

Abstract
The enantioselective synthesis of 4-fluoroisochromanones via chiral aryl iodide-catalyzed fluorolactonization is reported. This methodology uses HF•pyridine as a nucleophilic fluoride source with a peracid stoichiometric oxidant, and provides access to lactones containing fluorine-bearing stereogenic centers in high enantio- and diastereoselectivity. The regioselectivity observed in these lactonization reactions is complementary to that obtained with established asymmetric electrophilic fluorination protocols.
Graphical abstract
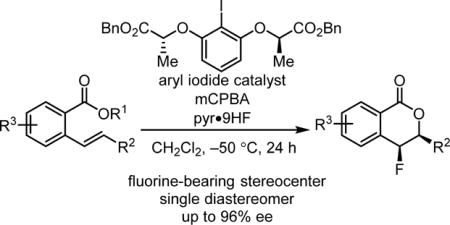
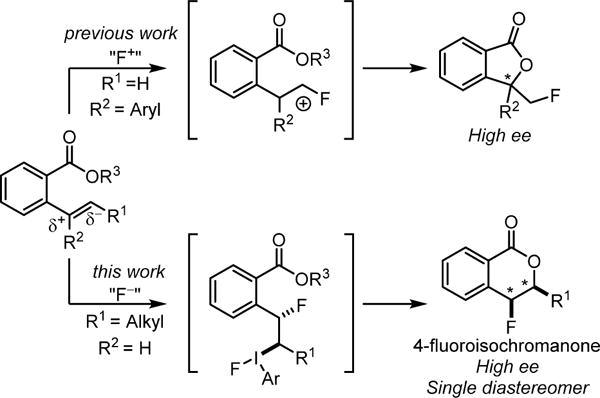
The stereocontrolled construction of C–F bonds represents a frontier endeavor in synthetic chemistry, motivated in large part by the important ways that fluorine incorporation is known to modulate the physical and biological properties of organic molecules.1 Electrophilic fluorine sources (“F+”) such as Selectfluor, N-fluoropyridinium salts, and N-fluorobenzenesulfonimide (NFSI) have been used extensively in the enantiocontrolled generation of fluorine-bearing stereogenic centers,2 most often via the intermediacy of enolate equivalents to produce α-fluorocarbonyl compounds.2a,3 Fluorofunctionalization of alkenes using F+ sources is another powerful approach to the enantioselective synthesis of fluorine-containing chiral compounds that allows for the synthesis of highly functionalized products from simple olefin-containing starting materials.4,5 In reported efforts directed toward enantioselective fluorolactonization reactions, electrophilic fluorinating reagents have been shown to afford γ-butyrolactone products with exocyclic fluoromethyl substituents in moderate-to-high enantioselectivity (Scheme 1, top).6 We hypothesized that a hypervalent iodine catalysis of a nucleophilic fluorination pathway (“F−”) could provide a complementary approach to regioisomeric fluorolactone products containing a C–F stereogenic center (Scheme 1, bottom), in a manner analogous to that observed in recently described enantioselective acetoxylactonization reactions.7 Herein, we report the development of an enantioselective, catalytic fluorolactonization reaction for the preparation of 4-fluoroisochromanones in high enantio- and diastereoselectivity.
Scheme 1.
Enantioselective fluorolactonizations with electrophilic and nucleophilic fluorinating agents
We reported recently a protocol for the aryl iodide-catalyzed diastereoselective 1,2-difluorination of both terminal and internal alkenes8,9 using HF-pyridine (pyr•9HF) as a nucleophilic fluorine source and meta-chloroperbenzoic acid (mCPBA) as a stoichiometric oxidant. These conditions were first developed by Shibata and coworkers in the context of catalytic aminofluorination reactions.10 Alkenes bearing proximal weakly Lewis basic groups were shown to undergo net anti-difluorination, while substrates lacking such functionality afforded syn-difluoride products. We proposed that properly positioned weakly nucleophilic groups (e.g. the o-NO2 group in Scheme 2) can displace the aryl iodide from intermediate I to form an unstable bridged intermediate II. Invertive displacement of the neighboring group by fluoride led to the anti-diastereomeric outcome. The propensity for such anchimeric assistance in these reactions suggested that styrenes containing other nucleophilic neighboring groups such as an ortho-carboxylic acid or ester might undergo fluorolactonization reactions.11 As outlined in the proposed catalytic pathway in Scheme 2, nucleophilic displacement of the aryliodo group in intermediate I with a carboxylate equivalent would lead to formation of 4-fluoroisochromanone products. This approach would produce a fluorine-bearing stereogenic center with a syn relationship between the two newly formed bonds,12 a stereochemical outcome distinct from prototypical bromo- and iodolactonizations.13 Furthermore, as members of the polyketide-derived 4-oxyisochromanone class of natural products are known to possess interesting biological activity,14 we were motivated by the possibility that an efficient, stereocontrolled route to 4-fluoro analogs may be of interest for biological or pharmacological applications.
Scheme 2.
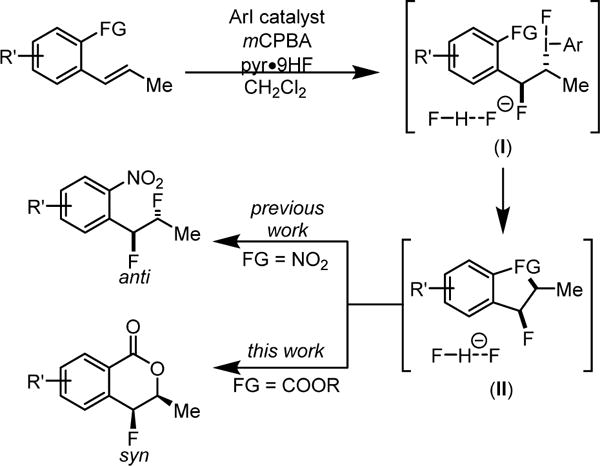
Anchimeric assistance in 1,2-difluorination of styrenes with ArI-catalysts and an analogous fluorolactonization pathway
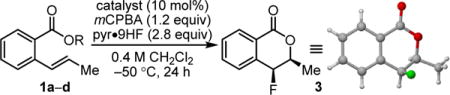
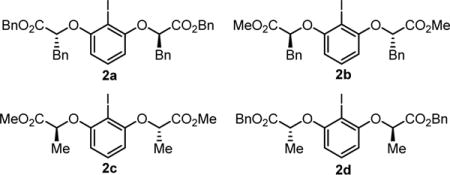
Methyl benzoate derivative 1a was evaluated as a model substrate for the proposed fluorolactonization reaction. Various classes of chiral aryl iodides were examined as potential catalysts, including chiral resorcinol derivatives such as 2a–2d,7,15 which have found application previously in a variety of enantioselective alkene oxidation reactions.16 In the presence of catalysts 2a–d, mCPBA as oxidant, and pyr•9HF as the fluorine source and acid promoter, 1a was observed to undergo cyclization to fluoroisochromanone 3 as a single observable diastereomer (Table 1).17 The syn relative configuration was determined via X-ray crystallographic analysis, consistent with the reaction pathway outlined in Scheme 2.18
Table 1.
Optimization of the fluorolactonization reaction
| |||||
|---|---|---|---|---|---|
| entry | substrate | R | catalyst | ee (%)a | yield (%)b |
| 1 | 1a | Me | 2a | 87 | 72 |
| 2 | 1a | Me | 2b | −87 | 50 |
| 3 | 1a | Me | 2c | −94 | 68 |
|
| |||||
| 4 | 1a | Me | 2d | 95 | 86 (68)c |
|
| |||||
| 5 | 1b | iPr | 2d | 95 | 72 |
| 6 | 1c | Bn | 2d | 87 | 42 |
| 7 | 1d | H | 2d | 86 | 95 (70)c |
Conditions: substrate (0.10 mmol), catalyst (10 mol%), mCPBA (0.12 mmol), pyr•9HF (2.5 mmol HF) in CH2CI2 (0.25 mL) cooled to −50 °C, 24 h.
Enantioselectivities were determined by GC or HPLC analysis with commercial chiral columns.
Yields were measured by GC and are based on an internal standard.
Isolated yield on a 1 mmol scale.
Catalysts 2c and 2d were found to impart significantly higher enantioselectivities than the corresponding benzyl-substituted analogs 2a–b (entries 1–4). This observation stands in contrast to results obtained in the migratory geminal difluorination of β-substituted styrenes reported recently by our group,19 where that trend was reversed and the polarizable benzylic groups in para-substituted analogs of 2a–b were shown to play a critical role in enhancing ee. It is evident that subtle yet fundamentally different factors are responsible for enantioinduction in these closely related reactions. Variation of the ester group of the catalyst had very little effect on the enantioselectivity of the fluorolactonization reaction, but measurably improved yields were obtained with benzyl ester catalysts 2a and 2d relative to their methyl ester counterparts. On the basis of these results, 2d was selected as the optimal catalyst for further study.
Variation of the carboxylate equivalent (1a–c) or use of the free carboxylic acid 1d resulted in formation of fluorolactonization product 3, albeit with discernible changes in ee and yield (entries 4–7) and with methyl ester 1a affording the best results.
The effect of arene substitution in the enantioselective fluorolactonization reaction catalyzed by 2d is illustrated in Table 2. Alkyl, halide, or trifluoromethoxy substitution at the 6, 5, and 4-positions of 1 is generally well tolerated, with the fluorolactone products obtained in 80–96% ee (entries 1–3, 5–10). However, substrates bearing more electron-deficient trifluoromethyl or carbomethoxy groups underwent reaction in reduced yields and enantioselectivities (entries 12–15). In all cases, the fluorolactone product was obtained as a single diastereomer.20
Table 2.
Fluorolactonization of aryl-substituted substrates

| |||||
|---|---|---|---|---|---|
| entry | 4 | R | 5 | ee (%)a | yield (%)b |
| 1 | 4a | 6-Me | 5a | 96 | 53 |
| 2 | 4b | 5-Me | 5b | 93 | 50 |
| 3 | 4c | 4-Me | 5c | 94 | 64 |
| 4 | 4d | 3-Me | 5d | 66 | 52 |
| 5 | 4e | 5-OCF3 | 5e | 89 | 55 |
| 6 | 4f | 5-Br | 5f | 86 | 48 |
| 7 | 4g | 5-Cl | 5g | 86 | 60 |
| 8 | 4h | 6-F | 5h | 83 | 67 |
| 9 | 4i | 5-F | 5i | 93 | 57 |
| 10 | 4j | 4-F | 5j | 80 | 61 |
| 11 | 4k | 3-F | 5k | 30 | 49 |
| 12 | 4l | 5-CF3 | 5l | 73 | 60 |
| 13 | 4m | 4-CF3 | 5m | 76 | 44 |
| 14 | 4n | 5-CO2Me | 5n | 70 | 35 |
| 15 | 4o | 4-CO2Me | 5o | 58 | 51 |
Conditions: substrate (1 mmol), catalyst (10 mol%), mCPBA (1.2 mmol), pyr•9HF (25 mmol HF) in CH2CI2 (2.5 mL) cooled to −50 °C, 24 h.
Enantioselectivities were determined by GC analysis with commercial chiral columns.
Isolated yields are reported.
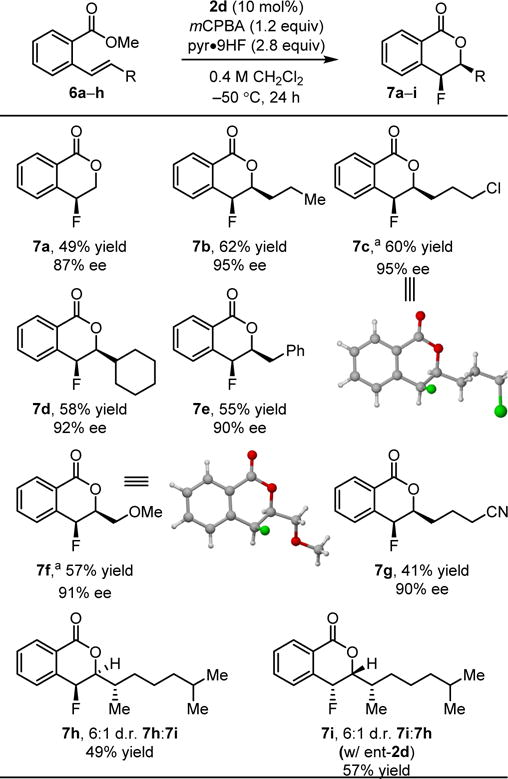
The new fluorolactonization reaction was also extended successfully to a variety of β-substituted styrene derivatives (Table 3). In general, substituents of varying size and functionality have little impact on reaction enantioselectivity. The Lewis basic ether and cyano substituents in 7f and 7g are observed not to alter the relative stereochemical outcome of the fluorolactonization reaction, despite the propensity toward anchimeric assistance pathways in these reactions as noted above. Catalyst control over stereoselectivity of the reaction was observed with 7h and 7i, with complementary diastereoselectivity obtained using 2d or ent-2d.
Table 3.
Fluorolactonization of substituted alkene substrates
|
Conditions: substrate (1 mmol), catalyst (10 mol%), mCPBA (1.2 mmol), pyr•9HF (25 mmol HF) in CH2Cl2 (2.5 mL) cooled to −50 °C, 24 h. Enantioselectivities were determined by HPLC or GC analysis with commercial chiral columns. Isolated yields are reported.
Absolute configuration was determined by X-ray crystallographic analysis; all other products are assigned by analogy.
As illustrated in Scheme 3, the regioselectivity observed in the fluorolactonization reactions with a nucleophilic fluoride source is opposite to that obtained with electrophilic reagents.21 Furthermore, as demonstrated with 1d, electrophilic fluorolactonizations of disubstituted styrenes are observed to be poorly diastereoselective. The successful introduction of C–F stereocenters in a highly stereocontrolled manner is thus a significant feature of the new catalytic protocol.
Scheme 3.
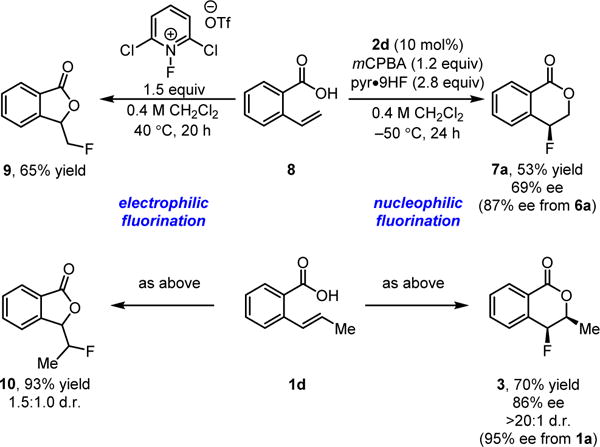
Comparison of the reactivity of electrophilic and nucleophilic fluorinating reagents
In conclusion, the enantio- and diastereoselective, catalytic synthesis of 4-fluoroisochromanones can be accomplished with HF-pyridine as a nucleophilic fluoride source. Readily accessible chiral aryl iodides catalyze fluorolactonization with generation of C–F stereogenic centers from simple styrene precursors. Ongoing efforts are directed toward exploring the scope of fluorofunctionalization reactions induced by hypervalent iodine(III) catalysis, as well as elucidating the basis of the subtle catalyst structural properties that control the enantioselectivity in these reactions.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM043214), by an NSF pre-doctoral fellowship to S.M.B., and by a Suzanne and Bob Wright postdoctoral fellowship to E.M.W. from the Damon Runyon Cancer Research Foundation (DRG-2180-14). We thank Dr. Shao-Liang Zheng for crystal structure determination and Jennifer Wang for mass spectrometry analysis.
Footnotes
Supporting Information. Experimental procedures and characterization data for new compounds are available free of charge via the internet at http://pubs.acs.org.
Funding Sources: No competing financial interests have been declared.
References
- 1.(a) Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA. J Med Chem. 2015;58:8315. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]; (b) Wang J, Sanchez-Rosello M, Acena JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]; (c) Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (d) Hagmann WK. J Med Chem. 2008;51:4359. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 2.For reviews on this topic, see:; (a) Yang X, Tao W, Phipps RJ, Toste FD. Chem Rev. 2015;115:826. doi: 10.1021/cr500277b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Champagne PA, Desroches J, Hamel JD, Vandamme M, Paquin JF. Chem Rev. 2015;115:9073. doi: 10.1021/cr500706a. [DOI] [PubMed] [Google Scholar]
- 3.For seminal examples utilizing organocatalysts, see:; (a) Shibata N, Suzuki E, Takeuchi Y. J Am Chem Soc. 2000;122:10728. [Google Scholar]; (b) Cahard D, Audouard C, Plaquevent JC, Roques N. Org Lett. 2000;2:3699. doi: 10.1021/ol006610l. [DOI] [PubMed] [Google Scholar]; (c) Kim DY, Park EJ. Org Lett. 2002;4:545. doi: 10.1021/ol010281v. [DOI] [PubMed] [Google Scholar]; (d) Enders D, Hüttl MR. Synlett. 2005;6:991. [Google Scholar]; (e) Marigo M, Fielenbach D, Braunton A, Kjærsgaard A, Jørgensen KA. Angew Chem Int Ed. 2005;44:3703. doi: 10.1002/anie.200500395. [DOI] [PubMed] [Google Scholar]; (f) Steiner DD, Mase N, Barbas CF., III Angew Chem Int Ed. 2005;44:3706. doi: 10.1002/anie.200500571. [DOI] [PubMed] [Google Scholar]; (g) Beeson TD, MacMillan DWC. J Am Chem Soc. 2005;127:8826. doi: 10.1021/ja051805f. [DOI] [PubMed] [Google Scholar]
- 4.For reviews on this topic, see:; (a) Wolstenhulme JR, Gouverneur V. Acc Chem Res. 2014;47:3560. doi: 10.1021/ar500282z. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Kuester WE, Burk MT. Angew Chem Int Ed. 2012;51:10938. doi: 10.1002/anie.201204347. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cahard D, Xu X, Couve-Bonnaire S, Pannecoucke X. Chem Soc Rev. 2010;39:558. doi: 10.1039/b909566g. [DOI] [PubMed] [Google Scholar]
- 5.For specific examples, see:; (a) Greedy B, Paris JM, Vidal T, Gouverneur V. Angew Chem Int Ed. 2003;42:3291. doi: 10.1002/anie.200351405. [DOI] [PubMed] [Google Scholar]; (b) Wilkinson SC, Lozano O, Schuler M, Pacheco M, Salmon R, Gouverneur V. Angew Chem Int Ed. 2009;48:7083. doi: 10.1002/anie.200901795. [DOI] [PubMed] [Google Scholar]; (c) Rauniyar V, Lackner AD, Hamilton GL, Toste FD. Science. 2011;334:1681. doi: 10.1126/science.1213918. [DOI] [PubMed] [Google Scholar]; (d) Wolstenhulme JR, Rosenqvist J, Lozano O, Ilupeju J, Wurz N, Engle KM, Pidgeon GW, Moore PR, Sandford G, Gouverneur V. Angew Chem Int Ed. 2013;52:9796. doi: 10.1002/anie.201304845. [DOI] [PubMed] [Google Scholar]
- 6.(a) Parmar D, Maji MS, Rueping M. Chem Eur J. 2014;20:83. doi: 10.1002/chem.201303385. [DOI] [PubMed] [Google Scholar]; (b) Egami H, Asada J, Sato K, Hashizume D, Kawato Y, Hamashima Y. J Am Chem Soc. 2015;137:10132. doi: 10.1021/jacs.5b06546. [DOI] [PubMed] [Google Scholar]
- 7.(a) Fujita M, Yoshida Y, Miyata K, Wakisaka A, Sugimura T. Angew Chem Int Ed. 2010;49:7068. doi: 10.1002/anie.201003503. [DOI] [PubMed] [Google Scholar]; (b) Shimogaki M, Fujita M, Sugimura T. Eur J Org Chem. 2013:7128. doi: 10.1021/acs.joc.7b01141. [DOI] [PubMed] [Google Scholar]
- 8.Banik SM, Medley JW, Jacobsen EN. J Am Chem Soc. 2016;138:5000. doi: 10.1021/jacs.6b02391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilmour and coworkers independently reported a similar protocol for the 1,2-difluorination of terminal alkenes:; Molnár IG, Gilmour R. J Am Chem Soc. 2016;138:5004. doi: 10.1021/jacs.6b01183. [DOI] [PubMed] [Google Scholar]
- 10.(a) Suzuki S, Kamo T, Fukushi K, Hiramatsu T, Tokunaga E, Dohi T, Kita Y, Shibata N. Chem Sci. 2014;5:2754. [Google Scholar]; For an additional report utilizing HF-pyridine and mCPBA in catalytic fluorination reactions, see:; (b) Kitamura T, Muta K, Oyamada J. J Org Chem. 2015;80:10431. doi: 10.1021/acs.joc.5b01929. [DOI] [PubMed] [Google Scholar]
- 11.For an example of an enantioselective fluorinative intramolecular cyclization reaction of alkenes with stoichiometric, chiral iodine(III) reagents, see:; Kong W, Feige P, de Haro T, Nevado C. Angew Chem Int Ed. 2013;52:2469. doi: 10.1002/anie.201208471. [DOI] [PubMed] [Google Scholar]
- 12.Alternatively, cyclization prior to nucleophilic fluorination would also provide 4-fluoroisochromanone products with syn-configurations Such a mechanism cannot be ruled out based on the data presented here For reactions where cyclization is proposed to precede fluorination, see; Sawaguchi M, Hara S, Fukuhara T, Yoneda N. J Fluorine Chem. 2000;104:277. [Google Scholar]
- 13.Chen J, Zhou L, Tan CK, Yeung YY. J Org Chem. 2012;77:999. doi: 10.1021/jo202221c. [DOI] [PubMed] [Google Scholar]
- 14.(a) Aldridge DC, Galt S, Giles D, Turner WB. J Chem Soc C. 1971:1623. doi: 10.1039/j39710003888. [DOI] [PubMed] [Google Scholar]; (b) Zhang W, Krohn K, Draegar S, Schultz B. J Nat Prod. 2008;71:1078. doi: 10.1021/np800095g. [DOI] [PubMed] [Google Scholar]
- 15.Uyanik M, Yasui T, Ishihara K. Angew Chem Int Ed. 2010;49:2175. doi: 10.1002/anie.200907352. [DOI] [PubMed] [Google Scholar]
- 16.For reviews on this topic, see:; (a) Yoshimura A, Zhdankin VV. Chem Rev. 2016;116:3328. doi: 10.1021/acs.chemrev.5b00547. [DOI] [PubMed] [Google Scholar]; (b) Romero RM, Woste TH, Muniz K. Chem Asian J. 2014;9:972. doi: 10.1002/asia.201301637. [DOI] [PubMed] [Google Scholar]; (c) Parra A, Reboredo S. Chem Eur J. 2013;19:17244. doi: 10.1002/chem.201302220. [DOI] [PubMed] [Google Scholar]
- 17.Reducing the number of equivalents of pyr•9HF resulted in lower yields of 3, albeit with consistent enantioselectivities (1.1 equiv. pyr•9HF: 93% ee and 28% yield; 5.6 equiv. pyr•9HF: 93% ee and 83% yield). Reactions conducted at −20 °C led to the generation of 3 in 86% ee and 71% yield (yields determined by GC analysis).
- 18.For examples of syn 1,2 additions to alkenes promoted by hypervalent iodine reagents, see references7, 8, and; Hara S, Nakahigashi J, Ishi-I K, Sawaguchi M, Sakai H, Fukuhara T, Yoneda N. Synlett. 1998:495. [Google Scholar]
- 19.Banik SM, Medley JW, Jacobsen EN. Science. 2016;353:51. doi: 10.1126/science.aaf8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The remainder of the mass balance in these reactions can be attributed to unreacted starting material and to the formation of several uncharacterized, but readily separable, byproducts. For a discussion of background reactions in closely related catalytic oxylactonization reactions, see reference7b
- 21.For an additional example of a racemic, regioselective fluorocyclization reaction with hypervalent iodine reagents, see:; Geary GC, Hope EG, Stuart AM. Angew Chem Int Ed. 2015;54:14911. doi: 10.1002/anie.201507790. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.