BCR), Toll-like receptors (TLRs), nucleotide oligomerization domain-like receptors, and TNF family receptors. Engagement of these receptors by ligands gives rise to activation of the heterotrimeric IκB kinase (IKK) complex, which consists of a, b, and c (NEMO) subunits [13]. Activated IKK directly phosphorylates IκBa, and, in its active form, starts the recognition and polyubiquitination by the bTrCP ubiquitin ligase, followed by proteasome degradation and release NF-κB to the nucleus [14] (Figure 1).
Figure 1.
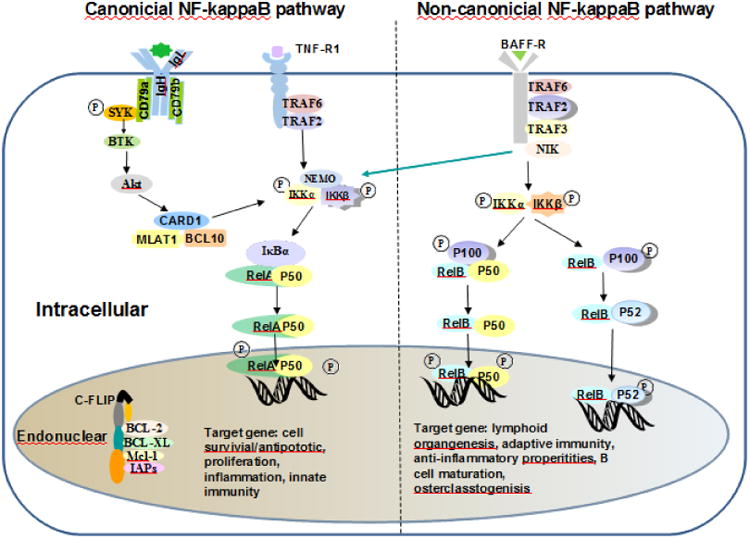
The canonical and non-canonical NF-κB pathways. The canonical and non-canonical NF-κB signaling pathways are shown in the left and right of the figure. The canonical pathway is activated by the C-like receptors 4, TNF receptors' family and the antigen receptors BCR and TCR, while the non-canonical pathway is activated by other receptors, such as BAFF-R, CD40, RANK, CD30, and LTβ-R. Arrows indicate activating steps.
Unlike the canonical pathway, the non-canonical NF-κB pathway depends on activation of the RelB subunit associated with p50 or p52. This pathway is activated preferentially by stimulation receptors, including the B-cell activating factor belonging to the TNF family receptors and CD40. These receptors activate NF-κB-inducing kinase, causing phosphorylation of a distinct inhibitor of the IKK complex that is composed of two IKKα subunits. This form of IKK phosphorylates p100, leading to its proteolytic processing into the NF-κB subunit p52, followed by the heterodimer, p52 and RelB, and translocation to the nucleus [13, 15] (Figure 1).
These two NF-κB pathways regulate several different functions dependent on the cell type and stimulus applied [16, 17]. The canonical pathway promotes inflammation, cell proliferation, and cell survival through the production of several inhibitors of apoptotic signaling, and also contributes to angiogenesis, tumor promotion and metastasis. The non-canonical pathway, instead, has anti-inflammatory activity and regulates lymphoid development and organization. In normal cells, regulation of NF-κB signaling is homeostatic and activation of these pathways is transient and stimulus dependent; various negative regulators modulate feedback mechanisms to terminate NF-κB signaling, such as re-accumulation of IκBα and induction of A20, an ubiquitin-editing enzyme. Interruption of homeostatic regulation of NF-κB signaling by a host of genetic aberrations in B cells plays an important role in the pathogenesis of lymphoid malignancies.
3. NF-κB function and pathway development in normal lymphoid tissues
Both canonical and non-canonical NF-κB pathways are essential for B-cell maturation at different differentiation stages. Compared with pro-B cells, pre-B cells have a more active canonical NF-κB pathway that promotes their transition from large to small pre-B cells [18]. The canonical and non-canonical NF-κB pathways control the production of λ-chain B-cells [19]. Those immature B-cells become transitional B cells through T1 and T2 stages, and develop into either mature follicular B cells or non-circulating marginal zone B-cells. At this stage, BCR cross-linking induces the activation of NF-κB and c-Rel to produce anti-apoptotic protein [20], induces the survival signaling mediated by BAFF, and increases the expression of Nfκb2. These events lead to generation of p100 protein [21], which, in turn, mediates the activation of the non-canonical NF-κB pathway. Notably, during B-cell generation and maintenance, NF-κB subunits have many physiological functions and play unique roles in the development and function of mature B lymphocytes (Table 1).
Table 1. Role of the components of the canonical and non-canonical NF-κB pathway in the differentiation and activation of B cells.
| subunits | B cell survival and differentiation | B cell activation | Mouse phonotype |
|---|---|---|---|
| RelA/p65 | Necessary for signal transduction pathways for lymphocyte proliferation;Required for proliferative responses to stimulation. | Essential to differentiate to IgM+ and to secrete immunoglobulin;Dispensable for the normal amount secretion of IgG1 and IgA. | Lethality: embryonic (days 15.5). Cause: fetal hepatocyte apoptosis. |
| RelB | Necessary for normal production of Ag-IgG in response to T cell-dependent and –independent stimuli. | Lethality: 3 months (50% mice). Cause: defects in acquired and innate immunity and multiple pathological lesions; multiorgan inflammation and hematopoietic abnormalities. |
|
| P52/p100/NF-Kb2 | Necessary for maintenance cell population of the peripheral B, bone marrow, and lymph nodes; Required for development of normal spleen architecture. | Required for response o stimulations and normal antibody production. | Mice development normally |
| Rel/c-Rel | Required for B cell survival, differentiation, and inducing anti-apoptotic gene. | Dispensable for isotype switching, cytokine and immune molecules' production | Mice are viable; Hematopoiesis is not affected, but defect in the immune system. |
| P50/p105/NF-Kb2 | Required for survival of non-activated B cells. | Dispensable for proliferate in response to bacterial lipopolysaccharide;Required for basal and specific antibody production. | Show no developmental abnormalities,Exhibit multifocal defects in immune responses involving B lymphocytes and nonspecific responses to infection. |
| RelB+P50 | Required for multiple functions in B cell development. | Dispensable for inflammatory infiltrates. | Lethality: premature death within three to four weeks after birth. Cause: myeloid hyperplasia and thymus atrophy |
| RelA+Rel | Essential for the survival of progenitors cells;Dispensable for regulating differentiation. | Dispensable for the late antigen-independent stages of B-cell maturation and the survival of peripheral B cells. | Lethality: embryonic (days 13.5). Cause: multiple hematopoietic cell defects. |
NF-κB activity is required for the functional activation of T-cell development and selected effector functions (Table 1). In the thymus, CD4-CD8- double negative thymocytes progress into CD4+CD8+ double positive thymocytes and eventually into the mature CD4+ or CD8+ T cells, which exit the thymus to enter the circulation. Constitutive NF-κB activity occurs at all stages of thymocyte differentiation throughout T cell development, and it is especially activated in the double negative phase during the pre-T cell receptor (TCR) assembly. NF-κB activity may be attributable to TCR that activates NF-κB to pro-apoptotic activities in negative section and anti-apoptotic/pro-survival activities in positive selection [22]. Additionally, NF-κB participates in the generation and suppressive functions of thymic T regulatory cells [23]. The NF-κB component of c-Rel, RelA or p50 subunits also play a role in the development of effector T-cells [24].
4. Antigen receptors and NF-κB in B-cell lymphoid malignancies
B-cell lymphomas are characterized by altered signaling of the BCR pathway. This pathway is the organizing principle of B-cell physiological behavior and is also involved in many obligatory cellular functions, including B-cell survival, development, humoral immunity and antigen-driven clonal selection. Neoplastic B cells of most lymphoma types generally retain BCR expression and have intact BCR complexes. Interaction of the BCR with self or environmental antigens provides neoplastic B cells with growth and/or survival signals that play a vital role in the pathogenesis of lymphoma. NF-κB is constitutively activated, more or less strongly, and plays a pathogenic role in many B-cell lymphomas [25, 26]. In large part, the NF-κB pathway is activated because it is a major downstream effector of the BCR pathway, conveying pro-survival and proliferation signaling [26], summarized in the following sections.
Bruton tyrosine kinase (BTK), primarily expressed in B cells, but not in T-cells or plasma cells, is a non-receptor tyrosine kinase that belongs to the Tec kinase family [27]. Upon activated by LYN or spleen tyrosine kinase (SYK), BTK phosphorylates phospholipase C-ɤ, which in turn forms a complex with BTK that activates protein kinase C (PKC), leading to recruitment of CARD11, B-cell lymphoma 10 (BCL10), and MALT1 into a multi-protein CBM complex. IKK is in conjunction with this complex just upstream of NF-κB, which transfers signaling toward the NF-κB effectors and plays a prominent role in B-cell malignancies [13, 15, 28, 29]. Ultimately, IKKβ is activated thereby triggering activation of the NF-κB pathway. A number of targeted inhibitors to BTK have been (or are being) developed and clinical trials have shown benefit in combination therapies for patients with relapsed or refractory lymphomas [30] (Figure 2).
Figure 2.
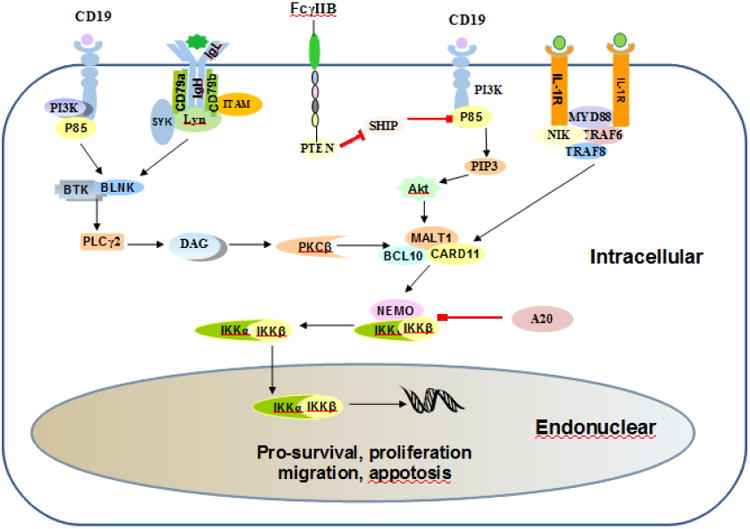
The activation modes of NF-κB in the pathogenesis of B cell lymphoma. There are three main modes: BCR, PI3K/AKT, and MYD88. (1). BCR is activated by antigen binding or cell autologous interaction, resulting in ITAMs phosphorylation in the cytoplasmic domains of CD79A and CD79B. Then SYK amplifies the initial activation signal by autophosphorylation and further ITAMs phosphorylation, which activates BTK and BLNK, leading to subsequent activation of PKCβ and CARD11-BCL10-MALT1 complex. (2). CD19 provides a docking site for the p85, followed by subsequent activation of BTK and BLNK. (3). MyD88 mutations are bound to IRAK kinase forming a helical protein, which activates the complex of CARD11-BCL10-MALT1. (4). A20 is a negative regulator of NF-κB. (5). IKK is phosphorylated to activate NF-κB transcription factors that regulate gene expression of several survival factors. The arrows indicate direction of signaling from plasma membrane toward effectors and bars indicate inhibitory steps.
PKCβ expression is essential for the pathogenesis of B cell malignancies. In the BCR pathway PKC functions downstream BTK through IKK to activate NF-κB transcription factors [2, 31, 32]. Of note, progression of chronic lymphocytic leukemia (CLL) in a murine TCL1 model depends on PKC expression [33], whereas disruption of PKC in stromal cells inhibits CLL cell survival in a murine TCL1 model [34]. On the other hand, overexpression of PKC-associated kinase (PKK) activates NF-κB signaling in diffuse large B-cell lymphoma (DLBCL) cells, whereas suppression of PKK expression inhibits NF-κB activity in these cells. In particular, the survival of DLBCL cells in vitro is impaired and tumor growth of xenografted DLBCL cells is inhibited, as PKK is knocked down in mice [35]. PKC inhibition has growth inhibitory effects due to inhibition of the NF-κB pathway, inducing G-phase cell-cycle arrest and/or cell death [36]. Additionally, enzastaurin, a PKCβ inhibitor that has been used in preclinical and clinical trials for B-cell malignancies, adds benefit in combination therapy approaches.
Phosphoinositol-3 kinase (PI3K), involved in a wide variety of cellular processes, is essential for B-cell development and serves as one of the drivers of lymphoma development [31]. PI3K can be activated by different factors, including many cell surface chemokines and cytokine receptors and BCR-related LYN-dependent phosphorylation of the immunereceptor tyrosine-based activation motifs (ITAM) in the cytoplasmic domain of CD19 [37-39]. PI3K catalyzes the production of phosphatidylinositol 3,4,5-triphosphate, which recruits and activates Akt thereby regulating downstream signaling including mammalian target of rapamycin, NF-JB, or other factors, eventually activating NF-κB [40]. Mice lacking PI3Kα and δ show severe defects in B-cell development [41], whereas constitutively active PI3Kα can rescue resting B cells lacking BCR expression from apoptosis [42]. In addition, PI3K and IKK1 synergistically drive peripheral B-cell differentiation and survival in a context-dependent manner [43]. In activated B-cell like (ABC) DLBCL, PI3K inhibition reduces NF-κB activity and decreases the expression of NF-κB target genes that promote survival of affected ABC-DLBCL cells [44]. Furthermore, chemical blockade of SYK can selectively induce apoptosis of BCR-dependent DLBCL cells through decreased BCR signaling including PI3K/AKT and NF-κB [45]. These data suggest an important role for the interaction of PI3K and NF-κB in the pathogenesis of B-cell malignances (Figure 2).
5. The pathogenic modes of activation of NF-κB in B-cell lymphomas
Frequent dysregulation of the NF-κB pathway influences survival, proliferation, and apoptosis of lymphoma cells. The first hint of the importance of NF-κB came from the discovery that p50 is homologous to v-rel, a highly transforming oncogene carried within the genome of an avian reticuloendotheliosis virus [46, 47]. Subsequently, it was discovered that NF-κB heterodimers constitutively accumulate in the nucleus of Hodgkin/Reed-Sternberg (HRS) cells in HL and that inhibition of NF-κB was shown to reduce the proliferation and survival of HL cell lines [48]. Subsequent investigations revealed truncating mutations in IκBα in HL cell lines and primary HRS cells [49-51]. These results showed that NF-κB pathway activation enables oncogenesis. There are three modes of activating NF-κB constitutively (Figure 2).
The first way lies in activation of BCR signaling through transition from extrinsic BCR activation into intrinsic activation. Acquired mutation or loss function mutations have an important role in antigenic drive in lymphomagenesis. For example, several ABC-DLBCL cell lines and about 20% of primary ABC-DLBCL tumors carry a mutation in the crucial tyrosine residue in the ITAM of CD79B [2]. This mutation increases the signaling response by preventing BCR internalization and by interfering with activation of LYN. However, this mutation, by itself, is not sufficient to initiate BCR activation; PI3K and BTK signaling remain essential for NF-κB activation for this subset of ABC-DLBCL cells [44].
CARD11, another BCR pathway component is a key scaffolding protein that connects BCR activation to NF-κB signaling and plays a vital role in some lymphomas. About 10% of ABC-DLBCL cases have activating mutations of CARD11 that are sufficient to intrinsically activate NF-κB signaling in malignant B cells, obviating the need for upstream BCR signaling in this subset of tumors [52]. Also, loss of function mutations of TNFAIP3 (A20), a negative regulator of NF-κB, contributes to NF-κB pro-survival signaling in ABC-DLBCL tumors [9, 53]. API2-MALT1, involved in a subset of MALT lymphomas, forms a complex with overexpressed BCL10, and can activate NF-κB independent of upstream BCR signaling [6, 54], responsible for failing to regress after eradication of the underlying infection (Figure 2, left panel).
MYD88 mutations represent a second mode of NF-κB activation. MYD88 mutations are one of the cytosolic adapters of Toll-like receptors (TLR) and are shared by all TLRs except TLR3. The interleukin-1 receptor-associated kinases (IRAK1, IRAK2, and IRAK4) link to MYD88 through hemophilic interactions involving their death domains, forming a helical protein complex [55]. Within this complex, IRAK4 phosphorylates IRAK1, then IRAK1 binds the ubiquitin ligase TRAF6, which, in turn, catalyzes lysine 63-linked polyubiquitination of the kinase TAK1, which forms complexes with the TAB2 and TAB3 zinc finger proteins to become enzymatically active. TAK1 phosphorylates IKKb and mitogen-activated protein kinases, which respectively triggers the NF-κB and c-Jun NH2-terminal kinase and p38/mitogen-activated protein kinase signaling pathways, leading to production of inflammatory cytokines and growth factors [56]. Although many different recurrent MYD88 mutations are reported, the most prevalent mutation substitutes a proline residue for a leucine residue at position 265 in the protein. MYD88 is a significant oncogenic mechanism in B-cell lymphoid malignancies (Figure 2, right panel).
A third mode by which NF-κB is activated constitutively appears to be a strong selective pressure to maintain a functional IgM-type for malignant B-cells. IgG signaling promotes plasmacytic differentiation whereeas IgM signaling promotes primarily cell proliferation as well as NF-κB activation. However, irrespective of pre- or post-germinal center origin, most mature B-cell malignancies express an IgM type BCR. Some evidence supports the concept that these malignancies derive a selection bias from antigen-dependent BCR signaling [57]. In ABC-DLBCL, acquired mutations in the IGH switch regions prevent isotype switching and thereby maintain the expression of an IgM type BCR by lymphoma cells [58]. For example, follicular lymphoma, characterized by t(14;18)(q32;q21) that leads to overexpression of BCL-2, has a productively rearranged Ig heavy chain locus that encodes the IgM type BCR expressed on the cell surface [59, 60].
6. NF-κB signaling and apoptotic pathways
The cellular commitment to apoptosis provides a fundamental benefit to the organism, and errors in this process may give rise to cancer. Recent studies have shown that NF-κB exerts its function mainly as an antagonist to p53 transactivation. p53, one of the key tumor suppressor genes, induces activation of the intrinsic apoptotic pathway. Furthermore, p53 tumor suppressor and transcription factor serve as one of the first lines of defense against the effects of genotoxic damage, oncogene activation, metabolic changes, and hypoxia [61]. Upon transcriptional induction of genes encoding anti-apoptotic and antioxidant proteins, NF-κB blocks cell apoptosis along the apoptotic pathways by regulating p53 [15]. Briefly, p53 up-regulates NOXA and PUMA (bbc3), two proteins of the Bcl-2 family that interact with Bax and Bak at the mitochondrial site where they form the signaling complex or apoptosome that initiates apoptosis. Subsequently, the effector caspases-3, -6 and -7 are activated to complete apoptosis (Figure 3).
Figure 3.
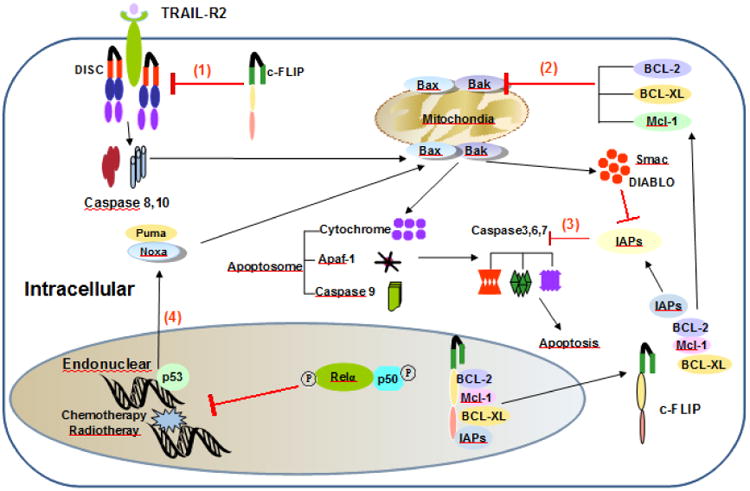
The apoptotic pathways of NF-κB: TRAIL mediating the extrinsic pathway and p53 mediating the intrinsic pathway. Numbers in circles indicate the steps of the apoptotic pathways inhibited by molecules that are induced by NF-κB activation including c-FLIP, Bcl-2 family's molecules, and IAPs. (1), the activation of procaspase 8 and 10 is inhibited by cFLIP. (2), the prosurvival signals, such as Bcl2, Bcl-XL, Mcl-1 inhibit the pro-apoptotic molecules Bak/Bax. (3), the inhibitors of apoptosis (IAPs) family proteins inhibit the effector caspases 3, 6 and 7. (4), NF-κB inhibits directly or indirectly the transcription factor p53. Arrows indicate activating steps, and bars indicate inhibitory steps.
The different units of the canonical and non-canonical NF-κB pathways participate in mediating pro-apoptotic effects through p53. In particular, the canonical pathway seems to have a major role. IKKβ has been shown to induce activation of NF-κB and expression of HDM2 (known as MDM2 in mice), which functions as an e3 ubiquitin ligase. As a result, p53 proteolysis is induced resulting in inhibition of the apoptotic pathway [62]. On the other side, RelA can antagonize the activation of p53 through the sequestration of co-activators, by binding p300 and CReB (CBP) [63, 64]. In addition, upon TRAIL-R2 induction, RelA is able to cooperate with pro-apoptotic signaling to stabilize p53 [65].
The non-canonical pathway also antagonizes p53. RelB can inhibit cell proliferation and tumor growth through p53 transactivation. IKKα switches the binding preference of CBP from p53 to NF-κB through phosphorylation of CBP [64]. Additionally, p52 has been shown to associate directly with the p53-regulated promoters to regulate p53 target genes such as PUMA, TRAIL-R2 [65].
The NF-κB pathway interacts with p53 through various mechanisms in different types of B-cell lymphoma. In CLL, NF-κB up-regulates anti-apoptosis BCL2-family members that function as a major downstream effector of p53-driven apoptosis [66], whereas in ABC-DLBCL and extranodal MZL, the forkhead transcription factor cooperates with NF-κB signaling to encode the p53 -regulatory proteins that repress transcription of pro-apoptotic genes, consequently promoting B-cell survival and contributing to B-cell lymphomagenesis [67]. Extranodal MZL, characterized by t(11;18)(q21;q21)/API2-MALT, depends on NF-κB to inhibit DNA damage-induced and p53-mediated apoptosis [68]. In ABC-DLBCL, NF-κB binds to the promoter regions of GADD45α and REDD1 and regulates these molecules through collaborating with p53 to mediate cyclin G2 expression, suggesting a central role for NF-κB in maintaining genomic integrity and prevention of apoptosis [69].
Recent studies have shown that microRNAs (miRNAs) participate in apoptosis in B-cell malignances by being involved in the NF-κB pathway and p53 [70]. P53 induces the expression of miR-214, functioned as tumor suppressor, which represses p65 expression, leading to inhibition of expression of MYC/BCL2 and suppression of cell survival in B-cell lymphoma [71].
7. Relevance of the Microenvironment in B-cell lymphomagenesis
The tissue microenvironment (TME) in which malignant cells arise and subsequently reside contains variable numbers of immune cells, blood vessels, stromal cells and extracellular matrix [3]. The molecular analysis of the B-cell lymphoma cells has resulted in a considerably improved understanding of the pathogenesis of lymphomas. By comparison, the importance of the TME in providing cellular contact, survival and proliferation signals, and immune evasion has been relatively neglected. Nevertheless, there is increasing evidence to support an important role for the TME plays in lymphomagenesis and progression [3], and the NF-κB pathway participates in interactions between lymphoma cells and TME cells.
The NF-κB pathway plays a crucial role in crosstalk with chemokine receptors and adhesion molecules, as has been highlighted in recent studies. For example, the CC-chemokine receptor 7 (CCR7) that is highly expressed by lymphoma cells functions in homing of lymphoma cells to lymph nodes. In several HL-derived cell lines and in HL tumors, constitutive NF-κB activity up-regulates CCR7 which might contribute to confinement of neoplastic cells and specific localization within target organs. Furthermore, NF-κB cooperates with AP-1 to stimulate expression of the cell-cycle regulator cyclin D2, proto-oncogene C-MET and the lymphocyte homing receptor CCR7 in primary HRS cells [72]. The mechanisms underlying the observed relationships between the chemokine receptor and NF-κB pathway in lymphomas is not fully understood.
Several studies have shown that the NF-κB pathway is involved in bidirectional crosstalk with stromal cells in B-cell lymphomas. By co-culturing CLL bone marrow-derived B-cells and bone marrow-derived stromal cells, one study has shown that the presence of high RelB activity, together with RelA activity, confers survival advantages to bone marrow CLL cells [73]. Another study reported that expression of PKC-βII and subsequent activation of NF-κB in stromal cells of bone marrow plays an essential role in support of survival for CLL, acute lymphoblastic leukemia and mantle cell lymphoma (MCL) [34], indicating that NF-κB involves mechanisms that promote the survival and growth of B-cell lymphomas. Moreover, bone marrow stromal cells protect the non-Hodgkin lymphoma cell lines SUDH-4 and SUDH-10 cells against apoptosis, partly by activation of NF-κB dependent mechanisms that up-regulate NF-κB mediated anti-apoptotic proteins [74].
Cooperation between neutrophils and fibroblast involves the NF-κB pathway and plays an essential role in the proliferation of lymphomas. With mutual contact directly or indirectly among lymphoma cells, neutrophils and fibroblasts, contact-dependent supportive effects trigger and sustain the survival and growth of follicular lymphoma (FL) and DLBCL, and among them tumor-associated neutrophils can activate fibroblasts via the NF-κB pathway, resulting in the recruitment of additional monocytes and neutrophils through the release of CCL2 and IL-8 [75]. On the other hand, blocking antibodies that target CXCR4 and VLA-4, molecules involved in environment-mediated drug resistance in the Jeko-1 (MCL) cell line, can induce Jeko-1 cells to migrate into protective stromal cells and also enhance sensitivity to a chemotherapy agent that targets NF-κB, associated with decreased phosphorylation of ERK1/2, AKT and NF-κB [76].
In addition, there is increasing evidence that immune regulation mediates the proliferation of B-cell malignancies via the NF-κB pathway. The cells of CLL with unmutated IGH variable regions shows a high rate of spontaneous apoptosis, but peripheral blood mononuclear cells can protect the survival of CLL cells through recovered expression of NF-κB pathway components and Bcl-2 [77]. In Burkitt lymphoma (BL) cell lines, RelB transcriptional activity directly increases antigen presentation of MHC class I and CD40 synthesis [78]. The molecular chaperone heat shock protein 90 (HSP90) has both immune regulatory activity and direct antitumor activity in HL in vitro and in vivo, whereas blockade of HSP90 inhibits constitutive activity of NF-κB, independent of IκB mutations [79].
The summarized data support that the concept that the NF-κB pathway plays an important role in the cell growth and survival of the lymphoma TME. Despite the recent focus on mechanisms of NF-κB activation in B-cell malignancies, increasing evidence has shown that NF-κB participates in interactions between stromal elements and non-malignant cells that constitute the TME, in both initiation and maintenance of B-cell lymphoma growth and survival, as well as immune escape strategies.
8. NF-κB pathway and MicroRNAs
MiRNAs are important regulators of gene expression. MiRNA regulation has subtle roles in buffering stochastic fluctuations in gene expression as well as in cell developmental fate decisions [80]. MiRNA participates in pathways fundamental to B-cell development [70, 81] and also has essential functions in the initiation of B-cell neoplasia and development [70]. As one of the important signaling pathways, NF-κB has been shown to have an essential role in miRNA regulation of B-cell malignancies [70], however the association between expression levels of various miRNAs and the NF-κB pathway are not yet clearly defined.
Using ChIP assays in BL and DLBCL, it was shown that the p65 subunit of NF-κB associates with a transcription start site (TSS) of miRNA, which occurs together with changes at histone H3K27me3 and H3K4me3, highlighting the relationship between NF-κB and epigenetically mediated miRNA control in B-cell transformation and DLBCL [85].
Among these miRNAs, miR-155 is strongly associated with NF-κB activation in lymphocyte development and B-cell malignancies, including DLBCL, FL, CLL and MCL [82-86]. MiR-155 is aberrantly up-regulated and present at a significantly higher level in ABC-DLBCL compared with germinal center B cell-like (GCB) DLBCL [82, 84, 87]. miR-155 is important in DLBCL pathogenesis, proven by the fact that high-grade lymphomas in Eμ-miR-155 transgenic mice can be caused by the dysregulation of B-cell miRNA alone [88]. In these mice, miR-155 down-regulates SHIP-1, which in turn phosphorylates C/EBPβ, blocking B-cell differentiation and initiating a chain of events that results in a polyclonal B-cell neoplasia [89]. Through their promoters, NF-κB and MYB can directly activate miR-155 expression, which leads to lower PU.1 expression and reduced levels of CD10 [90]. Overexpression of miR-155 contributes to the inferior survival of patients with ABC-DLBCL. Overexpression of miR-155 can down-regulate SMAS5, identified as a modulator of the TFG-β signaling way, rendering DLBCL cells resistant to growth-inhibitory effects of TFG-β1 and BMP through impaired cell arrest and defective p21 induction. Additionally, miR-155 can promote lymphomagenesis and cell migration by targeting HGAL, a lymphocyte motility inhibitor [86, 91]. These effects are related to the more aggressive behavior of B lymphoma cells in miR-155-expressing ABC-DLBCL (Figure 4).
Figure 4.
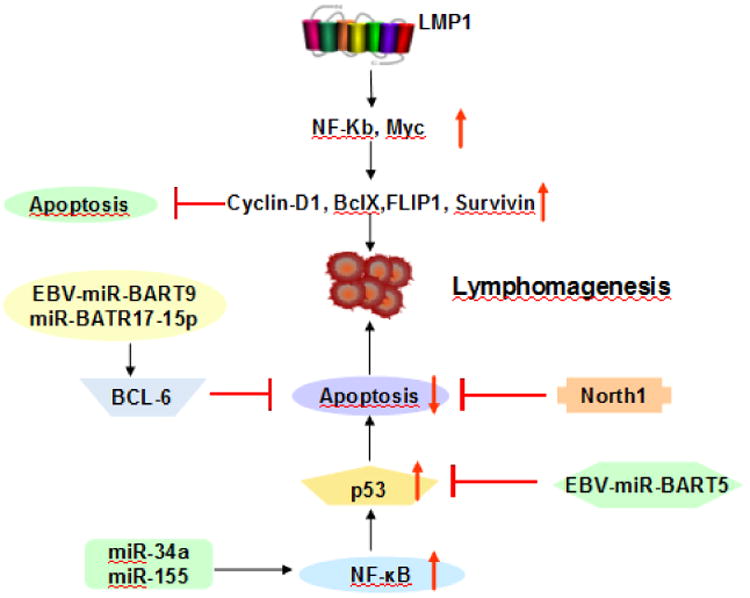
EBV-mediated critical cellular signaling is shown in HL and EBV-positive DLBCL of the elderly. LMP-1 gene polymorphisms associate with enhanced NF-κB activation, which not only increases the expression of cycling D1, BclXL, FLIP1, and Surviving to induce lymphomagenesis, but also inhibits apoptosis mediated by up-regulation of different anti-apoptotic proteins. MicroRNA, such as miR-34a, miR-155, contribute to EBV-positive DLBCL of the elderly lymphomagenesis by up-regulation NF-κB activation, while North1 and BCL-6 down-regulate the NF-κB pathway activation.
B-cell transformation mediated by Epstein-Barr virus (EBV) infection involves significant changes in gene expression of cellular miRNAs, such as miR-155, miR-146a, miR-34a, miR-150, miR199 and miR193/miR365 [92-94]. It also has been shown that EBV latent membrane protein type 1 (LMP1) activates NF-κB and up-regulates miR-155 expression [92]. Upon NF-κB activation, LMP1 induces expression of miR-146 and miR-155 which is associated with the levels of LMP1; low LMP1 levels in EBV-latency type I BL cells have low levels of miRNA whereas higher LMP1 levels in EBV-latency type III BL cells have increased miRNA levels [92, 93]. MiR-146a plays a role in the induction or maintenance of EBV latency by modulating innate immune responses in virus infected host cells [95]. On the other hand, NF-κB activation inhibits miR-34a impairing the growth of EBV-transformed cells.
MiRNAs are also able to interact with NF-κB pathway acting as a feedback regulatory loop that can influence the pre- or post-transcriptional expression of multiple genes. For example, A20, known as a negative NF-κB regulator, is a direct target of miR-125a and miR-125b and these miRNAs are commonly over-expressed in DLBCL cell lines and patient tumors. NF-κB activity can be elevated by ectopic expression of miR125a/miR125b in these cells, whereas A20 inhibits genetic expression of these miRNA to suppress NF-κB signaling. However, miR-125 can terminate A20 function, thus promoting and prolonging NF-κB activity, suggesting the presence of a positive self-regulatory loop among miRNA-125, NF-κB and A20 [9].
There is another means of mutual negative feedback regulation between miRNAs and the NF-κB pathway. MiR-124, a negative regulator of MYC and BCL2 expression in B-cell lymphoma, serves as a miRNA-dependent regulatory circuit that links p53 to NF-κB. Concordantly, stable or transient ectopic expression of miR-124 inhibits cell proliferation and survival, whereas genetic suppression of this miRNA promotes tumor growth. A promoter region of miR-124 matches a functional binding site of wild-type p53, but not mutant, which can increase miR-124 levels and inhibit p65, MYC and BCL2. Overall, there is a mutual negative feedback loop among miR-124, p53 and NF-κB [71]. These data show that precise gene dose regulation by miRNAs has important regulatory roles in the pathogenesis of B-cell lymphomas and that NF-κB contributes to these regulatory circuits. However, a detailed understanding of the regulatory circuits between NF-κB and miRNAs remains largely unknown.
Recent studies have shown that B-cell lymphomas are exposed to a promiscuous signaling network of miRNAs and not a single miRNA. In DLBCL patients, loss of essential miRNAs, such as miR-200c, miR-203 and miR-31, are found and it affects multiple targets including NF-κB components. Simultaneous depletion of the key miRNAs promotes translation of multiple targets and causes chronic activation of the NF-κB, Ras-Erk, and PI3K/Akt pathways that cooperate in B-cell transformation [71]. In nodal marginal zone lymphoma, it also has been shown that enriched expression of multiple miRNAs rather than by a single miRNA ensure robustness of biological processes [96].
Those results suggest that identifying miRNA targets is difficult as a large number of genes and pathway can be influenced by various miRNAs. Through the NF-κB pathway, a number of miRNAs are intimately connected to the development and progression of neoplastic B-cells. Both NF-κB and its associated miRNAs participate in the construction of a complex biologic network in B-cell lymphomas, which includes miR-contribution to the regulation of critical signaling pathways, indicating their potential use as predictive and prognostic markers. The relevance of miRNAs and NF-κB for the regulation of normal and malignant B-cell physiology requires more and advanced investigations.
9. NF-κB pathway and different types of lymphoma
9.1. Chronic lymphocytic leukemia
Patients with CLL have an extremely variable clinical outcome, with a subset of patients who have an indolent clinical course and another subset who have rapidly progressive disease. Through the BCR and Toll-like receptor, the NF-κB pathway is ubiquitously activated in CLL cells by signaling cascades of cell surface receptors to a variable degree, regardless of the disease stage or treatment status. Clinical stratification of CLL patients has shown that NF-κB contributes to a small set of patients with tumors that harbor cytogenetic alterations and somatic mutations and is associated with a poor prognosis.
L265P MYD88 mutant occurs in 3% of CLL patients who are approximately 20 years younger than the average age of CLL patients and have advanced clinical stage at diagnosis [97]. Further studies have shown that CLL cells bearing the L265P MYD88 mutant have activation of both NF-κB and STAT3, which responds to TLR ligands and secretes greater amounts of cytokines and chemokines than CLL cells with wild type MYD88 [97] (Figure 5b).
Figure 5.
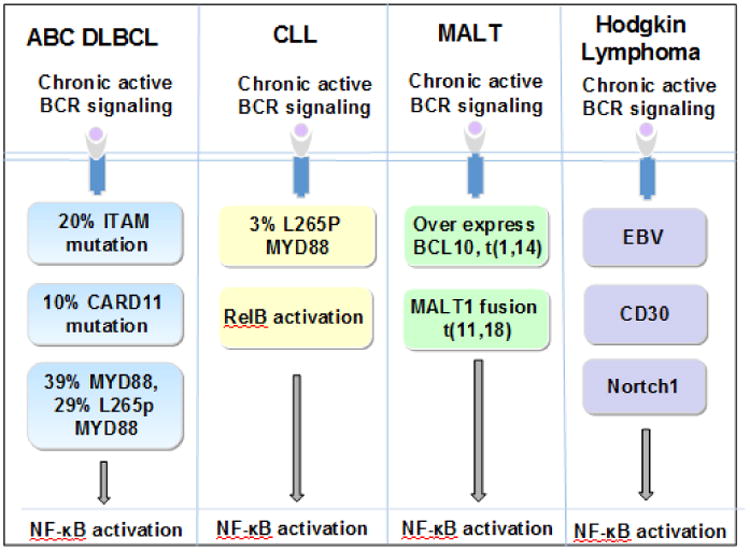
Extrinsic and intrinsic signaling of BCR signaling activates NF-κB in B-cell malignancies. a, In ABC-DLBCL, ITAM, CARD11, and MYD88 independence of BCR. b, In CLL, L265 MYD88 mutation and RelB activation. c, In MALT, overexpression of BCL10 or MALT1 fusion protein. d, In HL, EBV, CD30 and Notch1.
In a recent report, somatic mutations in nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor gene occur in 7% of CLL cases. This gene encodes IκBε, a negative regulator of NF-κB in normal B-cells, and increases p65 inhibition along with decreasing phosphorylation and nuclear translocation of p65, thereby sustaining the survival of CLL cells. This aberration occurs predominantly in poor-prognosis CLL patients [98, 99]. Consequently, this mutation may be a novel mechanism of NF-κB dysregulation contributing to disease progression.
The retinoblastoma (Rb) protein is a major route of crosstalk between the non-canonical NF-κB pathway and p53 in CLL. NF-κB up-regulates the activity of Rb, which induces expression of the polycomb protein EZH2 in CD40L stimulated cells from CLL patients. EZH2 antagonizes a group of p53 target genes, inhibiting tumor suppressor function and promoting tumorigenesis [100]. Therefore, there is a considerable magnification of the non-canonical NF-κB pathway and TP53 by dysregulated Rb and EZH2, strongly accelerating the development of CLL via their oncogenic cooperation, and responsible for poorer prognosis.
Cooperation of the NF-kB pathway with several other oncogenes, such as TCL-1, renders CLL moreaggressive clinically [101]; PKC-β-dependent activation of NF-κB in stromal cells is found to be essential for the survival of CLL cells in vivo [34]. Although only a small subset CLL cases overexpress NF-κB, they often show a poorer prognosis, suggesting that targeting the NF-κB pathway could be an attractive therapeutic strategy in this patient subset.
9.2. Marginal zone lymphoma
Marginal zone lymphomas represent a group of indolent small B-cell lymphomas that are subdivided into three subtypes: extranodal marginal zone lymphoma (MZL), also known as mucosa-associated lymphoid tissue (MALT) lymphoma; splenic MZL, and nodal MZL. Based on gene profiling and genome-wide sequencing, both the canonical and non-canonical NF-κB pathways can be activated among these subtypes, especially in MALT lymphoma and splenic MZL. NF-κB dysregulation can result from a variety of mechanisms, from molecular alterations and post transcriptional deregulation to epigenetic modifications.
9.2.1 Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue
MALT lymphoma commonly arises in the settingf chronic antigen stimulation, often as a result of extrinsic BCR activation attributable to an infectious agent or autoimmune stimulus. With tumor progression, the cell transitions to intrinsic activation and these advanced cases often fail to regress after eradication of the underlying extrinsic cause. There are several molecular mechanisms responsible for those advanced cases.
The first mechanism is a number of recurrent chromosomal translocations. In MALT lymphomas, there are four well-known translocations and others mentioned in case reports. The most common is t(11;18) which results in API2-MALT1 fusion. Other translocations include t (14;18)/IGH-MALT1, t (1;14)/BCL10-IGH and t (3;14)/FOXP3-IGH [6, 102]. BCL10 together with MALT1, a paracaspase, and CARD11 form a complex to recruit IKK leading to activation of the canonical NF-κB pathway. Moreover, NF-κB can be activated by the other translocations independent of upstream BCR signaling [8, 102-104].
In addition, receptor interacting protein-1, a novel API2-MALT1-associated protein, has been shown to be essential for activation of the canonical NF-κB pathway. API2-MALT1 promotes ubiquitination of receptor interacting protein-1 at lysine (K) 377, which is essential for full NF-κB activation, indicating that receptor interacting protein-1 is a critical component of API2-MALT1-dependent lymphomagenesis [7]. Another pathway involves NF-κB-inducing kinase (NIK) at arginine 325. API2-MALT1 induces proteolytic cleavage of NIK, which results in deregulated NIK activity and is associated with constitutive non-canonical NF-κB signaling, thereby leading to enhanced B-cell adhesion, and resistance to apoptosis [105] (Figure 5c).
9.2.2 Splenic marginal zone lymphoma
Splenic MZL is an indolent B-cell neoplasm that involves the spleen, abdominal lymph nodes, and the bone marrow and can have a leukemic phase. NF-κB pathway activation is very common in splenic MZL suggesting that active NF-κB is essential for the generation and/or maintenance of normal marginal zone B-cells [106]. Positive and negative mutations of NF-κB regulators are seen in 30-40% of splenic MZL patients, providing support for NF-κB dysregulation being a major contributor to the pathogenesis of this disease.
Canonical NF-κB pathway signaling is deregulated via a variety of mechanisms. TNFAIP (A20) is inactivated in 10-15% of SMZL due to nonsense or frame-shift mutations, which commonly occurs at a later event in the clonal evolution compared with KLF2 or NOTCH2 mutations [107, 108]. Lysine 171 mutations in IKBKB, which encodes the IKKβ protein, constitutively activate the canonical NF-κB pathway and occur in 10% of SMZL [109]. Additionally, the L265 MYD88 mutation occurs in 3-5% and CARD11 is mutated in 5-10% of SMZL [110].
Activation of the non-canonical NF-κB pathway accounts for 10-15% of SMZL cases. BIRC3 mutations are seen in 10% of SMZL, which, along with TRAF2 and TRAF3, activates the non-canonical NF-κB pathway [111]. Furthermore, TRAF3, as a central activator of the non-canonical NF-κB pathway, is mutated in 5% SMZL [107, 111].
9.3. Diffuse large B cell lymphoma
Diffuse large B-cell lymphoma (DLBCL) is the most common type of non-Hodgkin lymphoma, representing roughly 40% of all adult cases. There are three molecular subtypes of DLBCL distinguished by gene expression profiling signatures: activated B-cell (ABC), germinal center B-cell (GCB) and primary mediastinal B-cell lymphoma (PMBL). The activity of NF-κB is up-regulated in PMBL and ABC-DLBCL [112], but not in GCB-DLBCL. Compared with GCB-DLBCL patients with similar clinical risk factors, PMBL patients have a relatively favorable overall survival rate whereas patients with ABC-DLBCL patients have a worse outcome [113]. We have compared various clinical features between patients with DLBCL with or without expression of canonical or non-canonical pathway. DLBCL patients with predominantly activation of the non-canonical pathway of NF-κB showed a better response when treated with R-CHOP compared with DLBCL patients with canonical NF-κB pathway activation; this was true forboth the ABC and GCB subtypes. Among DLBCL patients with canonical pathway activation were more commonly men, for all patients and for ABC-DLBCL subtype, but not for GCB-DLBCL subtype.
Constitutive activation of the NF-κB pathway is hallmark of ABC-DLBCL and the canonical pathway acts as a major oncogenic event in ABC-DLBCL pathogenesis. It has been shown that various molecular mechanisms contribute to the activation of the NF-κB pathway in ABC-DLBCL. Chronic active BCR signaling h NF-κB as been identified as one of the mechanisms responsible for NF-κB activation in the ABC-DLBCL subtype. Approximately 10% of ABC-DLBCL cases have mutations in CARD 11 and 20% of cases have mutations in the ITAM of CD79B, both of which constitutively activate NF-κB [2]. When the NF-κB pathway is inhibited by using a non-degradable form of IκBa, or by treatment with a small molecule IKKβ inhibitor, the growth of ABC-DLBCL cells lines is suppressed [112]. Among the subunits of NF-κB, Iκb-ζ interaction with p50 and p52 plays an essential role in regulating a specific set of NF-κB target genes [114].
MYD88 mutations are the most frequently mutated oncogene in DLBCL. L265P MYD88 mutation is present in most ABC-DLBCL cell lines as well as about 30% of ABC-DLBCL biopsy specimens. An additional 10% of ABC-DLBCL cases have MYD88 mutations other than L265P, which occur at a comparable frequency in GCB-DLBCL.
Gene expression profiling has shown that about half of ABC-DLBCL cases express an NF-κB target gene signature, as well as a set of STAT3-dependent target genes that are associated with high IL-6 expression and STAT3 phosphorylation [115]. Once these cytokines bind to their receptors in an autocrine fashion, they activate JAK family kinases that phosphorylate the transcription factor STAT3. STAT3 can then interact with NF-κB heterodimers, subsequently enhancing transactivation of NF-κB target genes [116]. In addition, in ABC-DLBCL JAK kinase inhibitors can synergize with an IKKβ inhibitor in killing ABC-DLBCL cells indicating that both JAK kinases and NF-κB deliver survival signals [116] (Figure 5a).
Other mechanisms are involved in ABC-DLBCL pathogenesis. There are genetic alterations that interfere with terminal B cell differentiation. In normal B-cells, IRF4 is an NF-κB target gene that is activated by constitutive NF-κB activity, and which transactivates PRDM1 in antigen-stimulated normal B-cells to encode Blimp-1, a master transcriptional regulator that propels B-cells towards plasma cell differentiation [117, 118]. NF-κB and IRF4 are both up-regulated in ABC-, but not in GCB-DLBCL [117]. However, due to inactivating point mutations and deletions, epigenetic silencing or repression of transcription is induced by Bcl-6 and Spi-B [118-121]. Blimp-1 is not highly expressed in most ABC-DLBCL tumors; this leads to plasmacytic differentiation being arrested at the plasmablastic stage, eventually resulting in ABC-DLBCL [31]. This model has been supported by mice experiments [121]. In addition, miRNAs influence the development and correlate with aggressiveness of ABC-DLBCL. MiR-155, miR-21, miR-17-92, miR-15a, miR-29a/b/c, miR-34a, miR-150, miR-155, miR-21 and miR-17-92 are associated with lymphoma cell proliferation, invasion, and resistance to chemotherapy, whereas miR-15a, miR-29a/b/c, miR-34a and miR-150 are related to down-regulation of apoptosis. These data suggest that ABC-DLBCL cells are oncogenically addicted to high NF-κB activity for their survival and proliferation. Therefore, NF- κB appears to be an attractive therapeutic target in patients with ABC-DLBCL.
The NF-κB canonical and non-canonical pathways are expressed in both ABC- and GCB-DLBCL, but their prognostic impact differs significantly between this two subtypes. The expression of NF-κB, via the canonical or non-canonical pathway, is associated with a particularly poor outcome in patients with ABC-DLBCL compared with patients with GCB-DLBCL [122-124]. In ABC-DLBCL, dysregulation of the NF-κB canonical and non-canonical pathways is associated with inferior prognosis [123, 125]. In GCB-DLBCL, patients with P52+ and P52+/RelB+ which activate the non-canonical pathway are associated with better overall survival and progressive-free survival than patients with P52- and P52-/RelB- [123]; A worse overall survival occurs in patients with c-Rel+ DLBCL, associated with the canonical pathway, compared with the survival of patients c-Rel-tumors [126]. However, there are heterogeneous expression patterns in many patients and there are no well characterized studies correlating each of these subtypes. Based on our recent studies, we proposed that molecular heterogeneity responsible for canonical pathway activation is different between the DLBCL GCB and ABC subtypes. The difference is also apparent for non-canonical pathway activation between GCB and ABC subtypes in regards to signaling mechanisms. In GCB-DLBCL, canonical pathway activation predominates and is mediated through p50 activation, whereas in ABC-DLBCL p65 activation is more critical. For non-canonical pathway activation in GCB-DLBCL, RelA plays important role, whereas in ABC-DLBCL, p52 and c-REL are significantly dysregulated. These differences contribute to the underlying molecular events and likely explain the variation of response to the current NF-kB pathway inhibitors.
9.4. Epstein-Barr virus related lymphoma
Epstein-Barr virus (EBV), a ubiquitous c-herpes virus, has a tendency to preferentially infect B lymphocytes and canestablish lifelong latent infection in over 90% of the adult population worldwide. In vitro analysis has shown that human peripheral blood B lymphocytes can be transformed by EBV into continuously proliferating lymphoblastoid cell lines. Due to its preferred tendency to infect B lymphocytes, B-cell lymphomas related with EBV are common including lymphomatoid granulomatosis, lymphomas arising in immunosuppressed patients, BL, HL and EBV-positive DLBCL shown in the Table 2.
Table 2. EBV-associated B cell lymphomas and the corresponding forms of viral latency, miRNA and NF-κB subunits.
| Disease | % of EBV-related cases | Latency type | Viral proteins expressed | miRNA associated NF-κB | Involving NF-κB subunits |
|---|---|---|---|---|---|
| Burkitt lymphoma | Endemic > 95%,Sporadic 20-80%,AIDS-related 30-50%. | I | EBNA-1, EBERs, BARFO | miR-155 ↑, miR-146a ↑, miR-150 ↓. | c-Rel, RelB RelA/P65, p52/p100 |
| Hodgkin lymphoma | HIV-unrelated 20-90%b,HIV-associated≈ 100%. | II | EBNA-1, LMP-1, LMP-2A, EBERs, BARFO | miR-155 ↑, miR-17-92 ↑, miR-16 ↑, miR-21↑, miR-24↑, miR-150↓. | p50, Rel, NF-B2/p52, RelB |
| EBV+DLBCL of the elderly | ≈100% | II/III | EBNA-1, EBNA-2 (30%), LMP-1, LMP-2A, -2B, EBERs, BARF0 | miR-146b ↑, miR-222 ↑, miR-126 ↑, miR-150 ↑, miR-151 ↓. | p105/p50, P52/P100 |
| monomorphic B-cell post-transplant lymph proliferative disorder/DLBCL | 32-67% | III | LMP-1, EBNA-2, EBNA-3s | miR-155 ↑, miR-630 ↑, miR-1978 ↑, miR-211 ↑, miR-663b↑, miR-494 ↑, miR-1973 ↑, miR-21 ↑, miR-24 ↑, miR-1308 ↑, miR-222 ↑, miR-484 ↓, miR-1274b ↓. | p50, p52, RelA/P65 |
| Primary cutaneous DLBCL, leg type | 10% | EBER | miR-19b-3p ↑, miR-19a-3p↑. | P65 | |
| EBV+DLBCL | 3-15% | II/III | EBER, LMP1, EBNA2, EBNA3c | miR-155 ↑, miR-551 ↑, miR-34a ↑, miR-193b ↑, miR-365 ↑, EBV-miR-BART5 ↑, EBV-miR-BART9 ↑, BART17-5p ↑, miR-199a ↓, miR-223 ↓, miR-28 ↓, miR-150 ↓, miR-451↓. | RelA/P65 |
| Plasmablastic lymphoma | 53-68.5% | II | EBER | miR34b ↑, miR155 ↑, miR29a ↑, miR29b ↑. | c-REL |
| Immunodeficiency -associated B-cell lymphoma | 31-75% | II | EBER, LMP1, EBNA2, LMP2 | miR-34a ↑, miRs-9 ↑, miRs-221 ↑, miRs-222 ↑, miRs-146a ↑, miRs-146b ↑ and miRs-155 ↑. | REL |
Abbreviations: EBV, Epstein-Barr virus; EBNA, EBV nuclear antigens; EBER, EBV encoded RNA; AIDS, acquired immune deficiency syndrome; LMP, latent membrane protein type; DLBCL, diffuse large B-cell lymphoma.
EBV infection leads to the activation of specific viral gene expression patterns of latent infection that can be classified as types I, II, or III. Type III is characterized by the expression of all 6 EBV nuclear antigens (EBNA1, 2, 3A, 3B, 3C, and LP), latent membrane proteins (LMP1, 2A, and 2B), and EBV encoded RNA (EBER). Type II latency is related to expression of EBNA1, LMP1, LMP2A, and EBER and is common in classical HL, whereas only EBNA1 and EBER are expressed in type I latency as is characteristic of BL.
Most EBV-positive B-cell lymphomas are characterized by prominent NF-κB activation occurring via mechanisms that differ from other B-cell lymphomas. Viral antigens of EBV, in particular LMP1, is thought to be the primary pathogenic initiating factor. LMP1 functionally mimics CD40, a molecule that is expressed constitutively on the B-cell membrane and involved in B-cell activation and proliferation. The C-terminal tail of LMP1 has two distinct domains including the C-terminal activation regions 1 and 2 (CTAR1 and 2). LMP1 aggregation provides a platform for CTAR1 and CTAR2 to interact and activate the NF-κB pathway [127-129].
Our knowledge regarding the role of PD-1 in lymphomas is limited. In EBV associated malignancies such as nasopharyngeal carcinoma, LMP1 can up-regulate PD-L1 through the NF-κB pathways. In ovarian tumors, expression of PD-1 in myeloid dendritic cells paralyzes NF-κB leading to block a NF-κB-dependent cytokine release independent of adapter protein SHP-2 [130]. These findings show that PD-1 acts in a distinct manner in innate immune cells and indicating that further investigation of PD-1 and NF-κB pathways controlled in lymphomas will be essential [131].
Recent studies have shown that virally encoded miRNAs participate in EBV-mediated growth and transformation of B-cells and involve the NF-κB pathway. EBV and human miRNAs co-target oncogenic and apoptotic viral and human genes during latency [132]. NF-κB subunit p65 associated directly with a transcription start site mediates epigenetic deregulation of common miRNAs in EBV-mediated transformation of B-cells and lymphomas; this occurs together with a change at histone H3K27me3 and H3K4me3 [133] (Table 2 and Figure 4).
9.4.1 Burkitt lymphoma
Endemic (African) BL is a highly aggressive lymphoma that occurs in children and is almost always associated with EBV infection. In contrast, a sporadic form of BL occurs in developed nations and mostly lacks the virus. Restricted virus genes including EBNA-1, EBERs and distinct clusters of viral miRNA are expressed in BL cells [134] and are characterized by a very low basal level of NF-κB activity consisting of RelB-containing heterodimers.
NF-κB is activated by LMP-1 and ligand-independent signaling through overexpression CD30 in patients with BL. Most nuclei of BL cells contain the activated NF-κB subunits p50, c-Rel, RelB, and p52, but not p65, whereas sublines of EBV associated BL cells with activated p65 are resistant to anti-EBV therapy [135]. Through the CART1 and CART2, LMP-1 activates NF-κB and increases CD69 expression at the transcriptional level, which acts as a signal transducer in inflammatory processes and as an intrinsic negative modulator of T cell responses [136]. Moreover, NF-κB in combination with Myc is responsible for the phenotype, biological properties and growth pattern of cells driven into proliferation by EBV [137]. On the other hand, LMP-1 and NF-κB endow a net evolutionary benefit to the survival of BL cells. Furthermore, LMP-1 and overexpressed CD30 increase IκB-ζ expression at the transcriptional level in BL cell lines, whereas IκB- ζ inhibits NF-κB activation by LMP-1 and CD30, suggesting that NF-κB-induced IκB-ζ negatively modulates NF-κB hyper-activation to achieve homeostatic balance.
9.4.2 EBV-positive diffuse large B-cell lymphoma
EBV-associated DLBCL was included in the 2008 World Health Organization (WHO) classification as a provisional entity and designated as EBV+ DLBCL of the elderly. This entity was defined as an EBV-positive monoclonal large B-cell proliferation occurring in patients >50 years of age without known immunodeficiency or history of lymphoma. In the upcoming 2016 WHO classification, the designation of the elderly will be dropped as these tumors also occur in young patients.
The genomic features of EBV+ DLBCL remain sparsely characterized. Most of these tumors have an ABC immunophenotype and show prominent canonical and alternative NF-κB pathway activation, suggesting a strong association between NF-κB activation and the presence of EBV infection in DLBCL [138, 139]. Genes such as EZH2, CD79B, CARD11 and MYD88 are dysregulated and affect NF-κB pathway signaling in EBV-associated DLBCL [140]. EBV-mediated activation of NF-κB has an essential role in enforcing B-cell receptor signaling. CD30 expression may be another driver of EBV-associated DLBCL; CD30 is commonly expressed in EBV-associated DLBCLs and is associated with an adverse outcome [139].
Several studies have shown that cellular miRNAs, modulated by viral proteins, contribute to EBV-induced oncogenesis and activation of the NF-κB pathway. Mir-34a, induced by LMP1 through NF-κB signaling, promotes the growth of EBV-transformed cells [94]. Mir-155, especially shown in the ABC subtype, induces LMP1 via the NF-κB pathway [92]. MiR-155, through direct 3′UTR interactions, targets oncogenic and apoptotic genes as well as INPP5D (SHIP1), a key target implicated in lymphomagenesis [132, 141]. Some miRNAs indirectly contribute to NF-κB activation, for example, EBV-miR-BART5 targets and degrades p53-upregulated modulator of apoptosis [142], and EBV-miR-BART9 and BART17-5p can down-regulate BCL6 expression which represses the NF-κB pathway [143, 144] (Figure 4 and Table 2). Overall, considering the different heterogeneity of patients or cell lines in these studies, the precise function of miRNAs related to the NF-κB pathway in EBV-associated DLBCL and their prognostic value remain to be elucidated in a large series of patients.
9.5. Hodgkin lymphoma
In HL, the neoplastic HRS cells are characterized by constitutive activation of NF-κB. These cells usually represent 1% or less of all cells and are present in a reactive inflammatory cell background. The various inflammatory cell types in HL provide abundant cytokines that activate NF-κB. Jungnickel et al. found that HRS cells have deleterious IκB mutations, that play a role in the pathogenesis of HL [51].
Hodgkin lymphoma is often associated with EBV infection and EBV LMP-1 activates the canonical and non-canonical NF-κB signaling pathways. LMP1 promotes proteasome-mediated proteolysis of p100 NF-κB2 with the existence of IKKgamma/NEMO, leading to the activation of p52, which, in turn translocates to the nucleus in a complex with the p65 and RelB. In cases lacking IKKgamma/NEMO, LMP1 mediated proteolysis of p100 only results in nuclear translocation of p52 [28]. LMP-1 also induces overexpression of the polycomb group protein, Bmi-1, which down-regulates the ataxia telangiectasia-mutated (ATM) tumor suppressor, causing HL in transgenic mice through NF-κB signaling [145]. NF-κB constitutive activation also has been observed in EBV-negative HL cell lines where CD30 is responsible for its activation [146]. NF-κB mediated transcription, c-Jun activation and TRAF2 dependent signaling act as a common pathogenetic denominators of both EBV negative and EBV positive HL [147].
The non-canonical NF-κB pathway is responsible for maintaining the HL phenotype. Schwarzer et al. found that Notch1 induces the processing of the NF-κB2 gene product p100 into its p52 active form whereas Notch1 inhibition reduces NF-κB target genes as well as impairs the survival of HRS cells [148]. On the other side, p50/p50 dimers that are constitutively active in HL are induced by the overexpression of Bcl3, a protein that shares structural features with IκBα proteins, which have an anti-apoptotic effect in B and T lymphocytes and are tumorigenic in several malignancies [149] (Figure 5d, Figure 4 and Table 2).
10. Therapeutic approaches
Genetic abnormalities of NF-κB regulators result in many B-cell neoplasms becoming dependent on the NF-κB pathway. This pathway is a prime target for NF-κB therapies preferentially aimed at lymphoma cells compared with normal immune cells. However, it is predicted that long-term treatment of lymphoma patients with NF-κB inhibitors will eventually impair immune function considering its physiology, whereas short-term administration of such agents might be possible and manageable. Combining NF-κB inhibitors with cytotoxic chemotherapeutic drugs is a logical and rational therapeutic strategy that is being employed currently. Technically, the NF-κB pathway can be inhibited by targeting NF-κB components directly or indirectly by inhibiting upstream signaling pathway components. However, some agents aimed at NF-κB havenot shown significant benefit, for example, dasatinib and enzastaurin which target LYN and PKC respectively, have induced acomplete response in only 5% patients [33, 150-153]. Therefore, optimization of combination and personalized approaches is critical and needs to be well defined. Here, we focus on agents that have been used for the treatment of patients with B-cell lymphomas and have shown promising responses (Figure 6 and Table 3).
Figure 6.
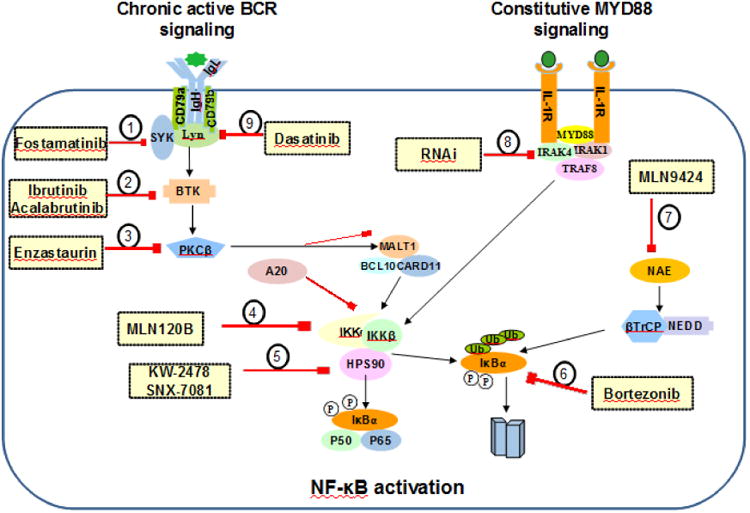
Therapeutic targets in B cell lymphoma of NF-κB activation. The pathogenesis of B cell lymphoma activation involves constitutive activation of BCR, MyD88, and NF-κB pathway promoting cell survival through NF-κB signaling pathways. Small molecule inhibitor is designed to target specific signals in these complex networks to inhibit NF-κB in B cell lymphomas, including: (1), SYK (Fostamatinib/R406). (2), BTK (Ibrutinib; Acalabrutinib). (3), PKCb (Enzastaurin). (4), IKK (MLN120B). (5), HSP90 (KW-2478; SNX-7081). (6), proteasome (Bortezomib). (7), NEDD8-activating enzyme (MLN4924). (8), IRAK4 (RNAi). (9), Src-family kinase (Dasatinib).
Table 3.
Targeted therapies for the NF-κB pathway in clinical development for B-cell malignancies directly and indirectly.
| Target/Agent | Pt Histology | Grade>3 adverse events | Clinical response | |
|---|---|---|---|---|
| NF-κB, Bortezomib (+EPOCH) | 12 ABC-DLBCL (R/R) | Platelet transfusion 52%, Thrombocytopenia 20%, Anemia 20%, Neutropenia 20%, Neutropenia and Fever 20%. | CR: 41.5%; PR: 41.5 %. | [154] |
|
| ||||
| NF-κB, Bortezomib (+rituximab) | 33 FL (untreated)5 MZL (untreated)3 SLL (untreated) | Neutropenia 5%, Fever 5%, Infection 5%, Infusion reaction 5%, Cardiac 5%, Fatigue 5%, Thrombocytopenia 2%, Diarrhea 2%, Hypokalemia 2%, Bowel obstruction 2%, Dehydration 2%. | CR: 25%; ORR:72% | [156] |
|
| ||||
| NF-κB, Bortezomib (+CHOP) | 40 DLBCL, 36MCL (untreated) | Neutropenia 25%, Thrombocytopenia 25%, Anemia 13%, Sepsis 8%, Febrile 12%. | DLBCL: CR 86%, PR14%. MCL: CR/CRu72 %, PR8 %. |
[155] |
|
| ||||
| LYN, Dasatinib | 11 CLL 17p (wt), 4 CLL 17p (del), (R/R) | Neutropenia 67%, Anemia 13%, Platelets 40%, infection 13%. | PR: 20%. | [150] |
|
| ||||
| LYN, Dasatinib+ Fludarabine | 20 CLL (fludarabine-refract ory CLL) | Monotherapy: Thrombocytopenia 4%, Hematoma 1%, Infectious 2%, Left ventricular dysfunction 2%, Toxicodermia1%, Vomiting 1%. | PR: 16.7%; Majority of patients obtained some reduction in lymph node size. | [151] |
|
| ||||
| PKC, Enzastaurin | 55 DLBCL (R/R) | Fatigue 4%; Mg 2%. | CR: 5%, SD >20 months: 7%. | [33] |
| 60 MCL (R/R) | Anemia 2%, Diarrhea 2%, Syncope 2%, Vomiting 2%. | ORR: 0%. | [152] | |
| grade 1 or 2 FL (untreated) | Grade 3 toxicity and one (1.5%) patient had grade 4 toxicity. | ORR: 26.4%, CR: 3.8%. | [153] | |
|
| ||||
| BTK, PCI-32765 | 578 (R/R) | Neutropenia 54%, Thrombocytopenia 15%, Febrile neutropenia 12%, Pneumonia 8%, Anemia 3%, Fatigue 3%. | CR 10.4%, PR72.3%. | [166] |
| 16 FL, 16 CLL/SLL, 9 MCL, 7DLBCL (R/R) | Pain 2%, Fever 2%, Respiratory nitric oxide 7%, Diarrhea 4%. | MCL: CR: 33%; PR: 44%. CLL/SLL: CR: 13%; PR: 63%. FL: CR: 19%; PR: 19%. DLBCL: PR: 29%. |
[167] | |
| 111 FL (R/R) | Neutropenia 16%, Thrombocytopenia 11%, Anemia 10%, Diarrhea 6%, Dyspnea4%. | CR: 21%; PR: 47%. | [168] | |
|
| ||||
| SYK, Fostamatinib disodium | 23DLBCL, 21FL, 11SLL/CLL, 9MCL (R/R). | Leukopenia 23%, Neutropenia 23%, Anemia 23%, Diarrhea 8%, Fatigue8%. | ORR: DLBCL22%; FL10%; SLL/CLL; 55%, MCL11%. | [161] |
Abbreviations: pt: patients; EPOCH, etoposide, prednisone, oncovin, cyclophosphamide, hydroxydaunorubicin; CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; ABC, activated B-cell like; DLBCL, diffuse large B-cell lymphoma; CLL, chronic lymphocytic leukemia; SLL, small lymphocytic lymphoma; FL, follicular lymphoma; MCL, mantle cell lymphoma; PKC, protein kinase C; BTK, Bruton tyrosine kinase; SYK, spleen tyrosine kinase; CR: complete remission, PR: partial remission, SD: stable disease, ORR: overall response rate, RR: relapsed /refractory
10.1. Bortezomib
Bortezomib, a proteasome inhibitor, suppresses NF-κB activity by preventing the degradation of phospho-IκBa. Treatment with bortezomib alone is ineffective in DLBCL patients, but combining bortezomib with cytotoxic chemotherapy was beneficial in a phase II study of patients with relapsed or refractory DLBCL; patients with ABC-DLBCL had a higher complete response rate and longer median and overall survival compared with patients with GCB-DLBCL [154, 155]. Bortezomib also has therapeutic efficacy and improves long-term survival rates in patients with untreated FL and high tumor burden [156]. Consequently, the combination of bortezomib with cytotoxic chemotherapy or other monoclonal antibody targeted agent may be a particularly effective way to improve the prognosis in patients with lymphoma associated with constitutive NF-κB activity.
10.2. MLN4924
MLN4924, a potent and selective small molecule inhibitor of NEDD8-activating enzyme (NAE), is capable of blocking phospho-IκBa degradation effectively causing NF-κB inhibition. MLN4924 can inhibit DLBCL cell line growth including ABC and GCB subtypes and can inhibit ABC-DLBCL tumors in xenograft mice models [157]. Another study found that MLN4924 is active in MCL and improves rituximab activity in vivo patient care [158]. In a phase I study, it was shown that 71% of 42 patients with relapsed and/or refractory lymphoma who had ≥1 post-baseline disease assessment achieved stable disease. Of these, 7 patients with lymphoma who received ≥5 cycles lasted stable disease 3.25 to 9.53 months [159]. Moreover, MLN4924 has dose-limited toxicities including febrile neutropenia, muscle cramps, transaminase elevations, and thrombocytopenia. Among them common grade ≥3 events were anemia (19%) and neutropenia (12%). These results provide preliminary evidence that MLN4924 may be potential for treatment in relapsed/refractory lymphoma.
10.3. Hsp90 inhibitor
Targeting HSP90 might be an alternative strategy for treating lymphoma patients. HSP90, a chaperone molecule that is an integral component of the IKK complex, prevents the proteasomal degradation of IKKα and IKKβ [29]. Several HSP90 inhibitors are now in clinical trials and early results suggest that HSP90 inhibitors are well tolerated and long-lasting stabilizations of disease can be obtained in patients with refractory solid tumor malignancies and lymphomas [160]. Further studies showed that KW-2478, a novel HSP90 inhibitor, had no dose-limiting toxicities and did not manifest retinal or ocular toxicity in patients with relapsed/refractory B-cell malignancies, whereas 24 of 25 (96%) evaluable patients showed stable disease, with 5 being free of disease progression for ≥6 months [161]. Furthermore, HSP90 inhibitors are synergistic with bortezomib and fludarabine to induce apoptosis of lymphoma cell lines [162]. The HSP90 inhibitor SNX-7081 overcomes bortezomib resistance in MCL in vitro and in vivo by down-regulation of the pro-survival ER chaperone [163], providing valuable insight into the synergistic effect and which may reduce the effective dose of the HSP90 inhibitor minimizing toxicity. However, HSP90 is dependent on numerous other client proteins for its proper function, making it difficult to combine HSP90 inhibitors with other drugs.
10.4. Fostamatinib
An attractive notion is to target upstream signaling pathways of NF-κB and other synergistic survival pathways simultaneously. Many interesting therapeutic targets, such as BTK, SYK, SRC-family kinases and PKCβ has been targeted by drugs in early phase clinical trials of patients with ABC-DLBCL. Fostamatinib is a SYK kinase inhibitor that has shown activity as a single agent in phase I/II trials involving patients with a variety of B-cell lymphoma types. Fostamatinib is generally well tolerated and produced complete and partial responses in 55% of relapsed/refractory patients in CLL/SLL [164], whereas 13% of DLBCL patients achieved linical benefit (at least stable disease). However, none of the patients with clinical benefit had an ABC signature [165].
10.5. BTK inhibitor
Ibrutinib, a highly selective BTK inhibitor, is toxic to ABC-DLBCL and CLL cells at low nanomolar concentrations. Ibrutinib has shown promising results in clinical trials for patients with relapsed or refractory DLBCL, MCL, and CLL. At a median follow-up of 17 months in phase III study of previously treated CLL/SLL patients, progression-free survival was significantly improved in the ibrutinib group, 17 months versus 13.3 months in the placebo group and rogression-free survival at 18 months was 79% in the ibrutinib group compared with 24% in the placebo group [166]. The treatment was well tolerated and the most frequent adverse events were neutropenia and nausea [166, 167]. In another trial of 111 relapsed/refractory patients with FL, 68% patients achieved an objective response and 21% had a complete response after follow-up of 15.3 months [168].
Acalabrutinib (ACP-196), a more selective and irreversible BTK inhibitor, has shown a 95% overall response rate in patients with relapsed CLL with promising safety and efficacy profiles. Notably, patients with chromosome 17p13.1 deletion had a 100% overall response rate [169]. The advantage of this BTK inhibitor lies in a cysteine residue in the active site of only 10 kinases in the human genome to covalently modify BTK, which insures highly selectivity for BTK, thereby endowing it with favorable pharmacodynamics properties. More clinical trials are in ongoing to better address the role of agents targeting BCR signaling in the future (Table 4).
Table 4.
Ongoing clinical trials of novel agents in patients of B-cell lymphomas association with NF-κB pathway inhibited.
| Agent | Targeting site | Phase | Type of patients | Clinical Trial.gov ID |
|---|---|---|---|---|
| Bortezomib | NF-κB | III | II-IV newly diagnosed T-Cell lymphoblastic lymphoma | NCT02112916 |
| NF-κB | II | High-risk II-IV stage FL | NCT01216683 | |
|
| ||||
| Dasatinib | LYN | II | Refractory DLBCL | NCT00918463 |
| LYN | I/II | Relapsed or refractory NHL | NCT00550615 | |
|
| ||||
| ACP 196 | BTK | I | Relapsed or refractory FL | NCT02180711 |
| BTK | III | Previously treated, high risk CLL | NCT02477696 | |
|
| ||||
| Ibrutinib | BTK | I/II | Refractory/recurrent primary or secondary central nervous system lymphoma | NCT02315326 |
| BTK | III | Newly diagnosed non-GCB DLBCL | NCT01855750 | |
| BTK | II | Newly diagnosed young patients with MCL |
NCT02427620 NCT02242097 |
|
| BTK | I | Treatment-naïve CLL/SLL | NCT02556892 | |
| BTK | II | Minimal residual disease in patients with CLL after front-line therapy | NCT02649387 | |
| BTK | II | Untreated young high risk DLBCL | NCT02670317 | |
| BTK | II | Advanced FL | NCT02451111 | |
| BTK | II | Newly diagnosed Epstein-Barr virus-positive DLBCL | NCT02670616 | |
| BTK | I/II | Relapsed/refractory MM | NCT02548962 | |
| BTK | I | Relapsed and refractory T-cell Lymphoma | NCT02309580 | |
|
| ||||
| Bbuparlisib | PI3K | I | Relapsed or refractory indolent B-cell lymphoma | NCT02049541 |
| PI3K | II | Refractory/recurrent primary/secondary central nervous system lymphoma | NCT01799889 | |
|
| ||||
| RP6530 | PI3K | I | Relapsed and refractory T-cell lymphoma | NCT02567656 |
|
| ||||
| CUDC-907 | PI3K | I | Refractory or relapsed lymphoma or MM | NCT01742988 |
|
| ||||
| Copanlisib | PI3K | II/III | Relapsed, indolent or aggressive NHL |
NCT01660451 NCT02626455 |
|
| ||||
| Entospletinib | SYK | II | Relapsed or refractory hematologic malignancies | NCT01799889 |
|
| ||||
| TGR-1202 and Ibrutinib | PI3K and BTK | I | CLL, MCL or other NHL |
NCT02268851 NCT02006485 |
|
| ||||
| Nelfinavir and Bortezomib | PI3K and NF-κB | I/II | Progressive MM | NCT01555281 |
|
| ||||
| Buparlisib and Ibrutinib | PI3K and BTK | I | Relapsed or refractory CLL | NCT02614508 |
|
| ||||
| Ibrutinib and Bortezomib | BTK and NF-κB | I/II | Relapsed and refractory MCL | NCT02356458 |
Abbreviations: DLBCL, diffuse large B-Cell Lymphoma; NHL, non-Hodgkin's lymphoma; CLL, chronic lymphocytic leukemia; SLL, small lymphocytic lymphoma; HL, follicular lymphoma; MCL, mantle cell lymphoma; MM, multiple myeloma.
10.6. Opportunities and challenges for targeted therapy
Concomitant inhibition of two pathways has been shown to be synergistically toxic for many lymphomas, as dual pathway blockage synergistically removes co-tumorigenisis signals and thus enhances antitumor effect. Superior antitumor activity has been shown in patients with B-cell lymphomas by combining bortezomib with cytotoxic chemotherapy or HSP90 inhibitor, as described. The combination of agents may prove particularly effective in selected patients with some positive mutant genes and this approach will likely have a promising future for the treatment of patients with B-cell malignancies. Based on promising results for trials designed to inhibit the NF-κB pathway many clinical trials of novel agents are ongoing in patients with B-cell lymphomas (Table 4).
Patients treated with these inhibitors typically experience an increase in lymphocytosis and a decrease in lymph node size, which may represent a disruption of the nodal microenvironment, thereby resulting in trafficking of lymphoma cells from the lymphoid compartment into blood. As a result, the true clinical benefit of such agents may be underestimated when judging by using traditional response criteria, as only a portion of patients may qualify as having achieved an objective response according to standard International Working Group criteria. Nevertheless the use of these inhibitors opens up new avenues for targeted therapies as they have apparent biologic and clinical activity to treat patients with B-cell lymphomas.
Each inhibitor will have off-target effects that actually might be responsible for its toxicity. Although agents are developed to inhibit a specific enzyme, it should be emphasized that more than one enzyme is often inhibited by these small molecules. Moreover, even though a drug may block only one particular enzyme, this blockage does not guarantee that effects of the drug can be ascribed soley to its ability of inhibiting that particular enzyme. Consequently, potential off-target effects of a drug may be understated.
11. Concluding remarks
The NF-κB pathway has an anti-apoptotic role and activation of this pathway is a common feature of many malignancies, independent of cell of origin. Although this statement seems simple enough, the reality is more complex.. Different B-cell lymphomas have specialized mechanisms for activating NF-κB. In addition, the subunits of the NF-κB pathway are closely associated with physiological functions and are preferentially expressed in lymphoid organs in which the development and activation of B-cell malignancies occur. The receptors that regulate the NF-κB pathway are strongly associated with the processes of immune response and lymphocyte survival. For these reasons, B-cell lymphomas act as a preferential site for the action of the NF-κB signaling. Further knowledge of the canonical and non-canonical NF-κB pathways in each of the B-cell lymphoma subtypes is needed to better define the relative role of NF-κB pathway and its components.
NF-κB is an Achilles heel of many B-cell neoplasms and therapies targeting this pathway have shown encouraging therapeutic effects with minimal toxicity in patients with these diseases. New therapies designed to specifically target NF-κB pathways are being developed. Combination chemotherapy with NF-κB pathway inhibitors has achieved a favorable outcome and may overcome tumor resistance and benefit patients. Furthermore, the multitude of available agents with various mechanisms creates diverse possibilities for combination therapy; each combination provides an opportunity to improve the therapeutic response for various types of lymphomas. Additionally, agents targeting the NF-κB pathway have shown less toxicity and appear to be well tolerated. Hence, the goal in treating patients with B-cell lymphoma should be to combine NF-κB inhibitors rationally with other targeted agents or chemotherapy to synergistically block several pathways and induce apoptosis, hopefully achieving more cures with less toxicity.
Many questions remain regarding the long-term administration of NF-κB inhibitors related to overwhelming infections. Moreover, it is also necessary to establish criteria for evaluating clinical response. Despite the preliminary results obtained from clinical trials for patients with relapsed/refractory DLBCL and CLL/SLL, it is currently unknown whether NF-κB inhibitors will be effective for patients with other types of B-cell lymphoma or as first-line therapy. Further knowledge and more clinical trials targeting NF-κB pathways among different B-cell lymphomas are needed to shed light upon the rationale for combination regiments. NF-κB inhibitors have a promising future for the treatment of patients with B-cell lymphomas.
12. Practice points.
Activation of the NF-κB pathway plays a vital role in the pathogenesis of lymphoma as a downstream effector of the BCR pathway to convey pro-survival and proliferation signals in the neoplastic B cells.
NF-κB blocks the intrinsic apoptotic pathway and is an antagonist to p53 transactivation by down-regulation Bcl-2 family.
MiRNAs, TME, and viral antigens of EBV in association with NF-κB participate in the construction of a complex biologic network in the context of B-cell lymphomas.
NF-κB pathway inhibitors have shown encouraging therapeutic effects in patients with relapsed/refractory lymphomas, including DLBCL, MCL, CLL/SLL, and FL.
NF-κB inhibitors, combined with immunotherapy agents, are under robust clinical evaluation and show great promise in cancer immunotherapy.
13. Research agenda.
Improved understanding the mechanisms of the canonical and non-canonical NF-κB pathways in each type of B-cell lymphoma.
Identify the genetic basis and possible environmental factors of NF-κB pathways in the regulation of normal and different malignant B-cell biology.
Assessment of the NF-κB pathway or its subunits in various types of B-cell lymphoma for improved risk stratification.
More clinical trials targeting NF-κB pathways among different B-cell lymphomas as first-line therapy or with combination regiments.
Acknowledgments
This study was supported by the National Cancer Institute/National Institutes of Health (R01CA138688 and 1RC1CA146299). LY and LL are the recipient of hematology/oncology scholarship award. The study is supported by The University of Texas MD Anderson Cancer Center Lymphoma Moonshot Program, Institutional Research and Development Fund, an Institutional Research Grant Award, an MD Anderson Cancer Center Lymphoma Specialized Programs on Research Excellence (SPORE) Research Development Program Award, an MD Anderson Cancer Center Myeloma SPORE Research Development Program Award, a Gunderson Lutheran Medical Foundation Award, and partially supported by the National Cancer Institute/National Institutes of Health (P50CA136411 and P50CA142509), and by MD Anderson's Cancer Center Support Grant CA016672. This research was also supported by funds from the University Cancer Foundation via the Sister Institution Network Fund at The University of Texas MD Anderson Cancer Center.
Footnotes
Conflict of interest: KHY receives research support from Roche Molecular System, Gilead Sciences Pharmaceutical, Seattle Genetics, Dai Sanyo Pharmaceutical, Adaptive Biotechnology, Incyte Pharmaceutical, and HTG Molecular Diagnostics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood. 2009;114:3367–3375. doi: 10.1182/blood-2009-06-225326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer. 2014;14:517–534. doi: 10.1038/nrc3774. [DOI] [PubMed] [Google Scholar]
- 4.Gires O, Zimber-Strobl U, Gonnella R, Ueffing M, Marschall G, Zeidler R, et al. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997;16:6131–6140. doi: 10.1093/emboj/16.20.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thorley-Lawson DA, Allday MJ. The curious case of the tumour virus: 50 years of Burkitt's lymphoma. Nat Rev Microbiol. 2008;6:913–924. doi: 10.1038/nrmicro2015. [DOI] [PubMed] [Google Scholar]
- 6.Maes B, Demunter A, Peeters B, De Wolf-Peeters C. BCL10 mutation does not represent an important pathogenic mechanism in gastric MALT-type lymphoma, and the presence of the API2-MLT fusion is associated with aberrant nuclear BCL10 expression. Blood. 2002;99:1398–1404. doi: 10.1182/blood.v99.4.1398. [DOI] [PubMed] [Google Scholar]
- 7.Rosebeck S, Rehman AO, Apel IJ, Kohrt D, Appert A, O'Donnell MA, et al. The API2-MALT1 fusion exploits TNFR pathway-associated RIP1 ubiquitination to promote oncogenic NF-kappaB signaling. Oncogene. 2014;33:2520–2530. doi: 10.1038/onc.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Q, Siebert R, Yan M, Hinzmann B, Cui X, Xue L, et al. Inactivating mutations and overexpression of BCL10, a caspase recruitment domain-containing gene, in MALT lymphoma with t(1;14)(p22;q32) Nat Genet. 1999;22:63–68. doi: 10.1038/8767. [DOI] [PubMed] [Google Scholar]
- 9.Kim SW, Ramasamy K, Bouamar H, Lin AP, Jiang D, Aguiar RC. MicroRNAs miR-125a and miR-125b constitutively activate the NF-kappaB pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20) Proc Natl Acad Sci U S A. 2012;109:7865–7870. doi: 10.1073/pnas.1200081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 11.Huang DB, Vu D, Ghosh G. NF-kappaB RelB forms an intertwined homodimer. Structure. 2005;13:1365–1373. doi: 10.1016/j.str.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 12.Hinz M, Arslan SC, Scheidereit C. It takes two to tango: IkappaBs, the multifunctional partners of NF-kappaB. Immunol Rev. 2012;246:59–76. doi: 10.1111/j.1600-065X.2012.01102.x. [DOI] [PubMed] [Google Scholar]
- 13.Liu F, Xia Y, Parker AS, Verma IM. IKK biology. Immunol Rev. 2012;246:239–253. doi: 10.1111/j.1600-065X.2012.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanarek N, Ben-Neriah Y. Regulation of NF-kappaB by ubiquitination and degradation of the IkappaBs. Immunol Rev. 2012;246:77–94. doi: 10.1111/j.1600-065X.2012.01098.x. [DOI] [PubMed] [Google Scholar]
- 15.Baldwin AS. Regulation of cell death and autophagy by IKK and NF-kappaB: critical mechanisms in immune function and cancer. Immunol Rev. 2012;246:327–345. doi: 10.1111/j.1600-065X.2012.01095.x. [DOI] [PubMed] [Google Scholar]
- 16.Pomerantz JL, Baltimore D. Two pathways to NF-kappaB. Mol Cell. 2002;10:693–695. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- 17.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Jimi E, Phillips RJ, Rincon M, Voll R, Karasuyama H, Flavell R, et al. Activation of NF-kappaB promotes the transition of large, CD43+ pre-B cells to small, CD43- pre-B cells. Int Immunol. 2005;17:815–825. doi: 10.1093/intimm/dxh263. [DOI] [PubMed] [Google Scholar]
- 19.Derudder E, Cadera EJ, Vahl JC, Wang J, Fox CJ, Zha S, et al. Development of immunoglobulin lambda-chain-positive B cells, but not editing of immunoglobulin kappa-chain, depends on NF-kappaB signals. Nat Immunol. 2009;10:647–654. doi: 10.1038/ni.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castro I, Wright JA, Damdinsuren B, Hoek KL, Carlesso G, Shinners NP, et al. B cell receptor-mediated sustained c-Rel activation facilitates late transitional B cell survival through control of B cell activating factor receptor and NF-kappaB2. J Immunol. 2009;182:7729–7737. doi: 10.4049/jimmunol.0803281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith SH, Cancro MP. Cutting edge: B cell receptor signals regulate BLyS receptor levels in mature B cells and their immediate progenitors. J Immunol. 2003;170:5820–5823. doi: 10.4049/jimmunol.170.12.5820. [DOI] [PubMed] [Google Scholar]
- 22.Mora AL, Stanley S, Armistead W, Chan AC, Boothby M. Inefficient ZAP-70 phosphorylation and decreased thymic selection in vivo result from inhibition of NF-kappaB/Rel. J Immunol. 2001;167:5628–5635. doi: 10.4049/jimmunol.167.10.5628. [DOI] [PubMed] [Google Scholar]
- 23.Oh H, Ghosh S. NF-kappaB: roles and regulation in different CD4(+) T-cell subsets. Immunol Rev. 2013;252:41–51. doi: 10.1111/imr.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molinero LL, Cubre A, Mora-Solano C, Wang Y, Alegre ML. T cell receptor/CARMA1/NF-kappaB signaling controls T-helper (Th) 17 differentiation. Proc Natl Acad Sci U S A. 2012;109:18529–18534. doi: 10.1073/pnas.1204557109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerondakis S, Banerjee A, Grigoriadis G, Vasanthakumar A, Gugasyan R, Sidwell T, et al. NF-kappaB subunit specificity in hemopoiesis. Immunol Rev. 2012;246:272–285. doi: 10.1111/j.1600-065X.2011.01090.x. [DOI] [PubMed] [Google Scholar]
- 26.Gasparini C, Celeghini C, Monasta L, Zauli G. NF-kappaB pathways in hematological malignancies. Cell Mol Life Sci. 2014;71:2083–2102. doi: 10.1007/s00018-013-1545-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohamed AJ, Yu L, Backesjo CM, Vargas L, Faryal R, Aints A, et al. Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228:58–73. doi: 10.1111/j.1600-065X.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 28.Eliopoulos AG, Caamano JH, Flavell J, Reynolds GM, Murray PG, Poyet JL, et al. Epstein-Barr virus-encoded latent infection membrane protein 1 regulates the processing of p100 NF-kappaB2 to p52 via an IKKgamma/NEMO-independent signalling pathway. Oncogene. 2003;22:7557–7569. doi: 10.1038/sj.onc.1207120. [DOI] [PubMed] [Google Scholar]
- 29.Broemer M, Krappmann D, Scheidereit C. Requirement of Hsp90 activity for IkappaB kinase (IKK) biosynthesis and for constitutive and inducible IKK and NF-kappaB activation. Oncogene. 2004;23:5378–5386. doi: 10.1038/sj.onc.1207705. [DOI] [PubMed] [Google Scholar]
- 30.Walter HS, Rule SA, Dyer MJ, Karlin L, Jones C, Cazin B, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood. 2016;127:411–419. doi: 10.1182/blood-2015-08-664086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niemann CU, Wiestner A. B-cell receptor signaling as a driver of lymphoma development and evolution. Semin Cancer Biol. 2013;23:410–421. doi: 10.1016/j.semcancer.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saijo K, Mecklenbrauker I, Santana A, Leitger M, Schmedt C, Tarakhovsky A. Protein kinase C beta controls nuclear factor kappaB activation in B cells through selective regulation of the IkappaB kinase alpha. J Exp Med. 2002;195:1647–1652. doi: 10.1084/jem.20020408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holler C, Pinon JD, Denk U, Heyder C, Hofbauer S, Greil R, et al. PKCbeta is essential for the development of chronic lymphocytic leukemia in the TCL1 transgenic mouse model: validation of PKCbeta as a therapeutic target in chronic lymphocytic leukemia. Blood. 2009;113:2791–2794. doi: 10.1182/blood-2008-06-160713. [DOI] [PubMed] [Google Scholar]
- 34.Lutzny G, Kocher T, Schmidt-Supprian M, Rudelius M, Klein-Hitpass L, Finch AJ, et al. Protein kinase c-beta-dependent activation of NF-kappaB in stromal cells is indispensable for the survival of chronic lymphocytic leukemia B cells in vivo. Cancer Cell. 2013;23:77–92. doi: 10.1016/j.ccr.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim SW, Oleksyn DW, Rossi RM, Jordan CT, Sanz I, Chen L, et al. Protein kinase C-associated kinase is required for NF-kappaB signaling and survival in diffuse large B-cell lymphoma cells. Blood. 2008;111:1644–1653. doi: 10.1182/blood-2007-05-088591. [DOI] [PubMed] [Google Scholar]
- 36.Naylor TL, Tang H, Ratsch BA, Enns A, Loo A, Chen L, et al. Protein kinase C inhibitor sotrastaurin selectively inhibits the growth of CD79 mutant diffuse large B-cell lymphomas. Cancer Res. 2011;71:2643–2653. doi: 10.1158/0008-5472.CAN-10-2525. [DOI] [PubMed] [Google Scholar]
- 37.Beckwith M, Jorgensen G, Longo DL. The protein product of the proto-oncogene c-cbl forms a complex with phosphatidylinositol 3-kinase p85 and CD19 in anti-IgM-stimulated human B-lymphoma cells. Blood. 1996;88:3502–3507. [PubMed] [Google Scholar]
- 38.Inabe K, Kurosaki T. Tyrosine phosphorylation of B-cell adaptor for phosphoinositide 3-kinase is required for Akt activation in response to CD19 engagement. Blood. 2002;99:584–589. doi: 10.1182/blood.v99.2.584. [DOI] [PubMed] [Google Scholar]
- 39.Herman SE, Johnson AJ. Molecular pathways: targeting phosphoinositide 3-kinase p110-delta in chronic lymphocytic leukemia. Clin Cancer Res. 2012;18:4013–4018. doi: 10.1158/1078-0432.CCR-11-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baracho GV, Miletic AV, Omori SA, Cato MH, Rickert RC. Emergence of the PI3-kinase pathway as a central modulator of normal and aberrant B cell differentiation. Curr Opin Immunol. 2011;23:178–183. doi: 10.1016/j.coi.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139:573–586. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jellusova J, Miletic AV, Cato MH, Lin WW, Hu Y, Bishop GA, et al. Context-specific BAFF-R signaling by the NF-kappaB and PI3K pathways. Cell Rep. 2013;5:1022–1035. doi: 10.1016/j.celrep.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kloo B, Nagel D, Pfeifer M, Grau M, Duwel M, Vincendeau M, et al. Critical role of PI3K signaling for NF-kappaB-dependent survival in a subset of activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci U S A. 2011;108:272–277. doi: 10.1073/pnas.1008969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen L, Monti S, Juszczynski P, Ouyang J, Chapuy B, Neuberg D, et al. SYK inhibition modulates distinct PI3K/AKT- dependent survival pathways and cholesterol biosynthesis in diffuse large B cell lymphomas. Cancer Cell. 2013;23:826–838. doi: 10.1016/j.ccr.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh S, Gifford AM, Riviere LR, Tempst P, Nolan GP, Baltimore D. Cloning of the p50 DNA binding subunit of NF-kappa B: homology to rel and dorsal. Cell. 1990;62:1019–1029. doi: 10.1016/0092-8674(90)90276-k. [DOI] [PubMed] [Google Scholar]
- 47.Kieran M, Blank V, Logeat F, Vandekerckhove J, Lottspeich F, Le Bail O, et al. The DNA binding subunit of NF-kappa B is identical to factor KBF1 and homologous to the rel oncogene product. Cell. 1990;62:1007–1018. doi: 10.1016/0092-8674(90)90275-j. [DOI] [PubMed] [Google Scholar]
- 48.Bargou RC, Leng C, Krappmann D, Emmerich F, Mapara MY, Bommert K, et al. High-level nuclear NF-kappa B and Oct-2 is a common feature of cultured Hodgkin/Reed-Sternberg cells. Blood. 1996;87:4340–4347. [PubMed] [Google Scholar]
- 49.Emmerich F, Meiser M, Hummel M, Demel G, Foss HD, Jundt F, et al. Overexpression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in Reed-Sternberg cells. Blood. 1999;94:3129–3134. [PubMed] [Google Scholar]
- 50.Cabannes E, Khan G, Aillet F, Jarrett RF, Hay RT. Mutations in the IkBa gene in Hodgkin's disease suggest a tumour suppressor role for IkappaBalpha. Oncogene. 1999;18:3063–3070. doi: 10.1038/sj.onc.1202893. [DOI] [PubMed] [Google Scholar]
- 51.Jungnickel B, Staratschek-Jox A, Brauninger A, Spieker T, Wolf J, Diehl V, et al. Clonal deleterious mutations in the IkappaBalpha gene in the malignant cells in Hodgkin's lymphoma. J Exp Med. 2000;191:395–402. doi: 10.1084/jem.191.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 53.Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–721. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu H, Ruskon-Fourmestraux A, Lavergne-Slove A, Ye H, Molina T, Bouhnik Y, et al. Resistance of t(11;18) positive gastric mucosa-associated lymphoid tissue lymphoma to Helicobacter pylori eradication therapy. Lancet. 2001;357:39–40. doi: 10.1016/S0140-6736(00)03571-6. [DOI] [PubMed] [Google Scholar]
- 55.Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12:229–243. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lenz G, Nagel I, Siebert R, Roschke AV, Sanger W, Wright GW, et al. Aberrant immunoglobulin class switch recombination and switch translocations in activated B cell-like diffuse large B cell lymphoma. J Exp Med. 2007;204:633–643. doi: 10.1084/jem.20062041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Staudt LM. A closer look at follicular lymphoma. N Engl J Med. 2007;356:741–742. doi: 10.1056/NEJMcibr067155. [DOI] [PubMed] [Google Scholar]
- 60.Vaandrager JW, Schuuring E, Kluin-Nelemans HC, Dyer MJ, Raap AK, Kluin PM. DNA fiber fluorescence in situ hybridization analysis of immunoglobulin class switching in B-cell neoplasia: aberrant CH gene rearrangements in follicle center-cell lymphoma. Blood. 1998;92:2871–2878. [PubMed] [Google Scholar]
- 61.Johnson RF, Perkins ND. Nuclear factor-kappaB, p53, and mitochondria: regulation of cellular metabolism and the Warburg effect. Trends Biochem Sci. 2012;37:317–324. doi: 10.1016/j.tibs.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 62.Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell. 2002;1:493–503. doi: 10.1016/s1535-6108(02)00068-5. [DOI] [PubMed] [Google Scholar]
- 63.Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999;19:3485–3495. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang WC, Ju TK, Hung MC, Chen CC. Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol Cell. 2007;26:75–87. doi: 10.1016/j.molcel.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schumm K, Rocha S, Caamano J, Perkins ND. Regulation of p53 tumour suppressor target gene expression by the p52 NF-kappaB subunit. EMBO J. 2006;25:4820–4832. doi: 10.1038/sj.emboj.7601343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Keimpema M, Gruneberg LJ, Mokry M, van Boxtel R, Koster J, Coffer PJ, et al. FOXP1 directly represses transcription of proapoptotic genes and cooperates with NF-kappaB to promote survival of human B cells. Blood. 2014;124:3431–3440. doi: 10.1182/blood-2014-01-553412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stoffel A, Chaurushiya M, Singh B, Levine AJ. Activation of NF-kappaB and inhibition of p53-mediated apoptosis by API2/mucosa-associated lymphoid tissue 1 fusions promote oncogenesis. Proc Natl Acad Sci U S A. 2004;101:9079–9084. doi: 10.1073/pnas.0402415101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramachandiran S, Adon A, Guo X, Wang Y, Wang H, Chen Z, et al. Chromosome instability in diffuse large B cell lymphomas is suppressed by activation of the noncanonical NF-kappaB pathway. Int J Cancer. 2015;136:2341–2351. doi: 10.1002/ijc.29301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Musilova K, Mraz M. MicroRNAs in B-cell lymphomas: how a complex biology gets more complex. Leukemia. 2015;29:1004–1017. doi: 10.1038/leu.2014.351. [DOI] [PubMed] [Google Scholar]
- 71.Jeong D, Kim J, Nam J, Sun H, Lee YH, Lee TJ, et al. MicroRNA-124 links p53 to the NF-kappaB pathway in B-cell lymphomas. Leukemia. 2015;29:1868–1874. doi: 10.1038/leu.2015.101. [DOI] [PubMed] [Google Scholar]
- 72.Mathas S, Hinz M, Anagnostopoulos I, Krappmann D, Lietz A, Jundt F, et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J. 2002;21:4104–4113. doi: 10.1093/emboj/cdf389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu J, Zhou P, Wang W, Sun A, Guo F. RelB, together with RelA, sustains cell survival and confers proteasome inhibitor sensitivity of chronic lymphocytic leukemia cells from bone marrow. J Mol Med (Berl) 2014;92:77–92. doi: 10.1007/s00109-013-1081-6. [DOI] [PubMed] [Google Scholar]
- 74.Lwin T, Hazlehurst LA, Li Z, Dessureault S, Sotomayor E, Moscinski LC, et al. Bone marrow stromal cells prevent apoptosis of lymphoma cells by upregulation of anti-apoptotic proteins associated with activation of NF-kappaB (RelB/p52) in non-Hodgkin's lymphoma cells. Leukemia. 2007;21:1521–1531. doi: 10.1038/sj.leu.2404723. [DOI] [PubMed] [Google Scholar]
- 75.Gregoire M, Guilloton F, Pangault C, Mourcin F, Sok P, Latour M, et al. Neutrophils trigger a NF-kappaB dependent polarization of tumor-supportive stromal cells in germinal center B-cell lymphomas. Oncotarget. 2015;6:16471–16487. doi: 10.18632/oncotarget.4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim YR, Eom KS. Simultaneous Inhibition of CXCR4 and VLA-4 Exhibits Combinatorial Effect in Overcoming Stroma-Mediated Chemotherapy Resistance in Mantle Cell Lymphoma Cells. Immune Netw. 2014;14:296–306. doi: 10.4110/in.2014.14.6.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Coscia M, Pantaleoni F, Riganti C, Vitale C, Rigoni M, Peola S, et al. IGHV unmutated CLL B cells are more prone to spontaneous apoptosis and subject to environmental prosurvival signals than mutated CLL B cells. Leukemia. 2011;25:828–837. doi: 10.1038/leu.2011.12. [DOI] [PubMed] [Google Scholar]
- 78.O'Sullivan BJ, MacDonald KP, Pettit AR, Thomas R. RelB nuclear translocation regulates B cell MHC molecule, CD40 expression, and antigen-presenting cell function. Proc Natl Acad Sci U S A. 2000;97:11421–11426. doi: 10.1073/pnas.97.21.11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boll B, Eltaib F, Reiners KS, von Tresckow B, Tawadros S, Simhadri VR, et al. Heat shock protein 90 inhibitor BIIB021 (CNF2024) depletes NF-kappaB and sensitizes Hodgkin's lymphoma cells for natural killer cell-mediated cytotoxicity. Clin Cancer Res. 2009;15:5108–5116. doi: 10.1158/1078-0432.CCR-09-0213. [DOI] [PubMed] [Google Scholar]
- 80.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc Natl Acad Sci U S A. 2007;104:7080–7085. doi: 10.1073/pnas.0702409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lawrie CH, Soneji S, Marafioti T, Cooper CD, Palazzo S, Paterson JC, et al. MicroRNA expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. Int J Cancer. 2007;121:1156–1161. doi: 10.1002/ijc.22800. [DOI] [PubMed] [Google Scholar]
- 83.Zhao JJ, Lin J, Lwin T, Yang H, Guo J, Kong W, et al. microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood. 2010;115:2630–2639. doi: 10.1182/blood-2009-09-243147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roehle A, Hoefig KP, Repsilber D, Thorns C, Ziepert M, Wesche KO, et al. MicroRNA signatures characterize diffuse large B-cell lymphomas and follicular lymphomas. Br J Haematol. 2008;142:732–744. doi: 10.1111/j.1365-2141.2008.07237.x. [DOI] [PubMed] [Google Scholar]
- 86.Vargova K, Curik N, Burda P, Basova P, Kulvait V, Pospisil V, et al. MYB transcriptionally regulates the miR-155 host gene in chronic lymphocytic leukemia. Blood. 2011;117:3816–3825. doi: 10.1182/blood-2010-05-285064. [DOI] [PubMed] [Google Scholar]
- 87.Malumbres R, Sarosiek KA, Cubedo E, Ruiz JW, Jiang X, Gascoyne RD, et al. Differentiation stage-specific expression of microRNAs in B lymphocytes and diffuse large B-cell lymphomas. Blood. 2009;113:3754–3764. doi: 10.1182/blood-2008-10-184077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Costinean S, Sandhu SK, Pedersen IM, Tili E, Trotta R, Perrotti D, et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood. 2009;114:1374–1382. doi: 10.1182/blood-2009-05-220814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thompson RC, Herscovitch M, Zhao I, Ford TJ, Gilmore TD. NF-kappaB down-regulates expression of the B-lymphoma marker CD10 through a miR-155/PU.1 pathway. J Biol Chem. 2011;286:1675–1682. doi: 10.1074/jbc.M110.177063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thompson RC, Vardinogiannis I, Gilmore TD. Identification of an NF-kappaB p50/p65-responsive site in the human MIR155HG promoter. BMC Mol Biol. 2013;14:24. doi: 10.1186/1471-2199-14-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gatto G, Rossi A, Rossi D, Kroening S, Bonatti S, Mallardo M. Epstein-Barr virus latent membrane protein 1 trans-activates miR-155 transcription through the NF-kappaB pathway. Nucleic Acids Res. 2008;36:6608–6619. doi: 10.1093/nar/gkn666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Motsch N, Pfuhl T, Mrazek J, Barth S, Grasser FA. Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) induces the expression of the cellular microRNA miR-146a. RNA Biol. 2007;4:131–137. doi: 10.4161/rna.4.3.5206. [DOI] [PubMed] [Google Scholar]
- 94.Forte E, Salinas RE, Chang C, Zhou T, Linnstaedt SD, Gottwein E, et al. The Epstein-Barr virus (EBV)-induced tumor suppressor microRNA MiR-34a is growth promoting in EBV-infected B cells. J Virol. 2012;86:6889–6898. doi: 10.1128/JVI.07056-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Contreras JR, Palanichamy JK, Tran TM, Fernando TR, Rodriguez-Malave NI, Goswami N, et al. MicroRNA-146a modulates B-cell oncogenesis by regulating Egr1. Oncotarget. 2015;6:11023–11037. doi: 10.18632/oncotarget.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arribas AJ, Campos-Martin Y, Gomez-Abad C, Algara P, Sanchez-Beato M, Rodriguez-Pinilla MS, et al. Nodal marginal zone lymphoma: gene expression and miRNA profiling identify diagnostic markers and potential therapeutic targets. Blood. 2012;119:e9–e21. doi: 10.1182/blood-2011-02-339556. [DOI] [PubMed] [Google Scholar]
- 97.Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mansouri L, Sutton LA, Ljungstrom V, Bondza S, Arngarden L, Bhoi S, et al. Functional loss of IkappaBepsilon leads to NF-kappaB deregulation in aggressive chronic lymphocytic leukemia. J Exp Med. 2015;212:833–843. doi: 10.1084/jem.20142009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tuveson D, Rai KR. Augmenting NF-kappaB in poor-risk CLL: A general paradigm for other cancers? J Exp Med. 2015;212:830–831. doi: 10.1084/jem.2126insight4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Iannetti A, Ledoux AC, Tudhope SJ, Sellier H, Zhao B, Mowla S, et al. Regulation of p53 and Rb links the alternative NF-kappaB pathway to EZH2 expression and cell senescence. PLoS Genet. 2014;10:e1004642. doi: 10.1371/journal.pgen.1004642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zanesi N, Balatti V, Riordan J, Burch A, Rizzotto L, Palamarchuk A, et al. A Sleeping Beauty screen reveals NF-kB activation in CLL mouse model. Blood. 2013;121:4355–4358. doi: 10.1182/blood-2013-02-486035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Willis TG, Jadayel DM, Du MQ, Peng H, Perry AR, Abdul-Rauf M, et al. Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell. 1999;96:35–45. doi: 10.1016/s0092-8674(00)80957-5. [DOI] [PubMed] [Google Scholar]
- 103.Akagi T, Motegi M, Tamura A, Suzuki R, Hosokawa Y, Suzuki H, et al. A novel gene, MALT1 at 18q21, is involved in t(11;18) (q21;q21) found in low-grade B-cell lymphoma of mucosa-associated lymphoid tissue. Oncogene. 1999;18:5785–5794. doi: 10.1038/sj.onc.1203018. [DOI] [PubMed] [Google Scholar]
- 104.McAllister-Lucas LM, Baens M, Lucas PC. MALT1 protease: a new therapeutic target in B lymphoma and beyond? Clin Cancer Res. 2011;17:6623–6631. doi: 10.1158/1078-0432.CCR-11-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rosebeck S, Madden L, Jin X, Gu S, Apel IJ, Appert A, et al. Cleavage of NIK by the API2-MALT1 fusion oncoprotein leads to noncanonical NF-kappaB activation. Science. 2011;331:468–472. doi: 10.1126/science.1198946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pillai S, Cariappa A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat Rev Immunol. 2009;9:767–777. doi: 10.1038/nri2656. [DOI] [PubMed] [Google Scholar]
- 107.Clipson A, Wang M, de Leval L, Ashton-Key M, Wotherspoon A, Vassiliou G, et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia. 2015;29:1177–1185. doi: 10.1038/leu.2014.330. [DOI] [PubMed] [Google Scholar]
- 108.Parry M, Rose-Zerilli MJ, Ljungstrom V, Gibson J, Wang J, Walewska R, et al. Genetics and Prognostication in Splenic Marginal Zone Lymphoma: Revelations from Deep Sequencing. Clin Cancer Res. 2015;21:4174–4183. doi: 10.1158/1078-0432.CCR-14-2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spina V, Rossi D. NF-kappaB deregulation in splenic marginal zone lymphoma. Semin Cancer Biol. 2016 doi: 10.1016/j.semcancer.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 110.Yan Q, Huang Y, Watkins AJ, Kocialkowski S, Zeng N, Hamoudi RA, et al. BCR and TLR signaling pathways are recurrently targeted by genetic changes in splenic marginal zone lymphomas. Haematologica. 2012;97:595–598. doi: 10.3324/haematol.2011.054080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rossi D, Deaglio S, Dominguez-Sola D, Rasi S, Vaisitti T, Agostinelli C, et al. Alteration of BIRC3 and multiple other NF-kappa B pathway genes in splenic marginal zone lymphoma. Blood. 2011;118:4930–4934. doi: 10.1182/blood-2011-06-359166. [DOI] [PubMed] [Google Scholar]
- 112.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194:1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–1947. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- 114.Nogai H, Wenzel SS, Hailfinger S, Grau M, Kaergel E, Seitz V, et al. IkappaB-zeta controls the constitutive NF-kappaB target gene network and survival of ABC DLBCL. Blood. 2013;122:2242–2250. doi: 10.1182/blood-2013-06-508028. [DOI] [PubMed] [Google Scholar]
- 115.Ya ng J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007;21:1396–1408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lam LT, Wright G, Davis RE, Lenz G, Farinha P, Dang L, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood. 2008;111:3701–3713. doi: 10.1182/blood-2007-09-111948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. 2006;7:773–782. doi: 10.1038/ni1357. [DOI] [PubMed] [Google Scholar]
- 118.Mandelbaum J, Bhagat G, Tang H, Mo T, Brahmachary M, Shen Q, et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell. 2010;18:568–579. doi: 10.1016/j.ccr.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tam W, Gomez M, Chadburn A, Lee JW, Chan WC, Knowles DM. Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood. 2006;107:4090–4100. doi: 10.1182/blood-2005-09-3778. [DOI] [PubMed] [Google Scholar]
- 120.Schmidlin H, Diehl SA, Nagasawa M, Scheeren FA, Schotte R, Uittenbogaart CH, et al. Spi-B inhibits human plasma cell differentiation by repressing BLIMP1 and XBP-1 expression. Blood. 2008;112:1804–1812. doi: 10.1182/blood-2008-01-136440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Calado DP, Zhang B, Srinivasan L, Sasaki Y, Seagal J, Unitt C, et al. Constitutive canonical NF-kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell. 2010;18:580–589. doi: 10.1016/j.ccr.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li L, Xu-Monette ZY, Ok CY, Tzankov A, Manyam GC, Sun RF, et al. Prognostic impact of c-Rel nuclear expression and REL amplification and crosstalk between c-Rel and the p53 pathway in diffuse large B-cell lymphoma. Oncotarget. 2015;6:23157–23180. doi: 10.18632/oncotarget.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ok CY, Xu-Monette ZY, Li L, Manyam GC, Montes-Moreno S, Tzankov A, et al. Evaluation of NF-kappa B subunit expression and signaling pathway activation demonstrates that p52 expression confers better outcome in germinal center B-cell-like diffuse large B-cell lymphoma in association with CD30 and BCL2 functions. Mod Pathol. 2015;28:1202–1213. doi: 10.1038/modpathol.2015.76. [DOI] [PubMed] [Google Scholar]
- 124.Odqvist L, Montes-Moreno S, Sanchez-Pacheco RE, Young KH, Martin-Sanchez E, Cereceda L, et al. NFkappaB expression is a feature of both activated B-cell-like and germinal center B-cell-like subtypes of diffuse large B-cell lymphoma. Mod Pathol. 2014;27:1331–1337. doi: 10.1038/modpathol.2014.34. [DOI] [PubMed] [Google Scholar]
- 125.Aad G, Abbott B, Abdallah J, Abdel Khalek S, Abdelalim AA, Abdesselam A, et al. Search for pair production of a new b′ quark that decays into a Z boson and a bottom quark with the ATLAS detector. Phys Rev Lett. 2012;109:071801. doi: 10.1103/PhysRevLett.109.071801. [DOI] [PubMed] [Google Scholar]
- 126.Curry CV, Ewton AA, Olsen RJ, Logan BR, Preti HA, Liu YC, et al. Prognostic impact of C-REL expression in diffuse large B-cell lymphoma. J Hematop. 2009;2:20–26. doi: 10.1007/s12308-009-0021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Eliopoulos AG, Young LS. LMP1 structure and signal transduction. Semin Cancer Biol. 2001;11:435–444. doi: 10.1006/scbi.2001.0410. [DOI] [PubMed] [Google Scholar]
- 128.Izumi KM, Kieff ED. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc Natl Acad Sci U S A. 1997;94:12592–12597. doi: 10.1073/pnas.94.23.12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mainou BA, Everly DN, Jr, Raab-Traub N. Unique signaling properties of CTAR1 in LMP1-mediated transformation. J Virol. 2007;81:9680–9692. doi: 10.1128/JVI.01001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fang W, Zhang J, Hong S, Zhan J, Chen N, Qin T, et al. EBV-driven LMP1 and IFN-gamma up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget. 2014;5:12189–12202. doi: 10.18632/oncotarget.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Karyampudi L, Lamichhane P, Krempski J, Kalli KR, Behrens MD, Vargas DM, et al. PD-1 Blunts the Function of Ovarian Tumor-Infiltrating Dendritic Cells by Inactivating NF-kappaB. Cancer Res. 2016;76:239–250. doi: 10.1158/0008-5472.CAN-15-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Riley KJ, Rabinowitz GS, Yario TA, Luna JM, Darnell RB, Steitz JA. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. 2012;31:2207–2221. doi: 10.1038/emboj.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vento-Tormo R, Rodriguez-Ubreva J, Lisio LD, Islam AB, Urquiza JM, Hernando H, et al. NF-kappaB directly mediates epigenetic deregulation of common microRNAs in Epstein-Barr virus-mediated transformation of B-cells and in lymphomas. Nucleic Acids Res. 2014;42:11025–11039. doi: 10.1093/nar/gku826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Qiu J, Cosmopoulos K, Pegtel M, Hopmans E, Murray P, Middeldorp J, et al. A novel persistence associated EBV miRNA expression profile is disrupted in neoplasia. PLoS Pathog. 2011;7:e1002193. doi: 10.1371/journal.ppat.1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kurokawa M, Ghosh SK, Ramos JC, Mian AM, Toomey NL, Cabral L, et al. Azidothymidine inhibits NF-kappaB and induces Epstein-Barr virus gene expression in Burkitt lymphoma. Blood. 2005;106:235–240. doi: 10.1182/blood-2004-09-3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ishikawa C, Mori N. Epstein-Barr virus latent membrane protein 1 induces CD69 expression through activation of nuclear factor-kappaB. Int J Oncol. 2013;42:1786–1792. doi: 10.3892/ijo.2013.1871. [DOI] [PubMed] [Google Scholar]
- 137.Faumont N, Durand-Panteix S, Schlee M, Gromminger S, Schuhmacher M, Holzel M, et al. c-Myc and Rel/NF-kappaB are the two master transcriptional systems activated in the latency III program of Epstein-Barr virus-immortalized B cells. J Virol. 2009;83:5014–5027. doi: 10.1128/JVI.02264-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Montes-Moreno S, Odqvist L, Diaz-Perez JA, Lopez AB, de Villambrosia SG, Mazorra F, et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod Pathol. 2012;25:968–982. doi: 10.1038/modpathol.2012.52. [DOI] [PubMed] [Google Scholar]
- 139.Ok CY, Li L, Xu-Monette ZY, Visco C, Tzankov A, Manyam GC, et al. Prevalence and clinical implications of epstein-barr virus infection in de novo diffuse large B-cell lymphoma in Western countries. Clin Cancer Res. 2014;20:2338–2349. doi: 10.1158/1078-0432.CCR-13-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gebauer N, Gebauer J, Hardel TT, Bernard V, Biersack H, Lehnert H, et al. Prevalence of targetable oncogenic mutations and genomic alterations in Epstein-Barr virus-associated diffuse large B-cell lymphoma of the elderly. Leuk Lymphoma. 2015;56:1100–1106. doi: 10.3109/10428194.2014.944522. [DOI] [PubMed] [Google Scholar]
- 141.O'Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A. 2009;106:7113–7118. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Choy EY, Siu KL, Kok KH, Lung RW, Tsang CM, To KF, et al. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J Exp Med. 2008;205:2551–2560. doi: 10.1084/jem.20072581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Martin-Perez D, Vargiu P, Montes-Moreno S, Leon EA, Rodriguez-Pinilla SM, Lisio LD, et al. Epstein-Barr virus microRNAs repress BCL6 expression in diffuse large B-cell lymphoma. Leukemia. 2012;26:180–183. doi: 10.1038/leu.2011.189. [DOI] [PubMed] [Google Scholar]
- 144.Perez-Rosado A, Artiga M, Vargiu P, Sanchez-Aguilera A, Alvarez-Barrientos A, Piris M. BCL6 represses NFkappaB activity in diffuse large B-cell lymphomas. J Pathol. 2008;214:498–507. doi: 10.1002/path.2279. [DOI] [PubMed] [Google Scholar]
- 145.Dutton A, Woodman CB, Chukwuma MB, Last JI, Wei W, Vockerodt M, et al. Bmi-1 is induced by the Epstein-Barr virus oncogene LMP1 and regulates the expression of viral target genes in Hodgkin lymphoma cells. Blood. 2007;109:2597–2603. doi: 10.1182/blood-2006-05-020545. [DOI] [PubMed] [Google Scholar]
- 146.Nonaka M, Horie R, Itoh K, Watanabe T, Yamamoto N, Yamaoka S. Aberrant NF-kappaB2/p52 expression in Hodgkin/Reed-Sternberg cells and CD30-transformed rat fibroblasts. Oncogene. 2005;24:3976–3986. doi: 10.1038/sj.onc.1208564. [DOI] [PubMed] [Google Scholar]
- 147.Knecht H, Berger C, McQuain C, Rothenberger S, Bachmann E, Martin J, et al. Latent membrane protein 1 associated signaling pathways are important in tumor cells of Epstein-Barr virus negative Hodgkin's disease. Oncogene. 1999;18:7161–7167. doi: 10.1038/sj.onc.1203177. [DOI] [PubMed] [Google Scholar]
- 148.Schwarzer R, Dorken B, Jundt F. Notch is an essential upstream regulator of NF-kappaB and is relevant for survival of Hodgkin and Reed-Sternberg cells. Leukemia. 2012;26:806–813. doi: 10.1038/leu.2011.265. [DOI] [PubMed] [Google Scholar]
- 149.Mathas S, Johrens K, Joos S, Lietz A, Hummel F, Janz M, et al. Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood. 2005;106:4287–4293. doi: 10.1182/blood-2004-09-3620. [DOI] [PubMed] [Google Scholar]
- 150.Amrein PC, Attar EC, Takvorian T, Hochberg EP, Ballen KK, Leahy KM, et al. Phase II study of dasatinib in relapsed or refractory chronic lymphocytic leukemia. Clin Cancer Res. 2011;17:2977–2986. doi: 10.1158/1078-0432.CCR-10-2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kater AP, Spiering M, Liu RD, Doreen Te Raa G, Slinger E, Tonino SH, et al. Dasatinib in combination with fludarabine in patients with refractory chronic lymphocytic leukemia: a multicenter phase 2 study. Leuk Res. 2014;38:34–41. doi: 10.1016/j.leukres.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 152.Morschhauser F, Seymour JF, Kluin-Nelemans HC, Grigg A, Wolf M, Pfreundschuh M, et al. A phase II study of enzastaurin, a protein kinase C beta inhibitor, in patients with relapsed or refractory mantle cell lymphoma. Ann Oncol. 2008;19:247–253. doi: 10.1093/annonc/mdm463. [DOI] [PubMed] [Google Scholar]
- 153.Schwartzberg L, Hermann R, Flinn I, Flora D, Hsi ED, Hamid O, et al. Open-label, single-arm, phase II study of enzastaurin in patients with follicular lymphoma. Br J Haematol. 2014;166:91–97. doi: 10.1111/bjh.12853. [DOI] [PubMed] [Google Scholar]
- 154.Dunleavy K, Pittaluga S, Czuczman MS, Dave SS, Wright G, Grant N, et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood. 2009;113:6069–6076. doi: 10.1182/blood-2009-01-199679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ruan J, Martin P, Furman RR, Lee SM, Cheung K, Vose JM, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol. 2011;29:690–697. doi: 10.1200/JCO.2010.31.1142. [DOI] [PubMed] [Google Scholar]
- 156.Evens AM, Smith MR, Lossos IS, Helenowski I, Millenson M, Winter JN, et al. Frontline bortezomib and rituximab for the treatment of newly diagnosed high tumour burden indolent non-Hodgkin lymphoma: a multicentre phase II study. Br J Haematol. 2014;166:514–520. doi: 10.1111/bjh.12915. [DOI] [PubMed] [Google Scholar]
- 157.Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116:1515–1523. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- 158.Czuczman NM, Barth MJ, Gu J, Neppalli V, Mavis C, Frys SE, et al. Pevonedistat, a NEDD8-activating enzyme inhibitor, is active in mantle cell lymphoma and enhances rituximab activity in vivo. Blood. 2016;127:1128–1137. doi: 10.1182/blood-2015-04-640920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shah JJ, Jakubowiak AJ, O'Connor OA, Orlowski RZ, Harvey RD, Smith MR, et al. Phase I Study of the Novel Investigational NEDD8-Activating Enzyme Inhibitor Pevonedistat (MLN4924) in Patients with Relapsed/Refractory Multiple Myeloma or Lymphoma. Clin Cancer Res. 2016;22:34–43. doi: 10.1158/1078-0432.CCR-15-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rajan A, Kelly RJ, Trepel JB, Kim YS, Alarcon SV, Kummar S, et al. A phase I study of PF-04929113 (SNX-5422), an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumor malignancies and lymphomas. Clin Cancer Res. 2011;17:6831–6839. doi: 10.1158/1078-0432.CCR-11-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yong K, Cavet J, Johnson P, Morgan G, Williams C, Nakashima D, et al. Phase I study of KW-2478, a novel Hsp90 inhibitor, in patients with B-cell malignancies. Br J Cancer. 2016;114:7–13. doi: 10.1038/bjc.2015.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Roue G, Perez-Galan P, Mozos A, Lopez-Guerra M, Xargay-Torrent S, Rosich L, et al. The Hsp90 inhibitor IPI-504 overcomes bortezomib resistance in mantle cell lymphoma in vitro and in vivo by down-regulation of the prosurvival ER chaperone BiP/Grp78. Blood. 2011;117:1270–1279. doi: 10.1182/blood-2010-04-278853. [DOI] [PubMed] [Google Scholar]
- 163.Kaufman KL, Jenkins Y, Alomari M, Mirzaei M, Best OG, Pascovici D, et al. The Hsp90 inhibitor SNX-7081 is synergistic with fludarabine nucleoside via DNA damage and repair mechanisms in human, p53-negative chronic lymphocytic leukemia. Oncotarget. 2015;6:40981–40997. doi: 10.18632/oncotarget.5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115:2578–2585. doi: 10.1182/blood-2009-08-236471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Flinn IW, Bartlett NL, Blum KA, Ardeshna KM, LaCasce AS, Flowers CR, et al. A phase II trial to evaluate the efficacy of fostamatinib in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) Eur J Cancer. 2015;54:11–17. doi: 10.1016/j.ejca.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 166.Chanan-Khan A, Cramer P, Demirkan F, Fraser G, Silva RS, Grosicki S, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): a randomised, double-blind, phase 3 study. Lancet Oncol. 2016;17:200–211. doi: 10.1016/S1470-2045(15)00465-9. [DOI] [PubMed] [Google Scholar]
- 167.Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Byrd JC, Harrington B, O'Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016;374:323–332. doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]