

Abstract
Protein homeostasis (proteostasis) requires the timely degradation of misfolded proteins and their aggregates by protein quality control (PQC), of which molecular chaperones are an essential component. Compared with other cell types, PQC in neurons is particularly challenging because they have a unique cellular structure with long extensions. Making it worse, neurons are postmitotic, i.e., cannot dilute toxic substances by division, and, thus, are highly sensitive to misfolded proteins, especially as they age. Failure in PQC is often associated with neurodegenerative diseases, such as Huntington's disease (HD), Alzheimer's disease (AD), Parkinson's disease (PD), and prion disease. In fact, many neurodegenerative diseases are considered to be protein misfolding disorders. To prevent the accumulation of disease-causing aggregates, neurons utilize a repertoire of chaperones that recognize misfolded proteins through exposed hydrophobic surfaces and assist their refolding. If such an effort fails, chaperones can facilitate the degradation of terminally misfolded proteins through either the ubiquitin (Ub)-proteasome system (UPS) or the autophagy-lysosome system (hereafter autophagy). If soluble, the substrates associated with chaperones, such as Hsp70, are ubiquitinated by Ub ligases and degraded through the proteasome complex. Some misfolded proteins carrying the KFERQ motif are recognized by the chaperone Hsc70 and delivered to the lysosomal lumen through a process called, chaperone-mediated autophagy (CMA). Aggregation-prone misfolded proteins that remain unprocessed are directed to macroautophagy in which cargoes are collected by adaptors, such as p62/SQSTM-1/Sequestosome-1, and delivered to the autophagosome for lysosomal degradation. The aggregates that have survived all these refolding/degradative processes can still be directly dissolved, i.e., disaggregated by chaperones. Studies have shown that molecular chaperones alleviate the pathogenic symptoms by neurodegeneration-causing protein aggregates. Chaperone-inducing drugs and anti-aggregation drugs are actively exploited for beneficial effects on symptoms of disease. Here, we discuss how chaperones protect misfolded proteins from aggregation and mediate the degradation of terminally misfolded proteins in collaboration with cellular degradative machinery. The topics also include therapeutic approaches to improve the expression and turnover of molecular chaperones and to develop anti-aggregation drugs.
Keywords: proteolysis, protein aggregation, ubiquitin-proteasome system, autophagy-lysosome system, chaperon-mediated autophagy, macroautophagy, protein quality control
Introduction
Proteins may lose their folding when cells are exposed to stresses, such as oxidative stress, heat, and toxic chemicals. Misfolded proteins and their aggregates grow into intracellular or extracellular amyloid plaques or neurofibrillary tangles (Taylor et al., 2002). Eukaryotic cells operate the PQC system to remove these cytotoxic agents in a timely fashion. The excessive formation of protein aggregates and their fibrillar structures are universally observed in at least 30 different human diseases (Taylor et al., 2002; Broersen et al., 2010). These protein misfolding disorders include various neurodegenerative diseases, such as Alzheimer' disease (AD), Parkinson' disease (PD), Huntington disease' (HD), transmissible spongiform encephalopathies (TSE), and amyotrophic lateral sclerosis (ALS) (Moreno-Gonzalez and Soto, 2011; Doyle et al., 2013; Hetz and Mollereau, 2014; Valastyan and Lindquist, 2014).
One essential component of PQC is molecular chaperones that enhance the refolding of misfolded proteins and, thus, counteract their aggregation (Hartl et al., 2011; Kim et al., 2013). Molecular chaperones constitute up to 10% of the proteome and play important functions in proteostasis under normal conditions as well as during cellular stress responses (Kastle and Grune, 2012). The majority of molecular chaperones are called heat-shock proteins (HSPs) because they are induced by various stresses such as heat shock, oxidative stress, toxic chemical, and inflammation (Garrido et al., 2001). HSPs are divided into several subgroups based on their sizes, such as Hsp70, Hsp90, Hsp60, Hsp40 (DnaJ), and small HSPs. These molecular chaperones can assist the refolding of misfolded proteins through three distinct action modes. First, most chaperones such as Hsp70 can hold the clients in an unfolded state until spontaneous fold is achieved (Rudiger et al., 1997; Hartl et al., 2011; Kastle and Grune, 2012). Second, some molecular chaperons such as Hsp70 and Hsp60s can use ATP to unfold stable misfolded proteins and convert them into natively refoldable species (Ranford et al., 2000; Itoh et al., 2002; Tutar and Tutar, 2010). Third, some chaperones, such as yeast Hsp104 and human Hsp70 in complex with Hsp40 and Hsp110, can act as “disaggregases” because they use the energy of ATP hydrolysis to forcefully unfold and solubilize preformed aggregates into natively refolded proteins (Mosser et al., 2004; Shorter and Lindquist, 2004; Arimon et al., 2008; Lo Bianco et al., 2008; DeSantis et al., 2012). Despite distinct action modes, they share general properties to recognize and bind the hydrophobic sequences which are not normally exposed in the native folding (Buchner, 1996). Their binding to and dissociation from clients can be driven by adenosine-5′-triphosphate (ATP) hydrolysis. The ATPase and chaperone activity are typically regulated through their cooperation with cochaperones. In addition to ATP-dependent chaperones, neurons express ATP-independent chaperons that bind misfolded proteins and promote refolding (D'Andrea and Regan, 2003). These chaperones typically form a coordinated network with cochaperones and the machinery in proteolytic pathways.
While the primary function of molecular chaperones is to assist misfolded or unfolded proteins to regain or acquire the normal folding, they can facilitate the degradation of terminally misfolded proteins in collaboration with proteolytic machinery (Hoffmann et al., 2004; Ellis, 2006, 2007; Ellis and Minton, 2006; Pauwels et al., 2007). Eukaryotic cells operate two major proteolytic systems, the UPS and autophagy. In principle, terminally misfolded proteins are ubiquitinated by E3 Ub ligases and processively degraded by the proteasome. If the substrates are prone to aggregation or escape the surveillance of the UPS, however, they are redirected to macroautophagy in which cargoes are separated in the double membrane structure, called the autophagosome, and degraded by lysosomal hydrolases (Cha-Molstad et al., 2015). Some misfolded proteins carrying the KFERQ pentapeptide sequence can be sorted out by molecular chaperones and directly delivered to the lysosome through chaperone-mediated autophagy (CMA) (Chiang et al., 1989; Dice, 1990; Cuervo et al., 1997).
The UPS is an intracellular proteolytic system that mediates the degradation of more than 80% of normal and abnormal intracellular proteins (Wang and Maldonado, 2006). The importance of molecular chaperones in the UPS was initially proposed and demonstrated by Ciechanover and colleagues who showed that the molecular chaperone Hsc70 is required for Ub-dependent degradation of several substrates (Ciechanover et al., 1995; Bercovich et al., 1997). The UPS involves a cascade of E1, E2, and E3 enzymes whose cooperative activities mediate the conjugation of Ub to target proteins (Pickart, 2001). In this cascade, Ub with a size of 76 residues is activated by the Ub activating enzyme E1 and transferred to the Ub conjugating enzyme E2. The Ub moiety carried by E2 is conjugated to substrates, which requires the ubiquitination activity of the Ub ligase E3. In PQC, most E3s cannot recognize misfolded proteins and rather depend on molecular chaperones for substrate recognition. Ubiquitinated substrates are degraded by the proteasome into short peptides, typically with sizes of 8–12 amino acids. These peptides are displayed on the cell surface for immunosurveillance (Kloetzel and Ossendorp, 2004) or degraded into free amino acids by aminopeptidases. The UPS plays a pivotal role in proteostasis during neurodegeneration and prevents protein misfolding and aggregation (Morawe et al., 2012). In addition to PQC, the UPS regulates a variety of biological processes, including cell cycle, transcription, DNA repair, and apoptosis (Eldridge and O'Brien, 2010; Xie, 2010).
Autophagy is a process by which cytosolic materials are degraded by the lysosome. Depending on the mechanism of cargo delivery to the lysosome, autophagy can be divided into three pathways: microautophagy, CMA, and macroautophagy. In macroautophagy, terminally misfolded proteins in complex with molecular chaperones are collected by autophagy adaptors, such as p62 and NBR1. Cargo-loaded p62 undergoes self-polymerization and are deposited to the autophagosome through the interaction of p62 with LC3 (Lamark et al., 2009; Stolz et al., 2014). The autophagosome is fused with the lysosome to form the autolysosome wherein cargoes along with p62 are degraded by lysosomal hydrolases. Virtually all the misfolded proteins including those prone to aggregation in neurodegenerative diseases can be degraded by macroautophagy. In contrast to macroautophagy, CMA targets a subset of misfolded cytosolic proteins, especially those containing the KFERQ pentapeptide sequence (Fuertes et al., 2003; Massey et al., 2006; Kaushik and Cuervo, 2012). The substrates of the CMA are recognized by the molecular chaperone Hsc70 belonging to the Hsp70 family (Chiang et al., 1989). The cargo-Hsc70 complex is translocated into the lysosomal lumen and degraded by lysosomal hydrolases (Cuervo and Dice, 1996). Overall, lysosomal proteolysis through macroautophagy and CMA plays an important role in the removal of misfolded proteins that cannot be readily degraded by the UPS.
Misfolded proteins that survive the attempts of molecular chaperones to refold or degrade eventually form aggregates. As the last defense mechanism of PQC, molecular chaperons can directly resolve, i.e., disaggregate the already formed aggregates (Parsell et al., 1994; Mogk et al., 1999; Doyle et al., 2013). The disaggregation activity has been characterized in yeasts and mammals (Weibezahn et al., 2005; Hodson et al., 2012; Winkler et al., 2012). In yeasts, Hsp104 in collaboration with Hsp70, Hsp40, Hsp110, and sHSPs can directly disaggregate and reactivate proteins deposited in high order aggregates (Shorter, 2011; Torrente and Shorter, 2013). In mammals, Hsp110, Hsp105, Hsp100, and Hsp70/Hsp40 have been implicated in disaggregation (Lindquist and Kim, 1996; Glover and Lindquist, 1998; Doyle and Wickner, 2009). Through these multi-step defense processes, molecular chaperones play a key role in proteostasis.
Recent studies using mouse models suggest that molecular chaperones play a protective role in the pathogenesis of neurodegenerative disorders (Wyatt et al., 2012; Carman et al., 2013; Witt, 2013). By using disease models, HSPs have been shown to inhibit the aggregation of aggregation-prone proteins, such as Aβ, tau, HTT, and α-synuclein, and facilitate their degradation by the UPS or autophagy (Wyttenbach, 2004). As such, small molecule compounds that can modulate HSPs and proteolytic machinery are emerging as a means to treat neurodegenerative diseases. Below, we discuss the current understanding on the functions of HSPs in neurodegenerative diseases, including the recent results obtained from animal models of neurodegeneration.
Refolding of misfolded proteins by molecular chaperones
Neurons express various molecular chaperones which forms a complicated network of PQC to prevent aggregation. Their primary function is to assist the folding and assembly of newly synthesized polypeptides and the refolding of misfolded or damaged proteins. Depending on their sizes and action modes, molecular chaperones can be divided into several classes based on their sizes, such as Hsp70, Hsp90, Hsp60, Hsp40 (DnaJ), and small HSPs. Although the majority of molecular chaperones share similarity in action modes, such as substrate recognition and ATP hydrolysis-driven substrate binding, they are also different in substrate specificity, localization, and mechanistic details.
The Hsp70 family
The cytosolic chaperone Hsp70 is evolutionarily conserved and one of the most abundant chaperones. The homologs of Hsp70 are found in various subcellular compartments, including heat shock cognate 70 (Hsc70) in the cytosol and BiP/GRP78 in the ER. Hsp70 shows a broad range of activities in folding newly synthesized polypeptides, refolding misfolded proteins, the degradation of terminally misfolded proteins, and directly resolving already formed aggregates (Kastle and Grune, 2012). They commonly recognize diverse misfolded proteins through the interaction with a four to five residue stretch of hydrophobic amino acids exposed on the surface (Rudiger et al., 1997). The hydrophobic signatures occur on average every 30–40 residues in most misfolded proteins. Central to the chaperone activity of Hsp70 proteins is the transition between open and closed conformations of their substrate binding domain (SBD). In the ATP-bound open conformation, the SBD has low affinity to the client (Hartl et al., 2011). Once ATP hydrolysis is induced by cochaperones, Hsp70 acquires high affinity to the clients. The resulting ADP-bound form of Hsp70 facilitates the client' refolding by holding them in an unfolded state until spontaneous fold is achieved. The client that achieved the correct folding no longer has the exposed hydrophobic patches and, thus, is released from Hsp70. Extensive studies have shown that Hsp70 directly binds various pathogenic misfolded proteins in neurodegenerative diseases and facilitate their refolding. Such substrates of Hsp70 proteins include mutant huntingtin (mHTT) in HD and other polyQ diseases, α-synuclein in PD, amyloid-β (Aβ) and hyperphosphorylated tau in AD, and mutant SOD1 in ALS (Choo et al., 2004; Dedmon et al., 2005; Liu et al., 2005; Evans et al., 2006; Dompierre et al., 2007; Luk et al., 2008).
The Hsp40 (DnaJ) family
The Hsp40 proteins, also called J-proteins, form a large cochaperone family composed of 49 members (Odunuga et al., 2003). Amongst these, DnaJB6 and DnaJB8 are mainly expressed in neurons and can suppress polyglutamine aggregation and toxicity (Cheetham et al., 1992; Hageman et al., 2010). Although these cochaperones have the activity to bind and counteract protein aggregates or refold them, they also can modulate the ATP hydrolysis of Hsp70. The 70-residue J domain of Hsp40 binds misfolded proteins and interacts with the ATPase domain of Hsp70, which induces the ATP hydrolysis of Hsp70. ATP hydrolysis, in turn, brings the Hsp40-bound substrate close to the SBD of Hsp70 and increases Hsp70 affinity to the substrate, leading to Hsp40 release from the substrate and Hsp70 (Summers et al., 2009). As a consequence of this allosteric conformational change, the substrate is transferred from Hsp40 to Hsp70. Besides the conserved J domain, Hsp40 proteins carry diverse domains that mediate specific biological processes, such as intracellular localizations and client binding for proteolysis (Cheetham and Caplan, 1998; Kampinga and Craig, 2010). In neurodegenerative disease, Hsp40 proteins can act as cochaperones for Hsp70 proteins to assist the refolding of soluble misfolded proteins (Choo et al., 2004; Dedmon et al., 2005; Liu et al., 2005; Evans et al., 2006; Dompierre et al., 2007; Luk et al., 2008).
The Hsp90 family
The ATP-dependent chaperone Hsp90, which forms a dimer, is universally present in various cellular compartments, such as the cytosol, nucleus, ER, and mitochondria (Lindquist, 2009). Hsp90 is constitutively expressed in normal conditions, accounting for 1–2% of cellular proteins, and its level can increase to 4–6% if cells are exposed to stresses (Picard, 2002; Whitesell and Lindquist, 2005; Taipale et al., 2010; Finka and Goloubinoff, 2013). The activity of Hsp90 can be regulated by the HSR (heat-shock response) regulator HSF1 (heat-shock factor 1) (McLean et al., 2004; Putcha et al., 2010). Human neurons have a stress-inducible Hsp90α (Hsp90AA1) and a constitutively expressed Hsp90β (Hsp90AB1) that share 86% identity in protein sequence (Ammirante et al., 2008). These Hsp90 proteins bind a variety of clients and hold their folding, including kinases, nuclear receptors, transcription factors and cell surface receptors (Kastle and Grune, 2012). Remarkably, Hsp90 is thought to interact with approximately 2,000 proteins (Garnier et al., 2002), which accounts for up to 10% of total cellular proteins (Ratzke et al., 2010). Structural studies have shown that Hsp90 is composed of an N-terminal ATP-binding domain (N-domain), a mid-domain that binds the substrate (M-domain), and a C-terminal dimerization domain (C-domain) (Picard, 2002; Whitesell and Lindquist, 2005; Taipale et al., 2010; Finka and Goloubinoff, 2013). The substrate binding-release cycle of Hsp90 is regulated by ATP hydrolysis, which induces a large conformational transition between an open vs. closed form. In a free form, Hsp90 is in an open V-shaped conformation and, thus, binds clients. The ATP binding to the N-domain of client-loaded Hsp90 induces a conformational transition (Pearl and Prodromou, 2006). This results in a closed conformation where the N-domains of two Hsp90 molecules dimerize with each other. Following ATP hydrolysis, the substrate is released, and Hsp90 returns to an open conformation. The conformational transition of Hsp90 is regulated by various cochaperones, such as Hop, p23/Sba1, and Cdc37 (Picard, 2002; Whitesell and Lindquist, 2005; Taipale et al., 2010; Finka and Goloubinoff, 2013). Overall, the ability of Hsp90 to support the folding/refolding and stability of proteins is a double edge blade in neurodegeneration because it can also favor the accumulation of toxic protein aggregates (Schulte and Neckers, 1998; Boland et al., 2008; Eskelinen and Saftig, 2009; Chouraki and Seshadri, 2014).
The Hsp60 family
Hsp60, also called chaperonins, is a 60 kDa mitochondrial chaperone (Ranford et al., 2000; Itoh et al., 2002; Tutar and Tutar, 2010). GroEL, a well characterized bacterial chaperone, also belongs to this class. Although the primary location of Hsp60 is mitochondria, it can migrate to the cytosol under certain cellular stresses (Ranford et al., 2000; Itoh et al., 2002; Tutar and Tutar, 2010). Hsp60 forms a double ring complex, in which each rich is composed of seven subunits. Clients are fed into the central cavity of the double ring complex, in which their exposed hydrophobic residues are sequestered during refolding process (Ranford et al., 2000; Tutar and Tutar, 2010). The folding process by Hsp60 is modulated by a lid, which are formed by cochaperones such as Hsp10 in mitochondria. Following ATP hydrolysis, the unfolded client is released through the opening of the Hsp10 lid, now with a native folding (Ranford et al., 2000; Tutar and Tutar, 2010). Hsp60 works together with Hsp70 for protein folding of unfolded proteins. Neurons contain another type of chaperonins in the cytosol, which do not depend on cochaperones. They form a homotypic or heterotypic double ring complex, each of which is composed of eight subunits. The members of this group include the TCP-1 Ring Complex (TRiC), alternatively called TCP1 complex (CCT) (Lopez et al., 2015). Although, studies have shown that Hsp60 interacts with mutant α-synuclein in PD (Irizarry et al., 1998; Spillantini et al., 1998), the physiological importance of Hsp60 proteins in the refolding of pathogenic misfolded proteins in neurodegeneration remains poorly characterized.
The small HSP family
Different from other types of HSPs, small HSPs are ATP independent. To date, 10 small HSPs with sizes ranging from 12 to 42 kDa are known in humans. In mouse brain, five small HSPs are prominently expressed (Quraishe et al., 2008). Amongst these, the neuronal expression of Hsp27 and αB crystallin is selectively induced under stresses (Quraishe et al., 2008). Members of this family are characterized by a 100-residue α-crystallin domain flanked by variable N-terminal and C-terminal extensions. These extensions are responsible for substrate recognition and mediates the formation of oligomers. As holding factors, small HSPs bind to unfolded or misfolded proteins and prevent their aggregation until the clients are delivered to other chaperones, such as Hsp70 and Hsp40 system (Carra et al., 2012). Amongst these, Hsp27 is the most abundant and well characterized. Their expression is selectively induces by various stresses that perturb proteostasis (Sarto et al., 2000; Sun and MacRae, 2005).
Degradation of misfolded proteins by molecular chaperones through the UPS
While the primary functions of molecular chaperones relate refolding and unfolding of nascent and misfolded proteins, they can facilitate the degradation of terminally misfolded clients, either through the UPS or autophagy (Lanneau et al., 2010). The majority of these clients are tagged with Ub chains for degradation by the proteasome complex. However, the substrates prone to aggregation are redirected to autophagy. In either case, molecular chaperones are involved in the recognition and/or delivery of terminally misfolded substrates.
Ub-dependent selective proteolysis through the proteasome
The UPS mediates selective proteolysis of short-lived proteins by the proteasome and accounts for more than 80% of intracellular proteolysis (Hershko and Ciechanover, 1998). In the UPS, Ub is first activated by its conjugation to the ubiquitin activating enzyme E1. This conjugation involves the ATP-dependent formation of a thioester bond between the C-terminal glycine residue of Ub and an active site cysteine of E1 (Hershko and Ciechanover, 1998; Ciechanover, 2013). The activated Ub is transferred to the Ub conjugating enzyme E2 through a thioester bond. The Ub ligase E3 promotes the transfer of Ub from E2 to the lysine (Lys) residue of substrates. This generates an isopeptide bond between the C-terminal glycine residue of Ub and lysine residues on the substrate (Hershko and Ciechanover, 1998; Ciechanover, 2013). The human genome encodes more than 500 E3s. These E3s can be divided into three groups depending on their ubiquitination domains, including the really interesting new gene (RING) finger, the homologous to E6-AP (HECT) domain, and the U-box domain (Qian et al., 2006). Occasionally, E4 enzymes enhance the conjugation of additional Ub molecules to form a Ub chain, typically through the K48 linkage (Upadhya and Hegde, 2007). Once the first Ub is attached to substrate, subsequent Ub conjugations may use any of its seven Lys residues (Peng et al., 2003). This can generates a Ub chain with many different topologies, each of which has distinct functions. Amongst these topologies, the most widely used Lys48 linkage typically leads to proteasomal degradation. The Lys63 linkage mediates nonproteolytic processes, such as Ub-dependent protein-protein interactions (Hadian et al., 2011). Human cells also use the Lys11 linkage for cell cycle regulation and cell division as well as ERAD (Matsumoto et al., 2010) and K27 for ubiquitin fusion and degradation (Morawe et al., 2012). Ub moieties conjugated to substrates are reversible and can be detached and adjusted by deubiquitination enzymes (DUBs). The substrates conjugated with polyubiquitin are degraded by the 26S proteasome (Hershko and Ciechanover, 1998). This 2.5-MDa protease complex is composed of the 20S core particle with a size of 700 kDa associated with two 19S regulatory particles (Ravikumar et al., 2008; Douglas et al., 2009; Ciechanover and Kwon, 2015). The Ub chains conjugated to substrates are recognized by RPN10 and RPN13 of the 19S particle and stripped off by DUBs such as RPN11, USP14, and UCHL5 (ubiquitin C-terminal hydrolase L5; Ravikumar et al., 2008; Douglas et al., 2009; Ciechanover and Kwon, 2015). Deubiquitinated substrates are unfolded into a nascent polypeptide through ATP hydrolysis in the 19S particle and fed into the 20S particle for degradation, generating short peptides with average sizes of 8–12 amino acids (Hershko and Ciechanover, 1998). These peptides are degraded into amino acids by aminopeptidases, which are recycled for protein synthesis, or presented on the cell surface for immunosurveillance (Kloetzel and Ossendorp, 2004).
Molecular chaperones and Ub ligases work together in the UPS
Several cytosolic or nuclear Ub ligases are known to be involved in degradation of misfolded proteins in collaboration with molecule chaperones, including UBR1, UBR2, San1, Hul5, E6-AP, C-terminus of Hsc70-interacting protein (CHIP) and Parkin (Gardner et al., 2005; Heck et al., 2010; Kettern et al., 2010; Figure 1). CHIP is a 35 kDa protein that has dual functions, one as a cochaperone of Hsp70 and Hsp90, and the other as a Ub ligase that mediates ubiquitination of misfolded proteins using its the RING-like U-box domain (Ballinger et al., 1999; McDonough and Patterson, 2003). The E3 activity of CHIP requires the interaction with the E2 Ub conjugating enzyme UBCH5 (Cyr et al., 2002). When interacting with the Hsp70- or Hsp90-client complex, CHIP in collaboration with UBCH5 captures and ubiquitinates the misfolded clients for proteasomal degradation (Demand et al., 2001). To facilitate the delivery of the ubiquitinated substrates to the proteasome, CHIP also interacts with the S5a component (also known as Rpn10) of the 19S proteasome particle (Connell et al., 2001). In this process, CHIP indirectly recognize misfolded proteins through the interaction between its TPR (tetratricopeptide repeat) domain with Hsp70 or Hsp90 (Lanneau et al., 2010). The substrates of CHIP include hyperphosphorylated tau and mutant SOD1 (Lanneau et al., 2010). CHIP-mediated degradation of Hsp70 clients is further facilitated by the cochaperone BAG-1 (Takayama et al., 1997). BAG-1 uses its C-terminal region to bind the ATPase domain of Hsp70 and acts as a nucleotide exchange factor (NEF), inducing the release of substrates from Hsp70 (Takayama et al., 1997). On the other hand, BAG-1 has also a Ub-like (UBL) domain at its N-terminal region that supports the interaction with the proteasome (Alberti et al., 2003). BAG-1 directly interacts and cooperates with CHIP to guide the terminally misfolded clients to the UPS. In addition to BAG-1, Hsp27 belonging to the small HSP family can directly interact with the proteasome to modulate the ubiquitination of clients (Garrido et al., 2006). Hsp27 also binds the Ub chain of clients and, thus, increase the degradation of ubiquitinated proteins (Garrido et al., 2006).
Figure 1.

Protein degradation by molecular chaperones through the UPS. Molecular chaperones such as Hsp70 recognizes the hydrophobic sequences of misfolded proteins as degrons. The Ub ligase CHIP guides the chaperone-client complexes to the UPS and mediates the clients' ubiquitination. The UPS involves a cascade of E1, E2, and E3 enzymes whose cooperative activities mediate the conjugation of Ub to target proteins. In PQC, most E3s cannot recognize misfolded proteins and rather depend on molecular chaperones for substrate recognition. Ubiquitinated substrates are degraded by the proteasome into short peptides, typically with sizes of 8–12 amino acids.
The clients of Hsp90 can also be degraded through the UPS if they are no longer chaperoned by Hsp90, for example, owing to misfolding. These misfolded clients dissociated form Hsp90 are ubiquitinated by E3 ligases, such as CHIP, and degraded by the proteasome (Didelot et al., 2007). However, CHIP is mainly associated with Hsp70, and there should be additional E3 ligases that target the misfolded clients of Hsp90. One such candidate is the E3 ligase Triad3A which forms a complex with Hsp90 and receptor interacting protein 1 (RIP-1) and mediates the ubiquitination of RIP-1 and proteasomal degradation following Hsp90 inhibition by geldanamycin (Fearns et al., 2006).
The N-end rule pathway is a proteolytic system in which a single N-terminal residue acts as an essential component of a class of degrons, called N-degrons (Bachmair et al., 1986; Tasaki and Kwon, 2007; Sriram and Kwon, 2010; Sriram et al., 2011; Varshavsky, 2011). In mammals, these N-terminal degrons are recognized by the N-recognin family, including UBR1, UBR2, UBR4, UBR5, and p62 (Kwon et al., 1999a, 2002; Tasaki et al., 2005, 2009; An et al., 2006). Amongst these, the Ub ligases UBR1 and UBR2 have been shown to mediate the ubiquitination of misfolded cytosolic proteins, leading to proteasomal degradation (Eisele and Wolf, 2008; Heck et al., 2010; Prasad et al., 2010). These RING finger E3 ligases indirectly recognize misfolded proteins through molecular chaperones such as Hsp110 and Hsp70 (Heck et al., 2010; Nillegoda et al., 2010). Misfolded proteins targeted by N-recognins include TDP43 in ALS and tau and amyloid β in AD (Brower et al., 2013). Interestingly, in addition to the exposed hydrophobic residues, some of their misfolded clients are post-translationally conjugated with the amino acid L-Arg of Arg-tRNAArg by ATE1-encoded R-transferases (Grigoryev et al., 1996; Balogh et al., 2000, 2001; Kwon et al., 2000; Lee et al., 2005). The resulting N-terminal Arg residue acts as N-degron which is recognized by N-recognins such as UBR1 and UBR2 (Kwon et al., 1999b; Lee et al., 2008; Sriram et al., 2009; Meisenberg et al., 2012). In yeasts, the cytosolic E3 ligase Ubr1 has been shown to work with the nuclear E3 ligase San1 if cytosolic misfolded proteins overwhelm the capacity of E3 ligases (Heck et al., 2010; Prasad et al., 2010). In this collaboration between cytosolic and nuclear PQC systems, San1 associated with Hsp70 brings excessive cytosolic misfolded proteins to the nucleus for proteasomal degradation (Heck et al., 2010; Prasad et al., 2010). Different from other E3 ligases, San1 has many disordered structures and stretches of hydrophobic residues and, thus, can directly bind misfolded proteins (Rosenbaum et al., 2011). In mammals, the nuclear Ub ligase UHRF2 has been proposed to be a functional homolog of the yeast San1 (Nielsen et al., 2014).
Eukaryotic cells operate various degradative machinery designated to specific types of misfolded proteins. In yeasts, misfolded proteins generated by heat shock are specifically ubiquitinated by the E3 ligase Hul5 that has a HECT ubiquitination domain (Fang et al., 2011). In mammals, mislocalized membrane proteins are ubiquitinated by the Ub ligase RNF126 (RING finger 126) in collaboration with the BAG6 chaperone system (Rodrigo-Brenni et al., 2014). Proteins synthesized from aberrant mRNAs without stop codons are ubiquitinated by Listerin/Ltn1 (Bengtson and Joazeiro, 2010). Moreover, in the ER, membrane-associated misfolded proteins are ubiquitinated by the Ub ligase DOA10 (Nielsen et al., 2014). By contrast, misfolded proteins in the ER lumen are ubiquitinated by the Ub ligases Hrd1 and Gp78 mediates through a process called ERAD (ER-associated degradation) (Vembar and Brodsky, 2008). Except for San1 and Hul5, most of these E3s indirectly recognize misfolded proteins through cooperating molecular chaperones. Overall, the mechanistic details and clinical importance of these various PQC machinery in neurodegenerative diseases remains largely unexplored.
Deubiquitination enzymes (DUBs) in the degradation of misfolded proteins
DUBs detach Ub molecules from substrates and, thus, can modulate the proteasomal degradation of Ub-conjugated substrates. The proteasome is associated with DUBs such as RPN11, UCHL5, and USP14. RPN11 is a stoichiometric subunit of the proteasome and detaches Ub molecules en bloc from substrates (Hao et al., 2013). The free, unanchored Ub chains are deposited to aggresomes and recognized by HDAC which brings misfolded protein aggregates to aggresomes (Hao et al., 2013). The interaction between HDAC and unanchored Ub chains is essential for cargo-loaded HDAC to see where aggresomes are (Hao et al., 2013). In contrast to RPN11, USP14 is a conditionally recruited to the proteasome through its UBL domain. This enhances its activity up to 800-folds and, thus, modulates the degradation rate of substrates (Crosas et al., 2006). The treatment of the USP14 inhibitor IU1 has been shown to facilitate the degradation of aggregation-prone misfolded proteins such as tau and polyQ-expanded mutant ataxin-3 (Lee et al., 2010). The functions of DUBs in neurodegenerative diseases remain largely unexplored.
The UPS is impaired during neurodegeneration
The pathogenesis of most neurodegenerative diseases, such as AD, PD, ALS, HD, and prion diseases commonly involves the downregulation of the components of the UPS. One prominent risk factor is aging. The activities of UPS components, such as the proteasome, are often progressively declined as neurons age (Keller et al., 2000; Hwang et al., 2007; Tydlacka et al., 2008; Low, 2011). This may reduce the ability to degrade misfolded proteins, contributing to the accumulation of pathological protein aggregates. Making it worse, the accumulated aggregates as a consequence of reduced UPS activities now further inhibit the activities of UPS components, including the proteasome. The proteasome is particularly vulnerable to protein aggregates because its narrow chamber has a diameter of as small as 13 angstroms. Therefore, proteasome cannot digest protein aggregates that cannot be easily unfolded. For example, β-sheet-rich PrP aggregates were shown to block the opening of the 20S proteasome particle, further reducing proteasomal activity (Andre and Tabrizi, 2012). Following ubiquitination and aggregation, tau in AD binds the recognition site of the 19S catalytic particle and block its gate (Dantuma and Lindsten, 2010; Tai et al., 2012). Aggregates of many other pathogenic proteins in neurodegenerative disorders can directly inhibit proteasome activity (Gregori et al., 1995; Snyder et al., 2003; Lindersson et al., 2004; Kristiansen et al., 2007). The resulting proteotoxicity has adverse effects on neurons (Hegde and Upadhya, 2011). Indeed, the reduced UPS activity has been associated with neuronal damage in AD, HD, PD, ALS, ataxia, Angelman syndrome, Wallerian degeneration, and gracile axonal dystrophy (Hegde, 2010; Hegde and Upadhya, 2011).
Degradation of misfolded proteins by autophagy
Autophagy is a process by which cytosolic materials are degraded by the lysosome. Depending on the mechanism of cargo delivery to the lysosome, autophagy can be divided into three pathways: microautophagy, CMA, and macroautophagy. Terminally misfolded proteins in neurodegenerative diseases can be degraded through macroautophagy or CMA (Figure 2). The role of autophagy in proteostasis is vitally important for postmitotic neurons with long extensions, in which cytotoxic proteins cannot be diluted by cell division.
Figure 2.

The role of molecular chaperones in PQC. Molecular chaperones, such as Hsp70 in combination with the cochaperone Hsp40, facilitate the refolding of misfolded proteins. If the clients fail to refold, molecular chaperones can also mediate their degradation in collaboration with cellular proteolytic pathways. In principle, soluble misfolded proteins are targeted by the UPS, in which the clients are ubiquitinated by E3 Ub ligases followed by degradation through the 26S proteasome. However, if the clients are prone to aggregation or escape the surveillance of the UPS, they can be degraded by lysosomal hydrolases, either through macroautophagy or CMA. As the last step of PQC, molecular chaperones can disaggregate already formed aggregates. Also shown are misfolded proteins induced by oxidative stress in mitochondria.
Macroautophagy
Misfolded proteins prone to aggregation can be directed to macroautophagy for lysosomal degradation. These substrates, typically as a Ub-conjugated form, are collected by autophagy adaptors, such as p62 and NBR1 (Cha-Molstad et al., 2015). P62 is normally inactive and can be activated by binding to the N-terminally arginylated form of the molecular chaperone BiP/GRP78, the ER counterpart of cytosolic Hsp70 (Cha-Molstad et al., 2015). Upon the accumulation of non-degradable autophagic cargoes, BiP and other ER-residing chaperones, such as calreticulin and protein disulfide isomerase (PDI), are N-terminally arginylated by ATE1-encoded R-transferase. The resulting N-terminally arginylated BiP, R-BiP, locates in the cytosol where R-BiP binds the ZZ domain of p62 through its N-terminal arginine residue. This binding induces a conformational change in p62, facilitating the polymerization of p62 as well as the interaction of p62 with LC3-II which is anchored on the membrane of autophagosomes (Cha-Molstad et al., 2015). It is generally assumed that p62 and other autophagic adaptors recognize the Ub moieties conjugated to misfolded proteins and delivers them to the autophagosome through specific interaction with LC3 (Lamark et al., 2009; Stolz et al., 2014). The cargo-loaded autophagosome is fused with the lysosome to form the autolysosome wherein cargoes along with p62 are degraded by lysosomal hydrolases.
CMA
CMA is a selective proteolytic system and does not involve vesicle formation (Chiang et al., 1989; Dice, 1990; Cuervo et al., 1997). The selectivity enables the degradation of misfolded or damaged cytosolic proteins without interfering with the same kinds of proteins with normal functions (Fuertes et al., 2003; Massey et al., 2006; Kaushik and Cuervo, 2012). The target substrates of the CMA include cytosolic proteins that carry the KFERQ pentapeptide which functions as a degron. The CMA degron, found in approximately 30% of cytosolic proteins (Chiang and Dice, 1988; Dice, 1990), is recognized by chaperones associated with cochaperones such Hsc70 belonging to the Hsp70 family (Chiang et al., 1989) (Figure 3). The function of Hsc70 requires cochaperones, such as Hsp40, Hsp90, HIP, HOP, and BAG-1 (Agarraberes and Dice, 2001). The substrates associated with the Hsc70 chaperone system are translocated to the lysosomal membrane through the interaction of Hsc70 with LAMP2A, a single-span membrane protein (Cuervo and Dice, 1996). The stability of LAMP2A requires its association with Lys-Hsc70, a lysosomal homolog of Hsc70. Once the substrate is targeted to the lysosome, Lys-Hsc70 assists the active LAMP2A complex to be disassembled into the inactive monomeric form, which is now available for the next round of the CMA process (Bandyopadhyay et al., 2008). The levels of LAMP2A and Lys-Hsc70 are important underlying the rate of CMA degradation (Cuervo et al., 1995; Agarraberes et al., 1997; Cuervo and Dice, 2000). Although a large number of cytosolic proteins contain the CMA degron sequence, only a limited number of these proteins were demonstrated to be degraded by the CMA (Wing et al., 1991). Post-translational modifications can generate the substrates of the CMA (Chiang and Dice, 1988; Dice, 1990). Studies have shown that CMA is essential for the survival of neurons by degrading misfolded or damaged cytosolic proteins (Cuervo et al., 2004). The misregulation of the CMA has been shown to correlate to the pathogenesis of neurodegeneration (Cuervo et al., 2004).
Figure 3.
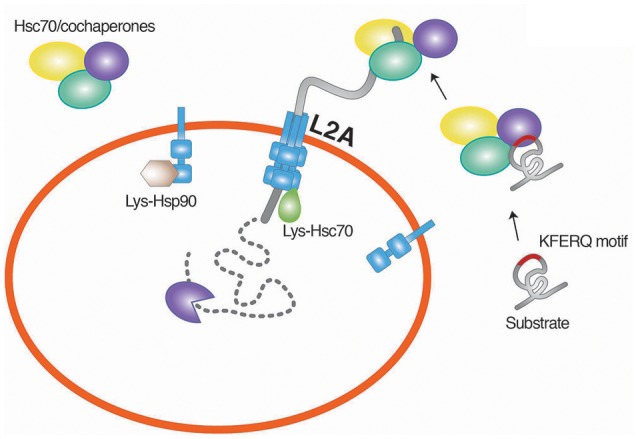
Chaperone-mediated degradation of misfolded proteins. CMA is a selective proteolytic system in which cytosolic proteins carrying the KFERQ pentapeptide are targeted by Hsc70. The function of Hsc70 requires cochaperones, such as Hsp40, Hsp90, HIP, HOP, and BAG-1. The substrates associated with the Hsc70 chaperone system are translocated to the lysosomal membrane through the interaction of Hsc70 with LAMP2A, a single-span membrane protein. L2A, LAMP2A.
Disaggregation of aggregates by molecular chaperones
Misfolded proteins may form aggregates if the PQC system is overwhelmed, for example, under severe stress conditions or by genetic mutations that allows the accumulation of non-degradable polypeptides. Yeast and mammalian cells have molecular chaperones (disaggregases) that can disaggregate the already formed aggregates (Parsell et al., 1994; Mogk et al., 1999; Doyle et al., 2013). Proteins recovered from aggregates are either refolded or degraded (Ravikumar et al., 2008; Douglas et al., 2009; Ciechanover and Kwon, 2015).
Yeast Hsp104 belonging to the Hsp100 family is a powerful AAA+ ATPase that has a hexameric ring structure with a central channel (Shorter, 2011; Torrente and Shorter, 2013). Once guided to protein aggregates by Hsp70, Hsp104 retrieves proteins from aggregates and threads them into nascent polypeptides (Seyffer et al., 2012; Lee et al., 2013; Lipinska et al., 2013; Carroni et al., 2014). During threading, Hsp70 and Hsp40 assist the unfolding of substrates to generate surface loops that are fed into the core of Hsp104 (Zietkiewicz et al., 2006). This disaggregation activity of Hsp104 was demonstrated to be effective for various aggregates (Mosser et al., 2004; Shorter and Lindquist, 2004; Arimon et al., 2008; Lo Bianco et al., 2008; DeSantis et al., 2012). Despite the disaggregase activities, Hsp104 exhibited the modest efficacy for the pathogenic misfolded proteins in human neurodegenerative diseases (DeSantis et al., 2012). The introduction of a few point mutations markedly increased its disaggregase activity for the preformed aggregates of α-synuclein in PD (Jackrel and Shorter, 2014), and TDP-43 and FUS in ALS (Jackrel and Shorter, 2014; Jackrel et al., 2014). Compared with wild-type Hsp104, the engineered form had increased ATPase activity with reduced dependence on the Hsp70/Hsp40 and, thus, exhibited enhanced activities in protein translocation and remodeling (Jackrel and Shorter, 2014; Jackrel et al., 2014).
In humans, Hsp70 and Hsp40 interact with the cochaperone Hsp110 to facilitate the disaggregation of protein aggregates (Gao et al., 2015; Nillegoda et al., 2015). Hsp110 belonging to the conserved Hsp70 superfamily has structural and functional similarity to Hsp70 including the nucleotide binding domain and acts as an NEF of Hsp70 (Polier et al., 2008). Although the action mechanism of the Hsp110-containing disaggregase complex remains unclear, it appears that the NEF activity of Hsp110 facilitates ADP release from Hsp70 (Rampelt et al., 2012). This ability to facilitate disaggregation in vitro has been equally observed with all three types of human Hsp110 isoforms: Hsp105α/HSPH1, Apg-2/HSPH2, and Apg-1/HSPH3 (Rampelt et al., 2012). The importance of Hsp110 in disaggregation has been demonstrates with various amyloids and prefibrillar oligomers and reactivate proteins from aggregates (Lo Bianco et al., 2008). Loss-of-function studies of Hsp110 have also shown its disaggregation activity in C. elegans (Rampelt et al., 2012), mouse cells (Yamagishi et al., 2010) and Plasmodium falciparum (Zininga et al., 2016).
Protective role of chaperones in neurodegeneration
Many neurodegenerative diseases are directly caused by the excessive accumulation of misfolded proteins and their aggregates. During the pathogenesis, molecular chaperones play a central role in the refolding, degradation, and disaggregation of these pathogenic protein species. Extensive studies have shown that molecular chaperones promote the removal of pathogenic misfolded proteins and their aggregates.
HD and other polyQ diseases
HD is a progressive neurodegenerative disease associated with the accumulation of mutant huntingtin (mHTT) that has the excessive repetition of glutamine residues, called polyQ, which causes misfolding (Shastry, 2003; Lee et al., 2011). These misfolded mHTT causes selective neuronal damage and death, leading to cognitive and motor defects (Gusella and MacDonald, 1998; Ramaswamy et al., 2007; Roos, 2010). Studies have shown that Hsp70 in a complex with Hsp40 plays a major role in inhibiting the formation of mHTT aggregates. Specifically, the Hsp70/Hsp40 machinery binds misfolded mHTT and holds its folding state to attenuate the formation of mHTT oligomers (Jana et al., 2000). The neuroprotective role of Hsp70 in the pathogenesis of PD is highlighted by a genetic screening of Drosophila PD model, which identified Hsp70 and Hsp40 as two major suppressors of the neurotoxicity caused by mHTT (Kazemi-Esfarjani and Benzer, 2000). Consistently, the knockout of Hsp70 in R6/2 transgenic HD mice has been shown to aggravate the symptoms in neurodegeneration (Wacker et al., 2009). A similar neuroprotective efficacy was observed with the neuronal chaperone HSJ1a (DNAJB2a) belonging to the Hsp40 family (Labbadia et al., 2012). In addition to the Hsp70-Hsp40 machinery, other members of the Hsp70 family have also been shown to counteract mHTT cytotoxicity. Specifically, the cytosolic chaperone Hsc70 binds and directly delivers mHTT to the lysosome via CMA, leading to selective degradation of mHTT and reduced toxicity (Bauer et al., 2010). This finding is further supported by in vivo studies using mice (Bauer et al., 2010) as well as flies (Gunawardena et al., 2003) overexpressing Hsc70. An ER counterpart of Hsp70, BiP/GRP78, has also been shown to counteract the accumulation of mHTT aggregates and apoptosis (Jiang et al., 2012). Besides the Hsp70 family members and their cochaperones, several other chaperones have been implicated in the refolding and/or degradation of polyQ protein aggregates, including Hsp84 (Mitsui et al., 2002), Hsp104 (Vacher et al., 2005), Hsp104/Hsp27 (Perrin et al., 2007), the chaperonin TRiC (Nollen et al., 2004; Behrends et al., 2006; Kitamura et al., 2006), and the cochaperone Prefoldin (Tashiro et al., 2013). Finally, HSPB7 belonging to small HSPs (Vos et al., 2010) and CHIP (Al-Ramahi et al., 2006) were shown to counteract the formation of polyQ aggregates in disease models.
PD
PD is the second most common neurodegenerative disease after AD, affecting up to 10% of humans over 65 years. This protein misfolding disorder is associated with the loss of dopaminergic neurons in the substantia nigra pars compacta of brain (Wirdefeldt et al., 2011). PD is characterized by the formation of insoluble α-synuclein aggregates which are deposited as nuclear inclusions (Goedert, 2001; Ross and Poirier, 2004; Hasegawa et al., 2016) as a ubiquitinated form (Hasegawa et al., 2002). These inclusion, called Lewy bodies, are mainly composed of α-synuclein aggregates (Irizarry et al., 1998; Spillantini et al., 1998) together with various components of PQC, including Ub (Kuzuhara et al., 1988) and molecular chaperones such as Hsp70, Hsp90, Hsp60, Hsp40, Hsp27, and CHIP (McLean et al., 2002). This co-aggregation pattern indicates that α-synuclein aggregates deposited in Lewy bodies are the remnants that survived the attempts of molecular chaperones to maintain proteostasis. Specifically, Hsp70 recognizes the hydrophobic degron of misfolded α-synuclein through its substrate binding domain (Dedmon et al., 2005; Luk et al., 2008). By holding the folding status, Hsp70 facilitates the refolding of misfolded α-synuclein and inhibits the formation of its oligomers (Outeiro et al., 2008). The in vivo efficacy of Hsp70 was demonstrated with overexpressed Hsp70 in flies (Auluck and Bonini, 2002; McLean et al., 2004; Zhou et al., 2004; Opazo et al., 2008; Danzer et al., 2011) and mice (Klucken et al., 2004). Moreover, the depletion of molecular chaperones was shown to aggravate the degeneration of neurons caused by proteotoxicity (Ebrahimi-Fakhari et al., 2011).
The ER chaperone BiP belonging to the Hsp70 family can interact with α-synuclein and reduce its neurotoxicity (Gorbatyuk et al., 2012). Overexpressed BiP has been shown to protect nigral dopaminergic neurons in a rat model of PD, which correlates to reduced ER stress mediators and apoptosis (Gorbatyuk et al., 2012). The anti-aggregation and neuroprotective activity of BiP was further demonstrated with photoreceptor cells expressing aggregation-prone mutant rhodopsin (Gorbatyuk et al., 2010; Athanasiou et al., 2012). In addition to Hsp70 proteins, αB-crystallin belonging to small SHPs can interact with α-synuclein and inhibit the elongation of its fibrillar seeds by forming nonfibrillar aggregates (Kudva et al., 1997; Stege et al., 1999; Rekas et al., 2004; Shammas et al., 2011). Another small HSP, Hsp27, can arrest the aggregation of α-synuclein in the initial phage, perhaps by binding to the partially folded monomers (Rekas et al., 2007; Bruinsma et al., 2011).
AD
AD is the most common neurodegenerative disorder caused by aggregation-prone proteins and selective loss, inactivation, or shrinkage in the mature nervous system (Regeur et al., 1994). The pathogenesis involves the deposit of amyloid-β (Aβ) both outside and inside the neurons as well as intracellular neurofibrillary tangles of hyperphosphorylated tau (Shankar et al., 2008; Honjo et al., 2012). Self-assembly of Aβ, which is not misfolded, generates its neurotoxic oligomers, which, in turn, grows into amyloid fibrils (Shankar et al., 2008; Honjo et al., 2012). Studies have shown that various molecular chaperones interact with intracellular Aβ species, which has been internalized by endocytosis, as well as tau and regulate their degradation. Specifically, αB-crystallin (HSPB5) and DnaJB6 bind to Aβ fibrils and inhibit their elongation and growth (Shammas et al., 2011; Mansson et al., 2014). In addition, Hsp70, Hsp40, and Hsp90 interact with the oligomer form of Aβ peptides (Evans et al., 2006). When overexpressed, Hsp70 and Hsp40 were shown to reduce the formation of Aβ aggregates and redirected it from growing into fibrillar to soluble circular structures (Evans et al., 2006). In contrast to Hsp70, Hsp90 supports the folding of tau and, thus, stabilizes this neurotoxic protein, facilitating tau pathology in AD model (Carman et al., 2013). The cochaperone BAG-1 forms a complex with Hsp70 and tau and can inhibit tau degradation in cultured cells, leading to the accumulation of both tau and APP (Elliott et al., 2007, 2009). Given the opposing roles of Hsp70 and Hsp90, the Hsp90 inhibitor 17-AAG was successfully used to induce the expression of various chaperones, such as Hsp70, Hsp40, and Hsp60 (Chen et al., 2014). The induction of these chaperones reduced Aβ toxicity in neurons (Chen et al., 2014). Another line of evidence supporting the protective role of chaperones in AD is provided by a study with UBB+1, a frameshift mutation of ubiquitin B (Hope et al., 2003). UBB+1 can inhibit the proteasome and, thus, can be deposited into intracellular protein inclusions in AD. The overexpression of UBB+1 induced the expression of HSPs, which, in turn, protected cells against oxidative stress.
ALS
ALS is the most common adult onset motor neuron disease that affects the brainstem, cortex and spinal cord. It is characterized by the atrophy, weakness, and paralysis of muscles, leading to death within 3–5 years post diagnosis (Robberecht and Philips, 2013). The majority of ALS patients are sporadic, whereas 5–10% are familial, i.e., linked to mutations in specific genes. Numerous genetic mutations are linked to ALS, either genetically and/or pathologically. Amongst these, the mutations of SOD1, a free radical scavenger enzyme, accounts for 20% of familial ALS cases (Rosen et al., 1993). Several other ALS-linked mutated proteins form intracellular aggregates, including C9ORF72 (DeJesus-Hernandez et al., 2011), transactive response DNA binding protein (TDP-43), fused in sarcoma/translocated in liposarcoma (FUS), vesicle associated protein B (VAPB), ubiquilin-2, optineurin, and protein disulphide isomerase 1 and 3 (PDIA1 and PDIA3) (Robberecht and Philips, 2013). The majority of ALS cases are considered protein misfolding disorder because these mutations cause the accumulation of misfolded proteins and their aggregates.
Various molecular chaperones are implicated in the formation of these protein aggregates in ALS. For example, Hsp70/Hsp40, Hsp27, Hsp25, and αB-crystalline can form complexes with an ALS-causing mutant form of SOD, SOD1G93A. However, the overexpression of Hsp70 alone was not sufficient to reduce mutant SOD1 toxicity in ALS mouse model (Liu et al., 2005). Instead, PDI proteins exhibit a protective role in ALS models (Walker et al., 2010; Jeon et al., 2014). PDI assists the rearrangement of incorrectly arranged disulfide bonds of ER clients. It can also act as a chaperone that not only counteracts the aggregation of proteins independent of disulfide bonds but also delivers terminally misfolded proteins to ERAD (Quan et al., 1995). Over 15 missense mutations of PDIA1 and ERp57/PDIA3 were linked to ALS (Yang and Guo, 2016). Various in vitro and animal studies showed that PDI is deposited to the aggregates formed by the mutant forms of TDP-43 and FUS (Honjo et al., 2011; Farg et al., 2012), TDP-43 (Honjo et al., 2011; Walker et al., 2013), and VAPB (Tsuda et al., 2008). The overexpression of PDI reduces mutant SOD1 inclusions in vitro whereas PDI knockdown facilitates the formation of ALS inclusions (Walker et al., 2010).
Hsp27 also plays a protective role in the pathogenesis of ALS. Hsp27 binds mutant SOD1 in vitro and inhibits its fibril elongation (Yerbury et al., 2013). The overexpression of Hsp27 was shown to inhibit mutant SOD1-induced cell death (Patel et al., 2005). Hsp27 exhibited a synergistic efficacy when Hsp70 was coexpressed (Patel et al., 2005). In addition to Hsp27, HSJ1a shows a similar protective activity against the formation of mutant SOD aggregates at the late stage of the disease (Novoselov et al., 2013). HSJ1a interacts with SOD1G93 and facilitates its ubiquitination and proteasomal degradation.
Therapeutic application targeting molecular chaperones in neurodegeneration
Given the protective role of molecular chaperones against pathogenic protein aggregates in neurodegenerative diseases, molecular chaperones are logical targets for drug development to modulate aggregation and clearance of the aggregates. Indeed, pharmaceutical induction of molecular chaperones has been demonstrated to effectively inhibit the formation of pathogenic aggregates in disease models.
Hsp90 supports in the folding/refolding and stability of a number of clients, including pathogenic misfolded protein aggregates in neurodegenerative diseases. While these activities are overall beneficial for refolding, however, Hsp90 also assists in the stability of neurotoxic proteins, favoring the accumulation of toxic protein aggregates (Schulte and Neckers, 1998; Boland et al., 2008; Eskelinen and Saftig, 2009; Chouraki and Seshadri, 2014). Therefore, one such strategy is the pharmaceutical inhibition of Hsp90. Geldanamycin competes with ATP and inhibits the folding and stabilization of neurotoxic proteins (Schulte and Neckers, 1998; Boland et al., 2008; Eskelinen and Saftig, 2009; Chouraki and Seshadri, 2014). In addition, upon binding to geldanamycin, Hsp90 releases a HSP-inducing transcript factor, HSF1 (McLean et al., 2004; Putcha et al., 2010). The dissociated HSF1 which otherwise would be sequestered by Hsp90 move to the nucleus and transcriptionally induces HSPs, such as Hsp70 (McLean et al., 2004; Putcha et al., 2010). Geldanamycin was successfully used to inhibit protein aggregation in the Drosophila (McLean et al., 2004; Putcha et al., 2010) and mouse PD model (Shen et al., 2005) and in a primary culture model of familial ALS (Batulan et al., 2006).
Despite its therapeutic efficacy, geldanamycin is toxic and cannot penetrate the blood brain barrier (BBB). A number of geldanamycin derivatives or the compounds that target HSF1 are now available, including geranylgeranylacetaone, celastrol, arimoclomol, withaferin A, 17-N-allylamino-17-demethoxygeldanamycin (17-AAG), and PU-DZ8 (Kieran et al., 2004; Niikura et al., 2006; Hoogstra-Berends et al., 2012; Khan et al., 2012; Kalmar et al., 2014; Sharma et al., 2015). Amongst these, celastrol is an anti-inflammatory and antioxidant compound extracted from a perennial creeping plant belonging to the Celastraceae family (Cleren et al., 2005). The treatment of celastrol in HD model mice resulted in the induction of Hsp70 expression associated with reduced loss of dopaminergic neurons induced by MPTP in the substantia nigra pars compacta (Cleren et al., 2005). Celastrol protected neurons against polyglutamine toxicity in vivo and in vitro (Zhang and Sarge, 2007) and reduced the β-amyloid level in mouse AD (Paris et al., 2010) and HD (Zhang and Sarge, 2007) models. BBB-permeable Hsp90 inhibitors, 17-AAG and PU-DZ8, were used to decrease the levels of phosphorylated tau in the AD model (Luo et al., 2007) and to inhibit neurodegeneration in a fly HD model (Fujikake et al., 2008). In addition, as Hsp90 inhibition causes undesirable proteotoxicity, HSF1A, a small benzyl pyrazole-based compound, has been developed to activate Hsf1 without inhibiting Hsp90 (Neef et al., 2010). Overall, studies using these HSP-inducing compounds in animal models of neurodegenerative diseases demonstrate that this strategy has potential for therapeutic application.
Concluding remarks
Neurodegenerative diseases are caused by failure in PQC, which can be attributed to genetic mutations or alternatively an age-related decline in proteolytic activities. Molecular chaperones are an essential component of PQC in that they recognize unfolded or misfolded proteins, hold their folding status, and release them for spontaneous refolding. These nanoscale molecular machines can also facilitate the degradation of terminally misfolded proteins either through the UPS and autophagy. As the last defense mechanism of PQC, molecular chaperons can disaggregate the already formed aggregates. Thus, molecular chaperones play a pivotal role to protect neurons from the accumulation of pathogenic protein aggregates. It is therefore not surprising that pharmaceutical means are exploited to modulate the activities and functions of molecular chaperones. Indeed, small molecule compounds that target molecular chaperones such as Hsp90 have been successfully demonstrated to be effective in various neurodegenerative diseases. There is now an emerging consensus that proteostasis in diseases could be restored by using small molecule compounds or RNA interference that modulates chaperone expression or activities. A better understanding of chaperone functions in neurons will help the development of therapeutic means to restore proteostasis.
Author contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ho Sun Kim (Seoul National University) for editorial assistance and Jung Woo Hwang (Design EDA) for drawing the figures presented in this manuscript. This work was supported by the Basic Science Research Program (NRF-2016R1A2B3011389 to YK) of the National Research Foundation (NRF) funded by the Ministry of Science, ICT and Future Planning (MSIP) of Korea, the Brain Korea 21 PLUS Program (to Seoul National University), SNU Nobel Laureates Invitation Program (to AC), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to AC), the Israel Science Foundation (ISF) (to AC), the I-CORE Program of the Planning and Budgeting Committee and the ISF (Grant 1775/12) (to AC), the Deutsch-Israelische Projektkooperation (DIP) (to AC), and a special fund for research in the Technion established by Albert Sweet (to AC). AC is an Israel Cancer Research Fund USA Professor.
Glossary
Abbreviations
- 17-AAG
17-N-allylamino-17-demethoxygeldanamycin
- Aβ
amyloid-β
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- BBB
blood brain barrier
- C-domain
C-terminal dimerization domain
- CHIP
C-terminus of Hsc70-interacting protein
- CMA
chaperone-mediated autophagy
- DUBs
deubiquitination enzymes
- ERAD
ER-associated degradation
- HD
Huntington's disease
- Hsc70
heat shock cognate 70
- HSF1
heat-shock factor 1
- HSP
heat-shock protein
- HSR
heat-shock response
- M-domain
mid-domain
- mHTT
mutant huntingtin
- N-domain
N-terminal ATP-binding domain
- NEF
nucleotide exchange factor
- PD
Parkinson's disease
- PDI
protein disulfide isomerase
- PQC
protein quality control
- RING
really interesting new gene
- RIP-1
receptor interacting protein 1
- SBD
substrate binding domain
- TDP-43
transactive response DNA binding protein
- TRiC
TCP-1 Ring Complex
- TSE
transmissible spongiform encephalopathies
- Ub
ubiquitin
- UBL
Ub-like
- UCHL5
ubiquitin C-terminal hydrolase L5
- UPS
Ub-proteasome system
- VAPB
vesicle associated protein B.
References
- Agarraberes F. A., Dice J. F. (2001). A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 114(Pt 13), 2491–2499. [DOI] [PubMed] [Google Scholar]
- Agarraberes F. A., Terlecky S. R., Dice J. F. (1997). An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J. Cell Biol. 137, 825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S., Esser C., Hohfeld J. (2003). BAG-1–a nucleotide exchange factor of Hsc70 with multiple cellular functions. Cell Stress Chaperones 8, 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ramahi I., Lam Y. C., Chen H. K., de Gouyon B., Zhang M., Perez A. M., et al. (2006). CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J. Biol. Chem. 281, 26714–26724. 10.1074/jbc.M601603200 [DOI] [PubMed] [Google Scholar]
- Ammirante M., Rosati A., Gentilella A., Festa M., Petrella A., Marzullo L., et al. (2008). The activity of hsp90α promoter is regulated by NF-β B transcription factors. Oncogene 27, 1175–1178. 10.1038/sj.onc.1210716 [DOI] [PubMed] [Google Scholar]
- An J. Y., Seo J. W., Tasaki T., Lee M. J., Varshavsky A., Kwon Y. T. (2006). Impaired neurogenesis and cardiovascular development in mice lacking the E3 ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 6212–6217. 10.1073/pnas.0601700103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre R., Tabrizi S. J. (2012). Misfolded PrP and a novel mechanism of proteasome inhibition. Prion 6, 32–36. 10.4161/pri.6.1.18272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimon M., Grimminger V., Sanz F., Lashuel H. A. (2008). Hsp104 targets multiple intermediates on the amyloid pathway and suppresses the seeding capacity of Aβ fibrils and protofibrils. J. Mol. Biol. 384, 1157–1173. 10.1016/j.jmb.2008.09.063 [DOI] [PubMed] [Google Scholar]
- Athanasiou D., Kosmaoglou M., Kanuga N., Novoselov S. S., Paton A. W., Paton J. C., et al. (2012). BiP prevents rod opsin aggregation. Mol. Biol. Cell 23, 3522–3531. 10.1091/mbc.E12-02-0168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck P. K., Bonini N. M. (2002). Pharmacological prevention of Parkinson disease in Drosophila. Nat. Med. 8, 1185–1186. 10.1038/nm1102-1185 [DOI] [PubMed] [Google Scholar]
- Bachmair A., Finley D., Varshavsky A. (1986). In vivo half-life of a protein is a function of its amino-terminal residue. Science 234, 179–186. [DOI] [PubMed] [Google Scholar]
- Ballinger C. A., Connell P., Wu Y., Hu Z., Thompson L. J., Yin L. Y., et al. (1999). Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19, 4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogh S. A., Kwon Y. T., Denenberg V. H. (2000). Varying intertrial interval reveals temporally defined memory deficits and enhancements in NTAN1-deficient mice. Learn. Mem. 7, 279–286. 10.1101/lm.33500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogh S. A., McDowell C. S., Kwon Y. T., Denenberg V. H. (2001). Facilitated stimulus-response associative learning and long-term memory in mice lacking the NTAN1 amidase of the N-end rule pathway. Brain Res. 892, 336–343. 10.1016/S0006-8993(00)03268-6 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay U., Kaushik S., Varticovski L., Cuervo A. M. (2008). The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 28, 5747–5763. 10.1128/MCB.02070-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batulan Z., Taylor D. M., Aarons R. J., Minotti S., Doroudchi M. M., Nalbantoglu J., et al. (2006). Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol. Dis. 24, 213–225. 10.1016/j.nbd.2006.06.017 [DOI] [PubMed] [Google Scholar]
- Bauer P. O., Goswami A., Wong H. K., Okuno M., Kurosawa M., Yamada M., et al. (2010). Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 28, 256–263. 10.1038/nbt.1608 [DOI] [PubMed] [Google Scholar]
- Behrends C., Langer C. A., Boteva R., Bottcher U. M., Stemp M. J., Schaffar G., et al. (2006). Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol. Cell 23, 887–897. 10.1016/j.molcel.2006.08.017 [DOI] [PubMed] [Google Scholar]
- Bengtson M. H., Joazeiro C. A. (2010). Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 467, 470–473. 10.1038/nature09371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bercovich B., Stancovski I., Mayer A., Blumenfeld N., Laszlo A., Schwartz A. L., et al. (1997). Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J. Biol. Chem. 272, 9002–9010. [DOI] [PubMed] [Google Scholar]
- Boland B., Kumar A., Lee S., Platt F. M., Wegiel J., Yu W. H., et al. (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J. Neurosci. 28, 6926–6937. 10.1523/JNEUROSCI.0800-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broersen K., Rousseau F., Schymkowitz J. (2010). The culprit behind amyloid β peptide related neurotoxicity in Alzheimer's disease: oligomer size or conformation? Alzheimers. Res. Ther. 2:12. 10.1186/alzrt36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower C. S., Piatkov K. I., Varshavsky A. (2013). Neurodegeneration-associated protein fragments as short-lived substrates of the N-end rule pathway. Mol. Cell 50, 161–171. 10.1016/j.molcel.2013.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinsma I. B., Bruggink K. A., Kinast K., Versleijen A. A., Segers-Nolten I. M., Subramaniam V., et al. (2011). Inhibition of α-synuclein aggregation by small heat shock proteins. Proteins 79, 2956–2967. 10.1002/prot.23152 [DOI] [PubMed] [Google Scholar]
- Buchner J. (1996). Supervising the fold: functional principles of molecular chaperones. FASEB J. 10, 10–19. [PubMed] [Google Scholar]
- Carman A., Kishinevsky S., Koren J., III., Lou W., Chiosis G. (2013). Chaperone-dependent Neurodegeneration: a Molecular Perspective on Therapeutic Intervention. J. Alzheimers Dis. Parkinson. 2013(Suppl 10):007. 10.4172/2161-0460.S10-007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carra S., Crippa V., Rusmini P., Boncoraglio A., Minoia M., Giorgetti E., et al. (2012). Alteration of protein folding and degradation in motor neuron diseases: implications and protective functions of small heat shock proteins. Prog. Neurobiol. 97, 83–100. 10.1016/j.pneurobio.2011.09.009 [DOI] [PubMed] [Google Scholar]
- Carroni M., Kummer E., Oguchi Y., Wendler P., Clare D. K., Sinning I., et al. (2014). Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. Elife 3:e02481. 10.7554/eLife.02481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha-Molstad H., Sung K. S., Hwang J., Kim K. A., Yu J. E., Yoo Y. D., et al. (2015). Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat. Cell Biol. 17, 917–929. 10.1038/ncb3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham M. E., Brion J. P., Anderton B. H. (1992). Human homologues of the bacterial heat-shock protein DnaJ are preferentially expressed in neurons. Biochem. J. 284(Pt 2), 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham M. E., Caplan A. J. (1998). Structure, function and evolution of DnaJ: conservation and adaptation of chaperone function. Cell Stress Chaperones 3, 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Wang B., Liu D., Li J. J., Xue Y., Sakata K., et al. (2014). Hsp90 chaperone inhibitor 17-AAG attenuates Aβ-induced synaptic toxicity and memory impairment. J. Neurosci. 34, 2464–2470. 10.1523/JNEUROSCI.0151-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang H. L., Dice J. F. (1988). Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J. Biol. Chem. 263, 6797–6805. [PubMed] [Google Scholar]
- Chiang H. L., Terlecky S. R., Plant C. P., Dice J. F. (1989). A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246, 382–385. [DOI] [PubMed] [Google Scholar]
- Choo Y. S., Johnson G. V., MacDonald M., Detloff P. J., Lesort M. (2004). Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 13, 1407–1420. 10.1093/hmg/ddh162 [DOI] [PubMed] [Google Scholar]
- Chouraki V., Seshadri S. (2014). Genetics of Alzheimer's disease. Adv. Genet. 87, 245–294. 10.1016/B978-0-12-800149-3.00005-6 [DOI] [PubMed] [Google Scholar]
- Ciechanover A. (2013). Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Bioorg. Med. Chem. 21, 3400–3410. 10.1016/j.bmc.2013.01.056 [DOI] [PubMed] [Google Scholar]
- Ciechanover A., Kwon Y. T. (2015). Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 47:e147. 10.1038/emm.2014.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A., Laszlo A., Bercovich B., Stancovski I., Alkalay I., Ben-Neriah Y., et al. (1995). The ubiquitin-mediated proteolytic system: involvement of molecular chaperones, degradation of oncoproteins, and activation of transcriptional regulators. Cold Spring Harb. Symp. Quant. Biol. 60, 491–501. [DOI] [PubMed] [Google Scholar]
- Cleren C., Calingasan N. Y., Chen J., Beal M. F. (2005). Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J. Neurochem. 94, 995–1004. 10.1111/j.1471-4159.2005.03253.x [DOI] [PubMed] [Google Scholar]
- Connell P., Ballinger C. A., Jiang J., Wu Y., Thompson L. J., Hohfeld J., et al. (2001). The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 3, 93–96. 10.1038/35050618 [DOI] [PubMed] [Google Scholar]
- Crosas B., Hanna J., Kirkpatrick D. S., Zhang D. P., Tone Y., Hathaway N. A., et al. (2006). Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell 127, 1401–1413. 10.1016/j.cell.2006.09.051 [DOI] [PubMed] [Google Scholar]
- Cuervo A. M., Dice J. F. (1996). A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503. [DOI] [PubMed] [Google Scholar]
- Cuervo A. M., Dice J. F. (2000). Regulation of lamp2a levels in the lysosomal membrane. Traffic 1, 570–583. 10.1034/j.1600-0854.2000.010707.x [DOI] [PubMed] [Google Scholar]
- Cuervo A. M., Dice J. F., Knecht E. (1997). A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J. Biol. Chem. 272, 5606–5615. [DOI] [PubMed] [Google Scholar]
- Cuervo A. M., Knecht E., Terlecky S. R., Dice J. F. (1995). Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. 269(5 Pt 1), C1200–C1208. [DOI] [PubMed] [Google Scholar]
- Cuervo A. M., Stefanis L., Fredenburg R., Lansbury P. T., Sulzer D. (2004). Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. 10.1126/science.1101738 [DOI] [PubMed] [Google Scholar]
- Cyr D. M., Hohfeld J., Patterson C. (2002). Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem. Sci. 27, 368–375. 10.1016/S0968-0004(02)02125-4 [DOI] [PubMed] [Google Scholar]
- D'Andrea L. D., Regan L. (2003). TPR proteins: the versatile helix. Trends Biochem. Sci. 28, 655–662. 10.1016/j.tibs.2003.10.007 [DOI] [PubMed] [Google Scholar]
- Dantuma N. P., Lindsten K. (2010). Stressing the ubiquitin-proteasome system. Cardiovasc. Res. 85, 263–271. 10.1093/cvr/cvp255 [DOI] [PubMed] [Google Scholar]
- Danzer K. M., Ruf W. P., Putcha P., Joyner D., Hashimoto T., Glabe C., et al. (2011). Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 25, 326–336. 10.1096/fj.10-164624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedmon M. M., Christodoulou J., Wilson M. R., Dobson C. M. (2005). Heat shock protein 70 inhibits α-synuclein fibril formation via preferential binding to prefibrillar species. J. Biol. Chem. 280, 14733–14740. 10.1074/jbc.M413024200 [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demand J., Alberti S., Patterson C., Hohfeld J. (2001). Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 11, 1569–1577. 10.1016/S0960-9822(01)00487-0 [DOI] [PubMed] [Google Scholar]
- DeSantis M. E., Leung E. H., Sweeny E. A., Jackrel M. E., Cushman-Nick M., Neuhaus-Follini A., et al. (2012). Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 151, 778–793. 10.1016/j.cell.2012.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dice J. F. (1990). Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 15, 305–309. [DOI] [PubMed] [Google Scholar]
- Didelot C., Lanneau D., Brunet M., Joly A. L., De Thonel A., Chiosis G., et al. (2007). Anti-cancer therapeutic approaches based on intracellular and extracellular heat shock proteins. Curr. Med. Chem. 14, 2839–2847. 10.2174/092986707782360079 [DOI] [PubMed] [Google Scholar]
- Dompierre J. P., Godin J. D., Charrin B. C., Cordelieres F. P., King S. J., Humbert S., et al. (2007). Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J. Neurosci. 27, 3571–3583. 10.1523/JNEUROSCI.0037-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P. M., Summers D. W., Cyr D. M. (2009). Molecular chaperones antagonize proteotoxicity by differentially modulating protein aggregation pathways. Prion 3, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle S. M., Genest O., Wickner S. (2013). Protein rescue from aggregates by powerful molecular chaperone machines. Nat. Rev. Mol. Cell Biol. 14, 617–629. 10.1038/nrm3660 [DOI] [PubMed] [Google Scholar]
- Doyle S. M., Wickner S. (2009). Hsp104 and ClpB: protein disaggregating machines. Trends Biochem. Sci. 34, 40–48. 10.1016/j.tibs.2008.09.010 [DOI] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D., Wahlster L., McLean P. J. (2011). Molecular chaperones in Parkinson's disease–present and future. J. Parkinsons. Dis. 1, 299–320. [PMC free article] [PubMed] [Google Scholar]
- Eisele F., Wolf D. H. (2008). Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase Ubr1. FEBS Lett. 582, 4143–4146. 10.1016/j.febslet.2008.11.015 [DOI] [PubMed] [Google Scholar]
- Eldridge A. G., O'Brien T. (2010). Therapeutic strategies within the ubiquitin proteasome system. Cell Death Differ. 17, 4–13. 10.1038/cdd.2009.82 [DOI] [PubMed] [Google Scholar]
- Elliott E., Laufer O., Ginzburg I. (2009). BAG-1M is up-regulated in hippocampus of Alzheimer's disease patients and associates with tau and APP proteins. J. Neurochem. 109, 1168–1178. 10.1111/j.1471-4159.2009.06047.x [DOI] [PubMed] [Google Scholar]
- Elliott E., Tsvetkov P., Ginzburg I. (2007). BAG-1 associates with Hsc70.Tau complex and regulates the proteasomal degradation of Tau protein. J. Biol. Chem. 282, 37276–37284. 10.1074/jbc.M706379200 [DOI] [PubMed] [Google Scholar]
- Ellis R. J. (2006). Molecular chaperones: assisting assembly in addition to folding. Trends Biochem. Sci. 31, 395–401. 10.1016/j.tibs.2006.05.001 [DOI] [PubMed] [Google Scholar]
- Ellis R. J. (2007). Protein misassembly: macromolecular crowding and molecular chaperones. Adv. Exp. Med. Biol. 594, 1–13. 10.1007/978-0-387-39975-1_1 [DOI] [PubMed] [Google Scholar]
- Ellis R. J., Minton A. P. (2006). Protein aggregation in crowded environments. Biol. Chem. 387, 485–497. 10.1515/BC.2006.064 [DOI] [PubMed] [Google Scholar]
- Eskelinen E. L., Saftig P. (2009). Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 1793, 664–673. 10.1016/j.bbamcr.2008.07.014 [DOI] [PubMed] [Google Scholar]
- Evans C. G., Wisen S., Gestwicki J. E. (2006). Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1-42) aggregation in vitro. J. Biol. Chem. 281, 33182–33191. 10.1074/jbc.M606192200 [DOI] [PubMed] [Google Scholar]
- Fang N. N., Ng A. H., Measday V., Mayor T. (2011). Hul5 HECT ubiquitin ligase plays a major role in the ubiquitylation and turnover of cytosolic misfolded proteins. Nat. Cell Biol. 13, 1344–1352. 10.1038/ncb2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farg M. A., Soo K. Y., Walker A. K., Pham H., Orian J., Horne M. K., et al. (2012). Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 33, 2855–2868. 10.1016/j.neurobiolaging.2012.02.009 [DOI] [PubMed] [Google Scholar]
- Fearns C., Pan Q., Mathison J. C., Chuang T. H. (2006). Triad3A regulates ubiquitination and proteasomal degradation of RIP1 following disruption of Hsp90 binding. J. Biol. Chem. 281, 34592–34600. 10.1074/jbc.M604019200 [DOI] [PubMed] [Google Scholar]
- Finka A., Goloubinoff P. (2013). Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones 18, 591–605. 10.1007/s12192-013-0413-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes G., Martin De Llano J. J., Villarroya A., Rivett A. J., Knecht E. (2003). Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem. J. 375(Pt 1), 75–86. 10.1042/BJ20030282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikake N., Nagai Y., Popiel H. A., Okamoto Y., Yamaguchi M., Toda T. (2008). Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 283, 26188–26197. 10.1074/jbc.M710521200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Carroni M., Nussbaum-Krammer C., Mogk A., Nillegoda N. B., Szlachcic A., et al. (2015). Human Hsp70 Disaggregase Reverses Parkinson's-Linked α-Synuclein Amyloid Fibrils. Mol. Cell 59, 781–793. 10.1016/j.molcel.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R. G., Nelson Z. W., Gottschling D. E. (2005). Degradation-mediated protein quality control in the nucleus. Cell 120, 803–815. 10.1016/j.cell.2005.01.016 [DOI] [PubMed] [Google Scholar]
- Garnier C., Lafitte D., Tsvetkov P. O., Barbier P., Leclerc-Devin J., Millot J. M., et al. (2002). Binding of ATP to heat shock protein 90: evidence for an ATP-binding site in the C-terminal domain. J. Biol. Chem. 277, 12208–12214. 10.1074/jbc.M111874200 [DOI] [PubMed] [Google Scholar]
- Garrido C., Brunet M., Didelot C., Zermati Y., Schmitt E., Kroemer G. (2006). Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle 5, 2592–2601. 10.4161/cc.5.22.3448 [DOI] [PubMed] [Google Scholar]
- Garrido C., Gurbuxani S., Ravagnan L., Kroemer G. (2001). Heat shock proteins: endogenous modulators of apoptotic cell death. Biochem. Biophys. Res. Commun. 286, 433–442. 10.1006/bbrc.2001.5427 [DOI] [PubMed] [Google Scholar]
- Glover J. R., Lindquist S. (1998). Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82. [DOI] [PubMed] [Google Scholar]
- Goedert M. (2001). α-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2, 492–501. 10.1038/35081564 [DOI] [PubMed] [Google Scholar]
- Gorbatyuk M. S., Knox T., LaVail M. M., Gorbatyuk O. S., Noorwez S. M., Hauswirth W. W., et al. (2010). Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. U.S.A. 107, 5961–5966. 10.1073/pnas.0911991107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk M. S., Shabashvili A., Chen W., Meyers C., Sullivan L. F., Salganik M., et al. (2012). Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. 20, 1327–1337. 10.1038/mt.2012.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregori L., Fuchs C., Figueiredo-Pereira M. E., Van Nostrand W. E., Goldgaber D. (1995). Amyloid β-protein inhibits ubiquitin-dependent protein degradation in vitro. J. Biol. Chem. 270, 19702–19708. [DOI] [PubMed] [Google Scholar]
- Grigoryev S., Stewart A. E., Kwon Y. T., Arfin S. M., Bradshaw R. A., Jenkins N. A., et al. (1996). A mouse amidase specific for N-terminal asparagine. The gene, the enzyme, and their function in the N-end rule pathway. J. Biol. Chem. 271, 28521–28532. [DOI] [PubMed] [Google Scholar]
- Gunawardena S., Her L. S., Brusch R. G., Laymon R. A., Niesman I. R., Gordesky-Gold B., et al. (2003). Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40, 25–40. 10.1016/S0896-6273(03)00594-4 [DOI] [PubMed] [Google Scholar]
- Gusella J. F., MacDonald M. E. (1998). Huntingtin: a single bait hooks many species. Curr. Opin. Neurobiol. 8, 425–430. [DOI] [PubMed] [Google Scholar]
- Hadian K., Griesbach R. A., Dornauer S., Wanger T. M., Nagel D., Metlitzky M., et al. (2011). NF-γB essential modulator (NEMO) interaction with linear and lys-63 ubiquitin chains contributes to NF-κB activation. J. Biol. Chem. 286, 26107–26117. 10.1074/jbc.M111.233163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman J., Rujano M. A., van Waarde M. A., Kakkar V., Dirks R. P., Govorukhina N., et al. (2010). A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 37, 355–369. 10.1016/j.molcel.2010.01.001 [DOI] [PubMed] [Google Scholar]
- Hao R., Nanduri P., Rao Y., Panichelli R. S., Ito A., Yoshida M., et al. (2013). Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 51, 819–828. 10.1016/j.molcel.2013.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl F. U., Bracher A., Hayer-Hartl M. (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- Hasegawa M., Fujiwara H., Nonaka T., Wakabayashi K., Takahashi H., Lee V. M., et al. (2002). Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076. 10.1074/jbc.M208046200 [DOI] [PubMed] [Google Scholar]
- Hasegawa M. T., Nonaka M., Masuda-Suzukake M. (2016). α-Synuclein: experimental pathology. Cold Spring Harb. Perspect. Med. 6. 10.1101/cshperspect.a024273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck J. W., Cheung S. K., Hampton R. Y. (2010). Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. U.S.A. 107, 1106–1111. 10.1073/pnas.0910591107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde A. N. (2010). The ubiquitin-proteasome pathway and synaptic plasticity. Learn. Mem. 17, 314–327. 10.1101/lm.1504010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde A. N., Upadhya S. C. (2011). Role of ubiquitin-proteasome-mediated proteolysis in nervous system disease. Biochim. Biophys. Acta 1809, 128–140. 10.1016/j.bbagrm.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A., Ciechanover A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. 10.1146/annurev.biochem.67.1.425 [DOI] [PubMed] [Google Scholar]
- Hetz C., Mollereau B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. 10.1038/nrn3689 [DOI] [PubMed] [Google Scholar]
- Hodson S., Marshall J. J., Burston S. G. (2012). Mapping the road to recovery: the ClpB/Hsp104 molecular chaperone. J. Struct. Biol. 179, 161–171. 10.1016/j.jsb.2012.05.015 [DOI] [PubMed] [Google Scholar]
- Hoffmann J. H., Linke K., Graf P. C., Lilie H., Jakob U. (2004). Identification of a redox-regulated chaperone network. EMBO J. 23, 160–168. 10.1038/sj.emboj.7600016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo K., Black S. E., Verhoeff N. P. (2012). Alzheimer's disease, cerebrovascular disease, and the β-amyloid cascade. Can. J. Neurol. Sci. 39, 712–728. 10.1017/S0317167100015547 [DOI] [PubMed] [Google Scholar]
- Honjo Y., Kaneko S., Ito H., Horibe T., Nagashima M., Nakamura M., et al. (2011). Protein disulfide isomerase-immunopositive inclusions in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 12, 444–450. 10.3109/17482968.2011.594055 [DOI] [PubMed] [Google Scholar]
- Hoogstra-Berends F., Meijering R. A., Zhang D., Heeres A., Loen L., Seerden J. P., et al. (2012). Heat shock protein-inducing compounds as therapeutics to restore proteostasis in atrial fibrillation. Trends Cardiovasc. Med. 22, 62–68. 10.1016/j.tcm.2012.06.013 [DOI] [PubMed] [Google Scholar]
- Hope A. D., de Silva R., Fischer D. F., Hol E. M., van Leeuwen F. W., Lees A. J. (2003). Alzheimer's associated variant ubiquitin causes inhibition of the 26S proteasome and chaperone expression. J. Neurochem. 86, 394–404. 10.1046/j.1471-4159.2003.01844.x [DOI] [PubMed] [Google Scholar]
- Hwang J. S., Hwang J. S., Chang I., Kim S. (2007). Age-associated decrease in proteasome content and activities in human dermal fibroblasts: restoration of normal level of proteasome subunits reduces aging markers in fibroblasts from elderly persons. J. Gerontol. A Biol. Sci. Med. Sci. 62, 490–499. [DOI] [PubMed] [Google Scholar]
- Irizarry M. C., Growdon W., Gomez-Isla T., Newell K., George J. M., Clayton D. F., et al. (1998). Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson's disease and cortical Lewy body disease contain α-synuclein immunoreactivity. J. Neuropathol. Exp. Neurol. 57, 334–337. [DOI] [PubMed] [Google Scholar]
- Itoh H., Komatsuda A., Ohtani H., Wakui H., Imai H., Sawada K., et al. (2002). Mammalian HSP60 is quickly sorted into the mitochondria under conditions of dehydration. Eur. J. Biochem. 269, 5931–5938. 10.1046/j.1432-1033.2002.03317.x [DOI] [PubMed] [Google Scholar]
- Jackrel M. E., DeSantis M. E., Martinez B. A., Castellano L. M., Stewart R. M., Caldwell K. A., et al. (2014). Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 156, 170–182. 10.1016/j.cell.2013.11.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackrel M. E., Shorter J. (2014). Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis. Model. Mech. 7, 1175–1184. 10.1242/dmm.016113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana N. R., Tanaka M., Wang G., Nukina N. (2000). Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 9, 2009–2018. 10.1093/hmg/9.13.2009 [DOI] [PubMed] [Google Scholar]
- Jeon G. S., Nakamura T., Lee J. S., Choi W. J., Ahn S. W., Lee K. W., et al. (2014). Potential effect of S-nitrosylated protein disulfide isomerase on mutant SOD1 aggregation and neuronal cell death in amyotrophic lateral sclerosis. Mol. Neurobiol. 49, 796–807. 10.1007/s12035-013-8562-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Lv H., Liao M., Xu X., Huang S., Tan H., et al. (2012). GRP78 counteracts cell death and protein aggregation caused by mutant huntingtin proteins. Neurosci. Lett. 516, 182–187. 10.1016/j.neulet.2012.03.074 [DOI] [PubMed] [Google Scholar]
- Kalmar B., Lu C. H., Greensmith L. (2014). The role of heat shock proteins in Amyotrophic Lateral Sclerosis: the therapeutic potential of Arimoclomol. Pharmacol. Ther. 141, 40–54. 10.1016/j.pharmthera.2013.08.003 [DOI] [PubMed] [Google Scholar]
- Kampinga H. H., Craig E. A. (2010). The HSP70 chaperone machinery: j proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592. 10.1038/nrm2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastle M., Grune T. (2012). Interactions of the proteasomal system with chaperones: protein triage and protein quality control. Prog. Mol. Biol. Transl. Sci. 109, 113–160. 10.1016/B978-0-12-397863-9.00004-3 [DOI] [PubMed] [Google Scholar]
- Kaushik S., Cuervo A. M. (2012). Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 22, 407–417. 10.1016/j.tcb.2012.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemi-Esfarjani P., Benzer S. (2000). Genetic suppression of polyglutamine toxicity in Drosophila. Science 287, 1837–1840. 10.1126/science.287.5459.1837 [DOI] [PubMed] [Google Scholar]
- Keller J. N., Huang F. F., Markesbery W. R. (2000). Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 98, 149–156. 10.1016/S0306-4522(00)00067-1 [DOI] [PubMed] [Google Scholar]
- Kettern N., Dreiseidler M., Tawo R., Hohfeld J. (2010). Chaperone-assisted degradation: multiple paths to destruction. Biol. Chem. 391, 481–489. 10.1515/BC.2010.058 [DOI] [PubMed] [Google Scholar]
- Khan S., Rammeloo A. W., Heikkila J. J. (2012). Withaferin A induces proteasome inhibition, endoplasmic reticulum stress, the heat shock response and acquisition of thermotolerance. PLoS ONE 7:e50547. 10.1371/journal.pone.0050547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D., Kalmar B., Dick J. R., Riddoch-Contreras J., Burnstock G., Greensmith L. (2004). Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat. Med. 10, 402–405. 10.1038/nm1021 [DOI] [PubMed] [Google Scholar]
- Kim Y. E., Hipp M. S., Bracher A., Hayer-Hartl M., Hartl F. U. (2013). Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355. 10.1146/annurev-biochem-060208-092442 [DOI] [PubMed] [Google Scholar]
- Kitamura A., Kubota H., Pack C. G., Matsumoto G., Hirayama S., Takahashi Y., et al. (2006). Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol. 8, 1163–1170. 10.1038/ncb1478 [DOI] [PubMed] [Google Scholar]
- Kloetzel P. M., Ossendorp F. (2004). Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. Curr. Opin. Immunol. 16, 76–81. 10.1016/j.coi.2003.11.004 [DOI] [PubMed] [Google Scholar]
- Klucken J., Shin Y., Masliah E., Hyman B. T., McLean P. J. (2004). Hsp70 Reduces α-Synuclein Aggregation and Toxicity. J. Biol. Chem. 279, 25497–25502. 10.1074/jbc.M400255200 [DOI] [PubMed] [Google Scholar]
- Kristiansen M., Deriziotis P., Dimcheff D. E., Jackson G. S., Ovaa H., Naumann H., et al. (2007). Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol. Cell 26, 175–188. 10.1016/j.molcel.2007.04.001 [DOI] [PubMed] [Google Scholar]
- Kudva Y. C., Hiddinga H. J., Butler P. C., Mueske C. S., Eberhardt N. L. (1997). Small heat shock proteins inhibit in vitro A β(1-42) amyloidogenesis. FEBS Lett. 416, 117–121. [DOI] [PubMed] [Google Scholar]
- Kuzuhara S., Mori H., Izumiyama N., Yoshimura M., Ihara Y. (1988). Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol. 75, 345–353. [DOI] [PubMed] [Google Scholar]
- Kwon Y. T., Balogh S. A., Davydov I. V., Kashina A. S., Yoon J. K., Xie Y., et al. (2000). Altered activity, social behavior, and spatial memory in mice lacking the NTAN1p amidase and the asparagine branch of the N-end rule pathway. Mol. Cell. Biol. 20, 4135–4148. 10.1128/MCB.20.11.4135-4148.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y. T., Kashina A. S., Davydov I. V., Hu R. G., An J. Y., Seo J. W., et al. (2002). An essential role of N-terminal arginylation in cardiovascular development. Science 297, 96–99. 10.1126/science.1069531 [DOI] [PubMed] [Google Scholar]
- Kwon Y. T., Kashina A. S., Varshavsky A. (1999a). Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol. Cell. Biol. 19, 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y. T., Levy F., Varshavsky A. (1999b). Bivalent inhibitor of the N-end rule pathway. J. Biol. Chem. 274, 18135–18139. [DOI] [PubMed] [Google Scholar]
- Labbadia J., Novoselov S. S., Bett J. S., Weiss A., Paganetti P., Bates G. P., et al. (2012). Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain 135(Pt 4), 1180–1196. 10.1093/brain/aws022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T., Kirkin V., Dikic I., Johansen T. (2009). NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8, 1986–1990. 10.4161/cc.8.13.8892 [DOI] [PubMed] [Google Scholar]
- Lanneau D., Wettstein G., Bonniaud P., Garrido C. (2010). Heat shock proteins: cell protection through protein triage. ScientificWorldJournal 10, 1543–1552. 10.1100/tsw.2010.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. H., Lee M. J., Park S., Oh D. C., Elsasser S., Chen P. C., et al. (2010). Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184. 10.1038/nature09299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Kim J. H., Biter A. B., Sielaff B., Lee S., Tsai F. T. (2013). Heat shock protein (Hsp) 70 is an activator of the Hsp104 motor. Proc. Natl. Acad. Sci. U.S.A. 110, 8513–8518. 10.1073/pnas.1217988110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. J., Pal K., Tasaki T., Roy S., Jiang Y., An J. Y., et al. (2008). Synthetic heterovalent inhibitors targeting recognition E3 components of the N-end rule pathway. Proc. Natl. Acad. Sci. U.S.A. 105, 100–105. 10.1073/pnas.0708465105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. J., Tasaki T., Moroi K., An J. Y., Kimura S., Davydov I. V., et al. (2005). RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. USA. 102, 15030–15035. 10.1073/pnas.0507533102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. J., Lim H. S., Masliah E., Lee H. J. (2011). Protein aggregate spreading in neurodegenerative diseases: problems and perspectives. Neurosci. Res. 70, 339–348. 10.1016/j.neures.2011.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindersson E., Beedholm R., Hojrup P., Moos T., Gai W., Hendil K. B., et al. (2004). Proteasomal inhibition by α-synuclein filaments and oligomers. J. Biol. Chem. 279, 12924–12934. 10.1074/jbc.M306390200 [DOI] [PubMed] [Google Scholar]
- Lindquist S. (2009). Protein folding sculpting evolutionary change. Cold Spring Harb. Symp. Quant. Biol. 74, 103–108. 10.1101/sqb.2009.74.043 [DOI] [PubMed] [Google Scholar]
- Lindquist S., Kim G. (1996). Heat-shock protein 104 expression is sufficient for thermotolerance in yeast. Proc. Natl. Acad. Sci. U.S.A. 93, 5301–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinska N., Zietkiewicz S., Sobczak A., Jurczyk A., Potocki W., Morawiec E., et al. (2013). Disruption of ionic interactions between the nucleotide binding domain 1 (NBD1) and middle (M) domain in Hsp100 disaggregase unleashes toxic hyperactivity and partial independence from Hsp70. J. Biol. Chem. 288, 2857–2869. 10.1074/jbc.M112.387589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Shinobu L. A., Ward C. M., Young D., Cleveland D. W. (2005). Elevation of the Hsp70 chaperone does not effect toxicity in mouse models of familial amyotrophic lateral sclerosis. J. Neurochem. 93, 875–882. 10.1111/j.1471-4159.2005.03054.x [DOI] [PubMed] [Google Scholar]
- Lo Bianco C., Shorter J., Regulier E., Lashuel H., Iwatsubo T., Lindquist S., et al. (2008). Hsp104 antagonizes α-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J. Clin. Invest. 118, 3087–3097. 10.1172/JCI35781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez T., Dalton K., Frydman J. (2015). The mechanism and function of Group II Chaperonins. J. Mol. Biol. 427, 2919–2930. 10.1016/j.jmb.2015.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low P. (2011). The role of ubiquitin-proteasome system in ageing. Gen. Comp. Endocrinol. 172, 39–43. 10.1016/j.ygcen.2011.02.005 [DOI] [PubMed] [Google Scholar]
- Luk K. C., Mills I. P., Trojanowski J. Q., Lee V. M. (2008). Interactions between Hsp70 and the hydrophobic core of α-synuclein inhibit fibril assembly. Biochemistry 47, 12614–12625. 10.1021/bi801475r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W., Dou F., Rodina A., Chip S., Kim J., Zhao Q., et al. (2007). Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc. Natl. Acad. Sci. U.S.A. 104, 9511–9516. 10.1073/pnas.0701055104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansson C., Arosio P., Hussein R., Kampinga H. H., Hashem R. M., Boelens W. C., et al. (2014). Interaction of the molecular chaperone DNAJB6 with growing amyloid-β 42 (Aβ42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J. Biol. Chem. 289, 31066–31076. 10.1074/jbc.M114.595124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey A. C., Kaushik S., Sovak G., Kiffin R., Cuervo A. M. (2006). Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. U.S.A. 103, 5805–5810. 10.1073/pnas.0507436103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M. L., Wickliffe K. E., Dong K. C., Yu C., Bosanac I., Bustos D., et al. (2010). K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol. Cell 39, 477–484. 10.1016/j.molcel.2010.07.001 [DOI] [PubMed] [Google Scholar]
- McDonough H., Patterson C. (2003). CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones 8, 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean P. J., Kawamata H., Shariff S., Hewett J., Sharma N., Ueda K., et al. (2002). TorsinA and heat shock proteins act as molecular chaperones: suppression of α-synuclein aggregation. J. Neurochem. 83, 846–854. 10.1046/j.1471-4159.2002.01190.x [DOI] [PubMed] [Google Scholar]
- McLean P. J., Klucken J., Shin Y., Hyman B. T. (2004). Geldanamycin induces Hsp70 and prevents α-synuclein aggregation and toxicity in vitro. Biochem. Biophys. Res. Commun. 321, 665–669. 10.1016/j.bbrc.2004.07.021 [DOI] [PubMed] [Google Scholar]
- Meisenberg C., Tait P. S., Dianova I. I., Wright K., Edelmann M. J., Ternette N., et al. (2012). Ubiquitin ligase UBR3 regulates cellular levels of the essential DNA repair protein APE1 and is required for genome stability. Nucleic Acids Res. 40, 701–711. 10.1093/nar/gkr744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui K., Nakayama H., Akagi T., Nekooki M., Ohtawa K., Takio K., et al. (2002). Purification of polyglutamine aggregates and identification of elongation factor-1α and heat shock protein 84 as aggregate-interacting proteins. J. Neurosci. 22, 9267–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A., Tomoyasu T., Goloubinoff P., Rudiger S., Roder D., Langen H., et al. (1999). Identification of thermolabile Escherichia coli proteins: prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 18, 6934–6949. 10.1093/emboj/18.24.6934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawe T., Hiebel C., Kern A., Behl C. (2012). Protein homeostasis, aging and Alzheimer's disease. Mol. Neurobiol. 46, 41–54. 10.1007/s12035-012-8246-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Gonzalez I., Soto C. (2011). Misfolded protein aggregates: mechanisms, structures and potential for disease transmission. Semin. Cell Dev. Biol. 22, 482–487. 10.1016/j.semcdb.2011.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D. D., Ho S., Glover J. R. (2004). Saccharomyces cerevisiae Hsp104 enhances the chaperone capacity of human cells and inhibits heat stress-induced proapoptotic signaling. Biochemistry 43, 8107–8115. 10.1021/bi0493766 [DOI] [PubMed] [Google Scholar]
- Nillegoda N. B., Kirstein J., Szlachcic A., Berynskyy M., Stank A., Stengel F., et al. (2015). Crucial HSP70 co–chaperone complex unlocks metazoan protein disaggregation. Nature 524, 247–251. 10.1038/nature14884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef D. W., Turski M. L., Thiele D. J. (2010). Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 8:e1000291. 10.1371/journal.pbio.1000291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S. V., Poulsen E. G., Rebula C. A., Hartmann-Petersen R. (2014). Protein quality control in the nucleus. Biomolecules 4, 646–661. 10.3390/biom4030646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikura Y., Ohta S., Vandenbeldt K. J., Abdulle R., McEwen B. F., Kitagawa K. (2006). 17-AAG, an Hsp90 inhibitor, causes kinetochore defects: a novel mechanism by which 17-AAG inhibits cell proliferation. Oncogene 25, 4133–4146. 10.1038/sj.onc.1209461 [DOI] [PubMed] [Google Scholar]
- Nillegoda N. B., Theodoraki M. A., Mandal A. K., Mayo K. J., Ren H. Y., Sultana R., et al. (2010). Ubr1 and Ubr2 function in a quality control pathway for degradation of unfolded cytosolic proteins. Mol. Biol. Cell 21, 2102–2116. 10.1091/mbc.E10-02-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nollen E. A., Garcia S. M., van Haaften G., Kim S., Chavez A., Morimoto R. I., et al. (2004). Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U.S.A. 101, 6403–6408. 10.1073/pnas.0307697101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoselov S. S., Mustill W. J., Gray A. L., Dick J. R., Kanuga N., Kalmar B., et al. (2013). Molecular chaperone mediated late-stage neuroprotection in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. PLoS ONE 8:e73944. 10.1371/journal.pone.0073944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odunuga O. O., Hornby J. A., Bies C., Zimmermann R., Pugh D. J., Blatch G. L. (2003). Tetratricopeptide repeat motif-mediated Hsc70-mSTI1 interaction. Molecular characterization of the critical contacts for successful binding and specificity. J. Biol. Chem. 278, 6896–6904. 10.1074/jbc.M206867200 [DOI] [PubMed] [Google Scholar]
- Opazo J. C., Hoffmann F. G., Storz J. F. (2008). Genomic evidence for independent origins of β-like globin genes in monotremes and therian mammals. Proc. Natl. Acad. Sci. U.S.A. 105, 1590–1595. 10.1073/pnas.0710531105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro T. F., Putcha P., Tetzlaff J. E., Spoelgen R., Koker M., Carvalho F., et al. (2008). Formation of toxic oligomeric α-synuclein species in living cells. PLoS ONE 3:e1867. 10.1371/journal.pone.0001867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris D., Ganey N. J., Laporte V., Patel N. S., Beaulieu-Abdelahad D., Bachmeier C., et al. (2010). Reduction of β-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer's disease. J. Neuroinflammation 7:17. 10.1186/1742-2094-7-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell D. A., Kowal A. S., Singer M. A., Lindquist S. (1994). Protein disaggregation mediated by heat-shock protein Hsp104. Nature 372, 475–478. 10.1038/372475a0 [DOI] [PubMed] [Google Scholar]
- Patel Y. J., Payne Smith M. D., de Belleroche J., Latchman D. S. (2005). Hsp27 and Hsp70 administered in combination have a potent protective effect against FALS-associated SOD1-mutant-induced cell death in mammalian neuronal cells. Brain Res. Mol. Brain Res. 134, 256–274. 10.1016/j.molbrainres.2004.10.028 [DOI] [PubMed] [Google Scholar]
- Pauwels K., Van Molle I., Tommassen J., Van Gelder P. (2007). Chaperoning Anfinsen: the steric foldases. Mol. Microbiol. 64, 917–922. 10.1111/j.1365-2958.2007.05718.x [DOI] [PubMed] [Google Scholar]
- Pearl L. H., Prodromou C. (2006). Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 75, 271–294. 10.1146/annurev.biochem.75.103004.142738 [DOI] [PubMed] [Google Scholar]
- Peng J., Schwartz D., Elias J. E., Thoreen C. C., Cheng D., Marsischky G., et al. (2003). A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21, 921–926. 10.1038/nbt849 [DOI] [PubMed] [Google Scholar]
- Perrin V., Regulier E., Abbas-Terki T., Hassig R., Brouillet E., Aebischer P., et al. (2007). Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington's disease. Mol. Ther. 15, 903–911. 10.1038/mt.sj.6300141 [DOI] [PubMed] [Google Scholar]
- Picard D. (2002). Heat-shock protein 90, a chaperone for folding and regulation. Cell. Mol. Life Sci. 59, 1640–1648. 10.1007/PL00012491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. 10.1146/annurev.biochem.70.1.503 [DOI] [PubMed] [Google Scholar]
- Polier S., Dragovic Z., Hartl F. U., Bracher A. (2008). Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell 133, 1068–1079. 10.1016/j.cell.2008.05.022 [DOI] [PubMed] [Google Scholar]
- Prasad R., Kawaguchi S., Ng D. T. (2010). A nucleus-based quality control mechanism for cytosolic proteins. Mol. Biol. Cell 21, 2117–2127. 10.1091/mbc.E10-02-0111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha P., Danzer K. M., Kranich L. R., Scott A., Silinski M., Mabbett S., et al. (2010). Brain-permeable small-molecule inhibitors of Hsp90 prevent α-synuclein oligomer formation and rescue α-synuclein-induced toxicity. J. Pharmacol. Exp. Ther. 332, 849–857. 10.1124/jpet.109.158436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian S. B., McDonough H., Boellmann F., Cyr D. M., Patterson C. (2006). CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature 440, 551–555. 10.1038/nature04600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan H., Fan G., Wang C. C. (1995). Independence of the chaperone activity of protein disulfide isomerase from its thioredoxin-like active site. J. Biol. Chem. 270, 17078–17080. [DOI] [PubMed] [Google Scholar]
- Quraishe S., Asuni A., Boelens W. C., O'Connor V., Wyttenbach A. (2008). Expression of the small heat shock protein family in the mouse CNS: differential anatomical and biochemical compartmentalization. Neuroscience 153, 483–491. 10.1016/j.neuroscience.2008.01.058 [DOI] [PubMed] [Google Scholar]
- Ramaswamy S., Shannon K. M., Kordower J. H. (2007). Huntington's disease: pathological mechanisms and therapeutic strategies. Cell Transplant. 16, 301–312. 10.3727/000000007783464687 [DOI] [PubMed] [Google Scholar]
- Rampelt H., Kirstein-Miles J., Nillegoda N. B., Chi K., Scholz S. R., Morimoto R. I., et al. (2012). Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 31, 4221–4235. 10.1038/emboj.2012.264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranford J. C., Coates A. R., Henderson B. (2000). Chaperonins are cell-signalling proteins: the unfolding biology of molecular chaperones. Expert Rev. Mol. Med. 2, 1–17. 10.1017/S1462399400002015 [DOI] [PubMed] [Google Scholar]
- Ratzke C., Mickler M., Hellenkamp B., Buchner J., Hugel T. (2010). Dynamics of heat shock protein 90 C-terminal dimerization is an important part of its conformational cycle. Proc. Natl. Acad. Sci. U.S.A. 107, 16101–16106. 10.1073/pnas.1000916107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B., Sarkar S., Rubinsztein D. C. (2008). Clearance of mutant aggregate-prone proteins by autophagy. Methods Mol. Biol. 445, 195–211. 10.1007/978-1-59745-157-4_13 [DOI] [PubMed] [Google Scholar]
- Regeur L., Jensen G. B., Pakkenberg H., Evans S. M., Pakkenberg B. (1994). No global neocortical nerve cell loss in brains from patients with senile dementia of Alzheimer's type. Neurobiol. Aging 15, 347–352. [DOI] [PubMed] [Google Scholar]
- Rekas A., Adda C. G., Andrew Aquilina J., Barnham K. J., Sunde M., Galatis D., et al. (2004). Interaction of the molecular chaperone αB-crystallin with α-synuclein: effects on amyloid fibril formation and chaperone activity. J. Mol. Biol. 340, 1167–1183. 10.1016/j.jmb.2004.05.054 [DOI] [PubMed] [Google Scholar]
- Rekas A., Jankova L., Thorn D. C., Cappai R., Carver J. A. (2007). Monitoring the prevention of amyloid fibril formation by α-crystallin. Temperature dependence and the nature of the aggregating species. FEBS J. 274, 6290–6304. 10.1111/j.1742-4658.2007.06144.x [DOI] [PubMed] [Google Scholar]
- Robberecht W., Philips T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. 10.1038/nrn3430 [DOI] [PubMed] [Google Scholar]
- Rodrigo-Brenni M. C., Gutierrez E., Hegde R. S. (2014). Cytosolic quality control of mislocalized proteins requires RNF126 recruitment to Bag6. Mol. Cell 55, 227–237. 10.1016/j.molcel.2014.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos R. A. (2010). Huntington's disease: a clinical review. Orphanet J. Rare Dis. 5:40. 10.1186/1750-1172-5-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. 10.1038/362059a0 [DOI] [PubMed] [Google Scholar]
- Rosenbaum J. C., Fredrickson E. K., Oeser M. L., Garrett-Engele C. M., Locke M. N., Richardson L. A., et al. (2011). Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol. Cell 41, 93–106. 10.1016/j.molcel.2010.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross C. A., Poirier M. A. (2004). Protein aggregation and neurodegenerative disease. Nat. Med. 10 (Suppl.), S10–S17. 10.1038/nm1066 [DOI] [PubMed] [Google Scholar]
- Rudiger S., Germeroth L., Schneider-Mergener J., Bukau B. (1997). Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 16, 1501–1507. 10.1093/emboj/16.7.1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarto C., Binz P. A., Mocarelli P. (2000). Heat shock proteins in human cancer. Electrophoresis 21, 1218–1226. [DOI] [PubMed] [Google Scholar]
- Schulte T. W., Neckers L. M. (1998). The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother. Pharmacol. 42, 273–279. 10.1007/s002800050817 [DOI] [PubMed] [Google Scholar]
- Seyffer F., Kummer E., Oguchi Y., Winkler J., Kumar M., Zahn R., et al. (2012). Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat. Struct. Mol. Biol. 19, 1347–1355. 10.1038/nsmb.2442 [DOI] [PubMed] [Google Scholar]
- Shammas S. L., Waudby C. A., Wang S., Buell A. K., Knowles T. P., Ecroyd H., et al. (2011). Binding of the molecular chaperone αB-crystallin to Aβ amyloid fibrils inhibits fibril elongation. Biophys. J. 101, 1681–1689. 10.1016/j.bpj.2011.07.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar G. M., Li S., Mehta T. H., Garcia-Munoz A., Shepardson N. E., Smith I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. 10.1038/nm1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K., Schmitt S., Bergner C. G., Tyanova S., Kannaiyan N., Manrique-Hoyos N., et al. (2015). Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 18, 1819–1831. 10.1038/nn.4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastry B. S. (2003). Neurodegenerative disorders of protein aggregation. Neurochem. Int. 43, 1–7. 10.1016/S0197-0186(02)00196-1 [DOI] [PubMed] [Google Scholar]
- Shen H. Y., He J. C., Wang Y., Huang Q. Y., Chen J. F. (2005). Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J. Biol. Chem. 280, 39962–39969. 10.1074/jbc.M505524200 [DOI] [PubMed] [Google Scholar]
- Shorter J. (2011). The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 6:e26319. 10.1371/journal.pone.0026319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J., Lindquist S. (2004). Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 304, 1793–1797. 10.1126/science.1098007 [DOI] [PubMed] [Google Scholar]
- Snyder H., Mensah K., Theisler C., Lee J., Matouschek A., Wolozin B. (2003). Aggregated and monomeric α-synuclein bind to the S6' proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 278, 11753–11759. 10.1074/jbc.M208641200 [DOI] [PubMed] [Google Scholar]
- Spillantini M. G., Crowther R. A., Jakes R., Hasegawa M., Goedert M. (1998). α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram S. M., Banerjee R., Kane R. S., Kwon Y. T. (2009). Multivalency-assisted control of intracellular signaling pathways: application for ubiquitin- dependent N-end rule pathway. Chem. Biol. 16, 121–131. 10.1016/j.chembiol.2009.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram S. M., Kim B. Y., Kwon Y. T. (2011). The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat. Rev. Mol. Cell Biol. 12, 735–747. 10.1038/nrm3217 [DOI] [PubMed] [Google Scholar]
- Sriram S. M., Kwon Y. T. (2010). The molecular principles of N-end rule recognition. Nat. Struct. Mol. Biol. 17, 1164–1165. 10.1038/nsmb1010-1164 [DOI] [PubMed] [Google Scholar]
- Stege G. J., Renkawek K., Overkamp P. S., Verschuure P., van Rijk A. F., Reijnen-Aalbers A., et al. (1999). The molecular chaperone αB-crystallin enhances amyloid β neurotoxicity. Biochem. Biophys. Res. Commun. 262, 152–156. 10.1006/bbrc.1999.1167 [DOI] [PubMed] [Google Scholar]
- Stolz A., Ernst A., Dikic I. (2014). Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 16, 495–501. 10.1038/ncb2979 [DOI] [PubMed] [Google Scholar]
- Summers D. W., Douglas P. M., Ramos C. H., Cyr D. M. (2009). Polypeptide transfer from Hsp40 to Hsp70 molecular chaperones. Trends Biochem. Sci. 34, 230–233. 10.1016/j.tibs.2008.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., MacRae T. H. (2005). The small heat shock proteins and their role in human disease. FEBS J. 272, 2613–2627. 10.1111/j.1742-4658.2005.04708.x [DOI] [PubMed] [Google Scholar]
- Tai H. C., Serrano-Pozo A., Hashimoto T., Frosch M. P., Spires-Jones T. L., Hyman B. T. (2012). The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 181, 1426–1435. 10.1016/j.ajpath.2012.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M., Jarosz D. F., Lindquist S. (2010). HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 11, 515–528. 10.1038/nrm2918 [DOI] [PubMed] [Google Scholar]
- Takayama S., Bimston D. N., Matsuzawa S., Freeman B. C., Aime-Sempe C., Xie Z., et al. (1997). BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J. 16, 4887–4896. 10.1093/emboj/16.16.4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T., Kwon Y. T. (2007). The mammalian N-end rule pathway: new insights into its components and physiological roles. Trends Biochem. Sci. 32, 520–528. 10.1016/j.tibs.2007.08.010 [DOI] [PubMed] [Google Scholar]
- Tasaki T., Mulder L. C., Iwamatsu A., Lee M. J., Davydov I. V., Varshavsky A., et al. (2005). A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol. Cell. Biol. 25, 7120–7136. 10.1128/MCB.25.16.7120-7136.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T., Zakrzewska A., Dudgeon D. D., Jiang Y., Lazo J. S., Kwon Y. T. (2009). The substrate recognition domains of the N-end rule pathway. J. Biol. Chem. 284, 1884–1895. 10.1074/jbc.M803641200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro E., Zako T., Muto H., Itoo Y., Sorgjerd K., Terada N., et al. (2013). Prefoldin protects neuronal cells from polyglutamine toxicity by preventing aggregation formation. J. Biol. Chem. 288, 19958–19972. 10.1074/jbc.M113.477984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J. P., Hardy J., Fischbeck K. H. (2002). Toxic proteins in neurodegenerative disease. Science 296, 1991–1995. 10.1126/science.1067122 [DOI] [PubMed] [Google Scholar]
- Torrente M. P., Shorter J. (2013). The metazoan protein disaggregase and amyloid depolymerase system: Hsp110, Hsp70, Hsp40, and small heat shock proteins. Prion 7, 457–463. 10.4161/pri.27531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda H., Han S. M., Yang Y., Tong C., Lin Y. Q., Mohan K., et al. (2008). The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell 133, 963–977. 10.1016/j.cell.2008.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutar L., Tutar Y. (2010). Heat shock proteins; an overview. Curr. Pharm. Biotechnol. 11, 216–222. [DOI] [PubMed] [Google Scholar]
- Tydlacka S., Wang C. E., Wang X., Li S., Li X. J. (2008). Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J. Neurosci. 28, 13285–13295. 10.1523/JNEUROSCI.4393-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya S. C., Hegde A. N. (2007). Role of the ubiquitin proteasome system in Alzheimer's disease. BMC Biochem. 8 (Suppl. 1):S12. 10.1186/1471-2091-8-S1-S12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacher C., Garcia-Oroz L., Rubinsztein D. C. (2005). Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington's disease. Hum. Mol. Genet. 14, 3425–3433. 10.1093/hmg/ddi372 [DOI] [PubMed] [Google Scholar]
- Valastyan J. S., Lindquist S. (2014). Mechanisms of protein-folding diseases at a glance. Dis. Model. Mech. 7, 9–14. 10.1242/dmm.013474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. (2011). The N-end rule pathway and regulation by proteolysis. Protein Sci. 20, 1298–1345. 10.1002/pro.666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar S. S., Brodsky J. L. (2008). One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957. 10.1038/nrm2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M. J., Zijlstra M. P., Kanon B., van Waarde-Verhagen M. A., Brunt E. R., Oosterveld-Hut H. M., et al. (2010). HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum. Mol. Genet. 19, 4677–4693. 10.1093/hmg/ddq398 [DOI] [PubMed] [Google Scholar]
- Wacker J. L., Huang S. Y., Steele A. D., Aron R., Lotz G. P., Nguyen Q., et al. (2009). Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington's disease. J. Neurosci. 29, 9104–9114. 10.1523/JNEUROSCI.2250-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker A. K., Farg M. A., Bye C. R., McLean C. A., Horne M. K., Atkin J. D. (2010). Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 133(Pt 1), 105–116. 10.1093/brain/awp267 [DOI] [PubMed] [Google Scholar]
- Walker A. K., Soo K. Y., Sundaramoorthy V., Parakh S., Ma Y., Farg M. A., et al. (2013). ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 8:e81170. 10.1371/journal.pone.0081170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Maldonado M. A. (2006). The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell. Mol. Immunol. 3, 255–261. [PubMed] [Google Scholar]
- Weibezahn J., Schlieker C., Tessarz P., Mogk A., Bukau B. (2005). Novel insights into the mechanism of chaperone-assisted protein disaggregation. Biol. Chem. 386, 739–744. 10.1515/BC.2005.086 [DOI] [PubMed] [Google Scholar]
- Whitesell L., Lindquist S. L. (2005). HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772. 10.1038/nrc1716 [DOI] [PubMed] [Google Scholar]
- Wing S. S., Chiang H. L., Goldberg A. L., Dice J. F. (1991). Proteins containing peptide sequences related to Lys-Phe-Glu-Arg-Gln are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem. J. 275 (Pt 1), 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J., Tyedmers J., Bukau B., Mogk A. (2012). Chaperone networks in protein disaggregation and prion propagation. J. Struct. Biol. 179, 152–160. 10.1016/j.jsb.2012.05.002 [DOI] [PubMed] [Google Scholar]
- Wirdefeldt K., Adami H. O., Cole P., Trichopoulos D., Mandel J. (2011). Epidemiology and etiology of Parkinson's disease: a review of the evidence. Eur. J. Epidemiol. 26 (Suppl. 1), S1–S58. 10.1007/s10654-011-9581-6 [DOI] [PubMed] [Google Scholar]
- Witt S. N. (2013). Molecular chaperones, α-synuclein, and neurodegeneration. Mol. Neurobiol. 47, 552–560. 10.1007/s12035-012-8325-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt A. R., Yerbury J. J., Dabbs R. A., Wilson M. R. (2012). Roles of extracellular chaperones in amyloidosis. J. Mol. Biol. 421, 499–516. 10.1016/j.jmb.2012.01.004 [DOI] [PubMed] [Google Scholar]
- Wyttenbach A. (2004). Role of heat shock proteins during polyglutamine neurodegeneration: mechanisms and hypothesis. J. Mol. Neurosci. 23, 69–96. 10.1385/JMN:23:1-2:069 [DOI] [PubMed] [Google Scholar]
- Xie Y. (2010). Structure, assembly and homeostatic regulation of the 26S proteasome. J. Mol. Cell Biol. 2, 308–317. 10.1093/jmcb/mjq030 [DOI] [PubMed] [Google Scholar]
- Yamagishi N., Goto K., Nakagawa S., Saito Y., Hatayama T. (2010). Hsp105 reduces the protein aggregation and cytotoxicity by expanded-polyglutamine proteins through the induction of Hsp70. Exp. Cell Res. 316, 2424–2433. 10.1016/j.yexcr.2010.06.003 [DOI] [PubMed] [Google Scholar]
- Yang Q., Guo Z. B. (2016). Polymorphisms in protein disulfide isomerase are associated with sporadic amyotrophic lateral sclerosis in the Chinese Han population. Int. J. Neurosci. 126, 607–611. 10.3109/00207454.2015.1050098 [DOI] [PubMed] [Google Scholar]
- Yerbury J. J., Gower D., Vanags L., Roberts K., Lee J. A., Ecroyd H. (2013). The small heat shock proteins αB-crystallin and Hsp27 suppress SOD1 aggregation in vitro. Cell Stress Chaperones 18, 251–257. 10.1007/s12192-012-0371-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Q., Sarge K. D. (2007). Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J. Mol. Med. 85, 1421–1428. 10.1007/s00109-007-0251-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Gu G., Goodlett D. R., Zhang T., Pan C., Montine T. J., et al. (2004). Analysis of α-synuclein-associated proteins by quantitative proteomics. J. Biol. Chem. 279, 39155–39164. 10.1074/jbc.M405456200 [DOI] [PubMed] [Google Scholar]
- Zietkiewicz S., Lewandowska A., Stocki P., Liberek K. (2006). Hsp70 chaperone machine remodels protein aggregates at the initial step of Hsp70-Hsp100-dependent disaggregation. J. Biol. Chem. 281, 7022–7029. 10.1074/jbc.M507893200 [DOI] [PubMed] [Google Scholar]
- Zininga T., Achilonu I., Hoppe H., Prinsloo E., Dirr H. W., Shonhai A. (2016). Plasmodium falciparum Hsp70-z, an Hsp110 homologue, exhibits independent chaperone activity and interacts with Hsp70-1 in a nucleotide-dependent fashion. Cell Stress Chaperones 21, 499–513. 10.1007/s12192-016-0678-4 [DOI] [PMC free article] [PubMed] [Google Scholar]