

Abstract
Background
Clustered regularly interspaced short palindromic repeats interference (CRISPRi) has provided an efficient approach for targeted gene inhibition. A non-model microorganism Halomonas species TD01 has been developed as a promising industrial producer of polyhydroxyalkanoates (PHA), a family of biodegradable polyesters accumulated by bacteria as a carbon and energy reserve compound. A controllable gene repression system, such as CRISPRi, is needed for Halomonas sp. TD01 to regulate its gene expression levels.
Results
For the first time CRISPRi was successfully used in Halomonas sp. TD01 to repress expression of ftsZ gene encoding bacterial fission ring formation protein, leading to an elongated cell morphology with typical filamentous shape similar to phenomenon observed with Escherichia coli. CRISPRi was employed to regulate expressions of prpC gene encoding 2-methylcitrate synthase for regulating 3-hydroxyvalerate monomer ratio in PHBV copolymers of 3-hydroxybutyrate (HB) and 3-hydroxyvalerate (HV). Percentages of HV in PHBV copolymers were controllable ranging from less than 1 to 13%. Furthermore, repressions on gltA gene encoding citrate synthase channeled more acetyl-CoA from the tricarboxylic acid (TCA) cycle to poly(3-hydroxybutyrate) (PHB) synthesis. The PHB accumulation by Halomonas sp. TD01 with its gltA gene repressed in various intensities via CRISPRi was increased by approximately 8% compared with the wild type control containing the CRISPRi vector without target.
Conclusions
It has now been confirmed that the CRISPRi system can be applied to Halomonas sp. TD01, a promising industrial strain for production of various PHA and chemicals under open and continuous fermentation process conditions. In details, the CRISPRi system was successfully designed in this study to target genes of ftsZ, prpC and gltA, achieving longer cell sizes, channeling more substrates to PHBV and PHB synthesis, respectively. CRISPRi can be expected to use for more metabolic engineering applications in non-model organisms.
Electronic supplementary material
The online version of this article (doi:10.1186/s12934-017-0655-3) contains supplementary material, which is available to authorized users.
Keywords: CRISPRi, PHBV, PHB, Synthetic biology, ftsZ, gltA, prpC
Background
The CRISPRi (clustered regularly interspaced short palindromic repeats interference) system provides an efficient method for targeted gene repression [1]. Deriving from the CRISPR/Cas9 system, the CRISPRi system contains a dCas9 protein co-expressed with a small guide RNA (sgRNA) [1–3]. The Cas9 protein is an RNA-guided DNA endonuclease. In the CRISPR system, the Cas9 protein binds to the sgRNA and form a protein-RNA complex, which will then bind to the targeted DNA sequence. The DNA will be cleaved by the catalytically active Cas9 protein [4]. Mutations in the Cas9 protein result in a catalytically dead dCas9 protein with DNA binding capability [1]. Therefore, the dCas9/sgRNA complex can bind to specific DNA target depending on the designed sequence of sgRNA, block transcriptional elongation, interfere RNA polymerase or transcriptional factor binding [1].
CRISPRi enables convenient and specific gene regulation in microbial metabolic engineering [1]. In our early study, CRISPRi was successfully used in Escherichia coli for regulating polyhydroxyalkanoates (PHA) production via simultaneously repressing multiple genes or multiple targets on one gene [5].
PHA are polyesters synthesized by a wide range of bacteria as carbon and energy source reserves [6]. PHA has been developed into various environmentally friendly plastic products, and it has been formed into an application value chain [7–9]. PHA can be classified into short-chain-length (scl) PHA and medium-chain-length (mcl) PHA [10]. Poly(3-hydroxybutyrate) (PHB) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) are common PHA already produced in large scale [9]. PHA production cost is still too high compared with petrochemical plastics that are not biodegradable [11].
Halomonas sp. TD01 is a halophile screened from Aydingol Lake in Xinjiang Province, China [12]. It can be grown under conditions of high salt concentrations and high pH, allowing a continuous and open fermentation without contamination. The genome of Halomonas sp. TD01 was sequenced and some genetic manipulation technologies have been developed for DNA manipulation [13, 14]. The absence of controllable repression system for gene expression has slowed down more applications for Halomonas sp. TD01.
FtsZ is a tubulin-like protein that is of great importance in the cell division process [15, 16]. FtsZ assembles to form Z rings in a dynamic state during the cell division process. FtsZ inhibition or deletion in E. coli leads to cell division repression and results in formation of filamentous cells from bar or spherical shapes [17, 18].
In Halomonas sp. TD01, propionic acid is transformed into propionyl-CoA, which can be further catalyzed by 2-methylcitrate synthase to form 2-methylcitrate and then enters the methyl citric acid cycle (MCC cycle) [19]. Propionyl-CoA can also enter the PHBV synthesis pathway to form 3-hydroxyvalerate monomers. 2-Methylcitrate synthase is encoded by prpC gene. It was expected that repressions on prpC should divert more propionyl-CoA to PHBV synthesis [20, 21].
The tricarboxylic acid (TCA) cycle provides energy and intermediates for synthesis of many important biological compounds [22]. In TCA cycle, acetyl-CoA and oxaloacetate are converted to citrate by citrate synthase encoded by gltA gene [22, 23]. Acetyl-CoA is the substrate for PHB synthesis, a high concentration of acetyl-CoA is of great importance to PHB production. Repressions of gltA gene should decrease acetyl-CoA consumption by the TCA cycle, therefore, improving substrate conversion to PHB synthesis.
In this study, it was aimed to exploit CRISPRi for enhanced PHA production by engineering Halomonas sp. TD01 to achieve regulation of its expression levels of various genes, including ftsZ, prpC and gltA.
Results
Feasibility study of the constructed CRISPRi for Halomonas sp. TD01
Gene ftsZ encoding bacterial fission ring protein was selected as a reporter gene for feasibility study of CRISPRi system for Halomonas sp. TD01. During the bacterial cell division process, FtsZ assembly leads to the formation of Z rings in the middle of a cell. FtsZ inhibitors interact with FtsZ in cytokinesis repressing the cell division progression, resulting in formation of filamentous cells [17, 18].
The sgRNAs were designed in the promoter region or near the ATG sequence in the targeted gene, and were right after a NGG sequence, namely, PAM sequence (protospacer adjacent motif sequence) [4]. All the sgRNAs could bind to the non-template DNA strand with sequence specificity. Thus, two sgRNAs were designed near the ATG sequence in ftsZ gene (Fig. 2b). CRISPRi inhibition systems pli-dCas9-ftsZ1 and pli-dCas9-ftsZ2 were constructed. The plasmids were then transferred via E. coli conjugation into Halomonas sp. TD01, forming the recombinants Halomonas sp. TD-ftsZ1 and TD-ftsZ2 strains. Halomonas sp. TD01 containing the non-target plasmid pli-dCa9-sgRNA, was named Halomonas sp. TD-sgRNA strain. Wild type Halomonas sp. TD01 and Halomonas sp. TD-sgRNA were used as control groups.
Fig. 2.
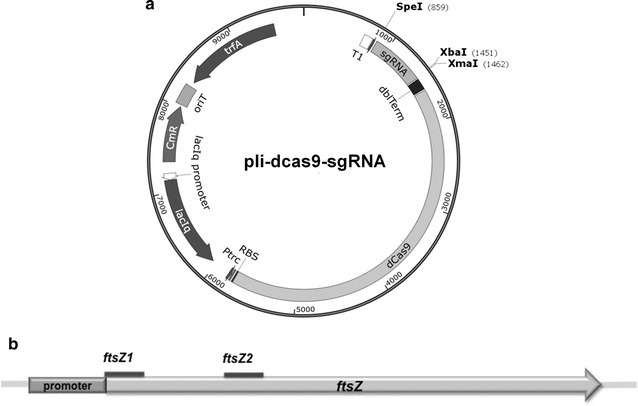
CRISPRi system used for Halomonas sp. TD01 (a) and relative binding positions of sgRNAs targeting ftsZ gene (b). Pli-dCas9-sgRNA, plasmid carrying the CRISPRi system; Ptrc, trc promoter; CmR, chloramphenicol resistance gene; oriT, origin of transfer; ftsZ, filamenting temperature-sensitive mutant Z. The length of ftsZ gene is 1179 bp, while the length of sgRNAs is around 20 bp. The promoter of ftsZ gene is 35 bp to 10 bp upstream of ftsZ gene. To inhibit ftsZ gene expression, ftsZ1 is designed 4 bp upstream from ATG sequence, from position −26 to −5, after the PAM sequence CGG (from position −29 to −27). FtsZ2 is designed 89 bp downstream of ATG sequence, from position 90 to 112, after the PAM sequence TGG (from position 87 to 89)
Growth curves of the strains were determined to observe whether cell growth was affected by inhibition of ftsZ gene (Additional file 1: Figure S1). Compared with the wild type Halomonas sp. TD01 and TD-sgRNA control groups, Halomonas sp. TD-ftsZ1 and TD-ftsZ2 exhibited long lag growth and lower cell density, implying that ftsZ gene inhibition decreased cell growth rate.
All the strains were cultured in the MM medium with 30 g/L glucose at 37 °C and 200 rpm. After 8 h cultivation, 1 mM IPTG was added for inducing the CRISPRi system. Cells were harvested after an overall cultivation of 48 h. Under an environmental scanning electron microscope (ESEM), Halomonas sp. TD-ftsZ1 and TD-ftsZ2 harboring the CRISPRi system showed elongated shapes compared with their controls Halomonas sp. TD01 and TD-sgRNA (Fig. 3a), demonstrating that the cell division process was effectively repressed via the CRISPRi. Lengths of bacteria in Halomonas sp. TD01 and TD-sgRNA control groups were approximately 1 μm, while lengths of bacteria in Halomonas sp. TD-ftsZ1 and TD-ftsZ2 groups varied from 20 to 70 μm, showing 20–70 folds increase in their cell lengths (Fig. 3b). The phenotype changes clearly indicated that the CRISPRi system had been successfully developed in Halomonas sp. TD01.
Fig. 3.
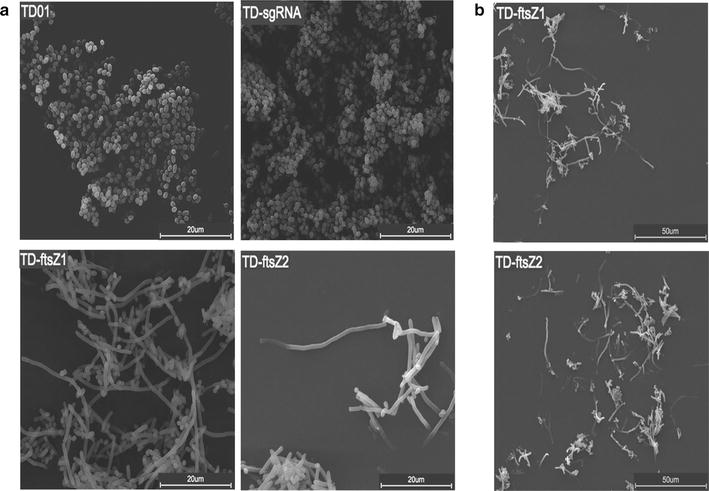
Scanning electron microscopy study on Halomonas sp. TD01 with its ftsZ gene repressed via CRISPRi under 5000 (a) and 2000 (b) times magnification. TD01: wild type Halomonas sp. TD used as a control group; TD-sgRNA, TD01 strain harboring the pli-dCas9-sgRNA plasmid without any DNA target site; TD-ftsZ1, TD-ftsZ2, TD01 strain harboring the CRISPRi plasmids pli-dCas9-ftsZ1 and pli-dCas9-ftsZ2 that regulated the expression level of fission ring protein ftsZ gene, respectively. Scale bars are 50 or 20 µm, as indicated
The uses of Halomonas CRISPRi system for controlling PHBV monomer ratios
Halomonas sp. TD01 is able to produce PHBV by adding the substrate propionic acid in the presence of glucose [14]. The PHBV synthesis involves the conversion of propionic acid to propionyl-CoA and subsequent transformation into 3-hydroxyvaleryl-CoA by β-ketothiolase (PhaA) and NADPH-dependent acetoacetyl-CoA reductase (PhaB). PHA synthase (PhaC) polymerizes 3-hydroxyvaleryl-CoA with 3-hydroxybutyryl-CoA to form PHBV (Fig. 1). Propionyl-CoA directly leads to the 3HV monomers in PHBV. In Halomonas sp. TD01, 3HV monomer amounts in PHBV was very low due to the rapid conversion of propionyl-CoA to 2-methylcitrate, which is converted to the methylcitric acid cycle (MCC cycle) [14]. In Halomonas sp. TD01, 2-methylcitrate synthase is encoded by prpC gene. PrpC knockout Halomonas sp. TD01 showed an accumulation of PHBV with a higher 3HV ratio when in presence of 1 g/L propionic acid. However, in this condition, the cells grew poorly and the cell dry weight (CDW) was very low [14]. Therefore, it is expected that the use of a CRISPRi repression system targeting prpC gene in Halomonas sp. TD01 could channel more propionic acid, or propionyl-CoA, to 3HV monomer in PHBV synthesis without impairing cell growth (Fig. 1).
Fig. 1.
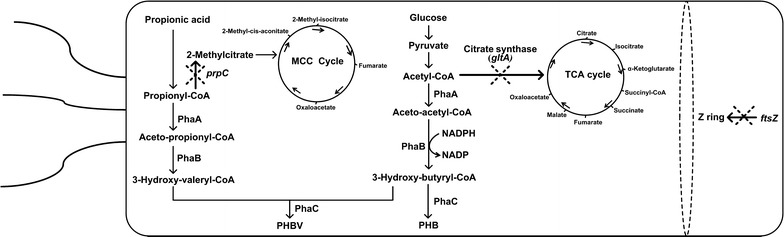
PHBV and PHB metabolic pathways. Dotted crosses show the repressed pathways in Halomonas sp. TD01 when targeting the indicated genes using the CRISPRi repression system. MMC, methylcitric acid cycle; prpC, 2-methylcitrate synthase; phaA, β-ketothiolase; phaB, NADPH-dependent acetoacetyl-CoA reductase; phaC, PHA synthase gene; TCA, tricarboxylic acid cycle; ftsZ, filamenting temperature-sensitive mutant Z
As shown in Fig. 4a, seven sgRNAs were designed along the prpC gene, mostly near the start site of the coding sequence or within 260 bp from the ATG sequence. CRISPRi plasmids are constructed and named as pli-dCas9-prpC1, pli-dCas9-prpC2, pli-dCas9-prpC3, pli-dCas9-prpC4, pli-dCas9-prpC5, pli-dCas9-prpC6 and pli-dCas9-prpC7, respectively (Additional file 1: Table S1; Fig. 4a). Various sgRNA binding sites led to different repressive effects. Two effective inhibition sites, namely prpC6 and prpC7, were combined together to form pli-dCas9-prpC6prpC7 to verify possible enhanced combinatory inhibition effect. All the plasmids were transferred via E. coli conjugation into Halomonas sp. TD01, forming strains TD-prpC1, TD-prpC2, TD-prpC3, TD-prpC4, TD-prpC5, TD-prpC6, TD-prpC7 and TD-prpC6prpC7, respectively.
Fig. 4.
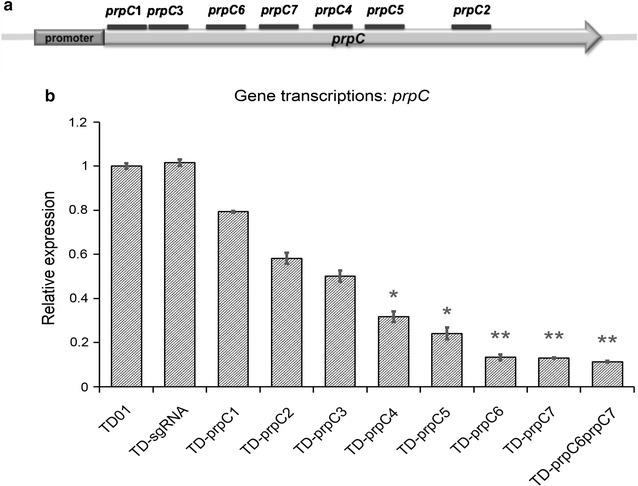
Controllable repression of prpC gene transcription in recombinant Halomonas sp. TD01. The relative binding positions of sgRNAs targeting prpC gene (a) and RT-PCR study of prpC transcription levels (b). All data were the average of three independent studies with standard deviations. Mean ± SE (n = 3). *p < 0.05 and **p < 0.01. TD01, Halomonas sp. TD wild type; TD-sgRNA, TD01 strain harboring the pli-dCas9-sgRNA plasmid without any target site; TD-prpC1, TD-prpC2, TD-prpC3, TD-prpC4, TD-prpC5, TD-prpC6, TD-prpC7, TD-prpC6prpC7, TD01 strains harboring the pli-dCas9-sgRNA plasmid with different DNA targets on gene prpC
To study the impact of prpC gene repression on cell growth, growth curves of the strains were established (Additional file 1: Figure S2). Wild type Halomonas sp. TD01 and TD-sgRNA were used as the control groups. The growth trends of the bacteria in all study groups were nearly the same, indicating that the CRISPRi system functioned without affecting cell growth.
All the strains were cultured in the MM medium in the presence of 1 g/L propionic acid and 30 g/L glucose at 37 °C and 200 rpm. After 12 h cultivation, 1 mM IPTG was added to induce the CRISPRi system. All bacteria were harvested after an overall cultivation time of 48 h (Table 1). All the strains grew well with CDW reaching approximately 14 g/L and accumulating approximately 75% PHBV. Meanwhile, Halomonas sp. TD01 and TD-sgRNA strains were used as controls, accumulating around 1% 3HV in PHBV copolymer. The percentage of 3HV monomer in PHBV copolymer varied from less than 1% to nearly 13% depending on the prpC repression intensity. In the single sgRNA inhibition system, Halomonas strains TD-prpC2, TD-prpC3, TD-prpC4 and TD-prpC5 all showed an obvious improvement in 3HV ratio compared to the controls. Halomonas strain TD-prpC2 accumulated nearly 5% 3HV in the PHBV, while 3HV ratios in PHBV copolymer accumulated by Halomonas strains TD-prpC3 and TD-prpC4 were around 6%. 3HV ratio in the PHBV produced by Halomonas strain TD-prpC5 was five times higher than that in the control groups, reaching 8% (Table 1). Halomonas strains TD-prpC6 and TD-prpC7 exhibited much higher repression efficiency, with around 12% 3HV in PHBV copolymer. In the two targets inhibition system, Halomonas strain TD-prpC6prpC7 produced PHBV copolymer consisting of 12.7% 3HV monomer (Table 1). The combination of prpC6 and prpC7 targets showed a slightly enhanced repression effect compared with that of the individual single sgRNA repression systems.
Table 1.
Shake flask PHBV production by recombinants Halomonas sp. TD01 controlling prpC gene expression via CRISPRi
| Recombinant TD01 strains | CDW (g/L) | PHBV (wt%) | 3HV (mol%) |
|---|---|---|---|
| TD01 | 13.22 ± 0.20 | 76.08 ± 2.81 | 0.83 ± 0.08 |
| TD-sgRNA | 13.95 ± 0.71 | 74.73 ± 2.25 | 1.29 ± 0.47 |
| TD-prpC1 | 14.99 ± 0.30 | 72.14 ± 2.37 | 1.79 ± 0.02 |
| TD-prpC2 | 14.43 ± 0.45 | 75.06 ± 3.20 | 4.78 ± 0.83 |
| TD-prpC3 | 14.08 ± 1.26 | 73.96 ± 7.18 | 5.72 ± 0.30 |
| TD-prpC4 | 13.58 ± 0.57 | 80.14 ± 8.62 | 6.44 ± 0.43 |
| TD-prpC5 | 13.59 ± 0.51 | 80.12 ± 3.27 | 8.16 ± 0.31 |
| TD-prpC6 | 14.17 ± 0.23 | 82.20 ± 3.82 | 11.94 ± 0.80 |
| TD-prpC7 | 13.54 ± 0.31 | 76.74 ± 0.80 | 12.15 ± 0.31 |
| TD-prpC6prpC7 | 14.67 ± 0.78 | 73.78 ± 4.82 | 12.70 ± 0.27 |
The recombinants harboring CRISPRi system were cultivated in MM medium containing 30 g/L glucose and 1 g/L propionic acid at 37 °C for 48 h as described in “Methods”. CDW, cell dry weight; PHBV (wt%), the weight percent of PHBV in CDW; TD01, Halomonas sp. TD wild type strain; TD-sgRNA, TD01 strain harboring the pli-dCas9-sgRNA plasmid without any DNA target site; TD-prpC1, TD-prpC2, TD-prpC3, TD-prpC4, TD-prpC5, TD-prpC6, TD-prpC7, TD-prpC6prpC7, TD01 strains harboring the pli-dCas9-sgRNA plasmid with different sgRNA targets of gene prpC, respectively. All data were the average of three independent studies with standard deviations. Mean ± SE (n = 3)
The mRNA expression levels RT-PCR agreed with the shake flask results (Fig. 4b). Wild type Halomonas sp. TD01 and the control TD-sgRNA showed a similar prpC mRNA expression level, indicating that the non-target system did not influence the gene expression in the strain Halomonas sp. TD-sgRNA. Strains Halomonas sp. TD-prpC1 and TD-prpC2 showed a higher mRNA expression level compared with strains TD-prpC3, TD-prpC4 and TD-prpC5, while strains TD-prpC6, TD-prpC7 and TD-prpC6prpC7 revealed a much lower mRNA expression level. This phenomenon again demonstrated the clear feasibility of the CRISPRi system for Halomonas sp. TD01. In addition, considering that prpC mRNA expression level in Halomonas strains TD-prpC6 and TD-prpC7 were already repressed to a very low level, it was hypothesized that further repression could hardly be expected when combining prpC6 and prpC7 inhibition targets. Thus the 3HV ratio in PHBV copolymer produced by Halomonas strain TD-prpC6prpC7 was almost the same as that in Halomonas strain TD-prpC6 or TD-prpC7.
The uses of Halomonas CRISPRi system for enhanced PHB synthesis
Halomonas sp. TD01 is able to produce PHB using glucose as substrate [13]. Acetyl-CoA, as a PHB substrate, is generated from pyruvate after glycolysis and oxidized in the TCA cycle. The citrate synthase catalyzes conversion of acetyl-CoA and oxaloacetate to citrate (Fig. 1). Gene gltA encoding citrate synthase, can not be completely repressed in Halomonas sp. TD01. However, partial repression on gltA should decrease the consumption of acetyl-CoA for TCA cycle and thus save some acetyl-CoA substrate for PHB synthesis (Fig. 1).
Four sgRNAs targeting gltA were designed. GltA1 was located in the promoter region, gltA2 sequence started with ATG, while gltA3 was just three base pairs away from gltA2. Finally, gltA4 was designed within 200 bp from the ATG sequence. The respective plasmids were constructed and named as pli-dCas9-gltA1, pli-dCas9-gltA2, pli-dCas9-gltA3 and pli-dCas9-gltA4. They were transferred into Halomonas sp. TD01 via E. coli conjugation, forming Halomonas strains TD-gltA1, TD-gltA2, TD-gltA3 and TD-gltA4.
Growth curves of the strains were established to investigate the effect of gltA inhibition on cell growth (Additional file 1: Figure S3). The bacterial growth curves showed that Halomonas strains TD-gltA1, TD-gltA2, TD-gltA3 and TD-gltA4 exhibited long lag growth phase in the beginning of the culture compared with the growth curves of Halomonas sp. TD01 and TD-sgRNA strains. Also, a similar cell density was reached after 15 h of cultivation, indicating that gltA repression prolonged the lag phase in cell growth, yet it did not affect cell density after the overnight cultivation.
All strains were cultured in the MM medium with 30 g/L glucose at 37 °C and 200 rpm. After 12 h cultivation, 1 mM IPTG was added to induce the CRISPRi system. Compared with the control Halomonas sp. TD-sgRNA, PHB content in Halomonas sp. TD-gltA2 showed a nearly 8% improvement, while strain TD-gltA3 had a 5% increase (Table 2). The mRNA expression levels from RT-PCR demonstrated that the gltA was repressed in the recombinant strains harboring the CRISPRi inhibition system targeting gltA gene (Additional file 1: Figure S4). Wild type Halomonas sp. TD01 and TD-sgRNA showed a similar gltA mRNA expression level, yet Halomonas strains TD-gltA1, TD-gltA2, TD-gltA3 and TD-gltA4 all showed a decreased gltA mRNA expression level. Therefore, partial CRISPRi repression on gltA had indeed reduced the consumption of acetyl-CoA thus improved PHB production in Halomonas sp. TD-sgRNA. Once again, gltA repressions demonstrated that the CRISPRi system was useful for metabolic engineering of Halomonas sp. TD01.
Table 2.
Shake flask PHB production by recombinants Halomonas sp. TD01 with controllable gltA gene expression via CRISPRi
| Recombinant TD01 strains | CDW (g/L) | PHB (wt%) |
|---|---|---|
| TD01 | 10.22 ± 0.25 | 77.68 ± 3.75 |
| TD-sgRNA | 13.28 ± 0.57 | 63.80 ± 3.08 |
| TD-gltA1 | 13.68 ± 0.21 | 66.79 ± 1.59 |
| TD-gltA2 | 13.53 ± 0.41 | 71.77 ± 7.16 |
| TD-gltA3 | 13.18 ± 0.33 | 69.22 ± 3.43 |
| TD-gltA4 | 13.10 ± 0.42 | 66.16 ± 8.11 |
All strains were cultivated in MM medium containing 30 g/L glucose at 37 °C for 48 h as described in “Methods”. CDW, cell dry weight; PHBV (wt%), the weight percent of PHBV in CDW; TD01, Halomonas sp. TD wild type strain; TD-sgRNA, TD01 strain harboring the pli-dCas9-sgRNA plasmid without any DNA target site; TD-gltA1, TD-gltA2, TD-gltA3, TD-gltA4, TD01 strains harboring the pli-dCas9-sgRNA plasmid with different sgRNA targets of gene gltA, respectively. All data were the average of three independent studies with standard deviations. Mean ± SE (n = 3)
Discussion
Halomonas sp. TD01 has been demonstrated to be a promising strain for PHA production due to its tolerance to high pH and high salt concentration. Therefore, allowing an open and continuous fermentation process without contamination [13]. This improves competitiveness of Halomonas sp. TD01 based PHA production process [24, 25]. However, as a non-model microorganism, Halomonas sp. TD01 still requires an inducible gene repression system for better performances.
CRISPRi has been used to regulate expression of desired genes without affecting the normal growth of engineered cells [1, 26–28]. In this study, an effective CRISPRi platform for genome editing in Halomonas sp. TD01 was developed. Pli-dCas9-sgRNA suitable for Halomonas sp. TD01 was designed to insert various sgRNAs, which form numerous CRISPRi repression plasmids. Multiple sgRNAs could also be inserted into pli-dCas9-sgRNA (Fig. 2a). The IPTG inducible CRISPRi system functioned well in Halomonas sp. TD01 for manipulating gene expression levels, achieving elongation of cell sizes, controllable PHBV copolymer monomer ratios, and enhanced PHB synthesis (Figs. 3, 4; Tables 1, 2).
FtsZ plays a crucial role in cytokinesis and assembles to form the Z rings during the cell division process [17]. Inhibition of ftsZ gene in Halomonas sp. TD01 resulted in formation of filamentous cells compared with short bar wild type of control groups (Fig. 3a). This phenomenon demonstrated the effectiveness of the established CRISPRi system for Halomonas sp. TD01, even though some Halomonas cells in the experimental groups still maintained their short bar shape possibly due to plasmid instability (Fig. 3a). Eventually, it is important to integrate the CRISPRi elements into the genome of Halomonas sp. TD01 to lower metabolic pressure on the bacteria, and to reduce the cost of antibiotics, as well as to improve the plasmid stability.
Halomonas sp. TD01 is able to produce PHBV using propionic acid as the precursor [14]. By increasing propionic acid concentration in the culture medium, Halomonas sp. TD01 can accumulate PHBV with a slightly improved ratio of 3HV (3% 3HV per 1 g/L propionic acid) [14]. The prpC gene knockout Halomonas sp. TD01 was very sensitive to 1 g/L propionic acid, growing slowly yet accumulated PHBV with higher 3HV content [14]. In this study, we achieved to regulate prpC gene at different expression levels without affecting cell growth (Table 1): all the recombinants and the controls grew well reaching approximately 14 g/L CDW that contains around 75% PHBV in the presence of 1 g/L propionic acid. Different CRISPRi inhibition targets resulted in variable repression effects (Table 1). Among the prpC gene inhibition targets, prpC6, prpC7 and their combined inhibition sites led to the highest improvement on 3HV ratio in PHBV copolymers (Table 1), reaching around 12–13% 3HV in the PHBV. Compared with prpC gene knockout approach which generates a fixed 3HV ratio in the PHBV, the CRISPRi platform provided a flexible regulation on 3HV contents in the PHBV (Table 1).
TCA cycle plays a crucial role in cell growth [22]. Genes involved in the TCA cycle are essential, as is the case of gltA gene, and therefore, they cannot be deleted from the genome of Halomonas sp. TD01. Thus, CRISPRi was employed to partially repress gltA expression under controlled intensities, allowing reduced consumption of acetyl-CoA for TCA cycle and diverging more acetyl-CoA to improve PHB production. Therefore, the constructed CRISPRi system can be used to regulate essential gene expression in Halomonas sp. TD01 for achieving multiple metabolic engineering goals.
Conclusions
A CRISPRi system dedicated to the non-model organism Halomonas sp. TD01 was successfully constructed and proven feasible, as evidenced by changing cell morphology, copolymer PHBV structures and homopolymer PHB synthesis when the genes ftsZ, prpC or gltA were repressed under different intensities. Considering the promising application prospect of Halomonas sp. TD01 for PHA and the chemical industry, the established CRISPRi system is expected to be useful for more metabolic engineering applications.
Methods
Strains, plasmids and culture conditions
Halomonas sp. TD01 was isolated from Aydingol Lake of Xinjiang Province, China, and stored in CGMCC (China General Micro-biological Culture Collection Center, Beijing). The collection number is 4353. E. coli S17-1 was used as a vector donor strain in conjugation. E. coli S17-1 was cultured in LB-20 medium. The ingredients of LB-20 medium are (g/L): 20 NaCl, 10 tryptone, 5 yeast extract. Halomonas sp. TD01 and its derivative strains were all cultivated in LB-60 medium. The ingredients of LB-60 medium are (g/L): 60 NaCl, 10 tryptone, 5 yeast extract. Chloramphenicol concentration used in this study was 25 µg/mL. All the strains and plasmids used in this study are listed (Additional file 1: Table S1).
Construction of recombinant strains
Plasmid construction
The plasmids used in this study are listed in Additional file 1: Table S1. Molecular cloning experiments were carried out according to manufacturers’ instructions or standard procedures. Kits for DNA purification and isolation of high quality plasmids were purchased from Qiagen (Shanghai, China). Restriction enzymes and DNA modification enzymes were provided by New England Biolabs (USA).
Based on plv-dCas9-sgRNA constructed in our previous study and pSEVA321 (kindly donated by Dr. Victor de Lorenzo of CSIC, Spain) [5, 29], a new plasmid termed pli-dCas9-sgRNA was successfully constructed, containing the dCas9 protein, restriction enzyme sites for sgRNA sequence insertion, Ptrc promoter, RK2 origin, chloramphenicol resistance selection marker, and the origin of transfer (oriT) for conjugation (Fig. 2a). Compared with the Ptet promoter induction system in our previous study [5], Ptrc promoter in plv-dCas9-sgRNA plasmid was found to be more effective as Ptet promoter could not function well in Halomonas sp. TD01. The IPTG inducible Ptrc promoter was more sensitive for induction.
To construct pli-dCas9-sgRNA, DNA fragments containing the dCas9 and sgRNA domain were amplified from PCR using plv-dCas9-sgRNA as the template, and then inserted into pSEVA321, forming pSEVA-dCas9-sgRNA. Multiple cloning sites (MCS) including XmaI, XbaI and SpeI were introduced into pSEVA-dCas9-sgRNA to form pli-dCas9-sgRNA. XmaI and XbaI were introduced upstream of the sgRNA expression cassette, while SpeI was inserted downstream of it. XbaI and SpeI are isocaudomers that create the same cohesive end after digestion and are required for sgRNA biobrick assembly (Fig. 2). By digesting pli-dCas9-sgRNA1 vector and PCR fragment containing sgRNA2 from pli-dCas9-sgRNA2 with XmaI/XbaI and XmaI/SpeI, respectively, pli-dCas9-sgRNA1sgRNA2 plasmid was formed after ligation. In addition, XmaI and XbaI restriction sites were reconstructed for the next round of sgRNA biobrick insertion (Fig. 2). In this way, multiple sgRNA biobricks could be inserted into one vector backbone for manipulating multiple genes simultaneously.
The 20–23 bp sgRNA complementary sequence was designed via primers (Additional file 1: Table S2). The forward and reverse primers were annealed to be a double-stranded DNA fragment precisely fitting the pli-dCas9-sgRNA vector, which was cleaved by BspQI enzyme. The reconstructed plasmid harboring the designed sgRNA sequence was formed after ligation. This technique allowed convenient changes in the complementary region to suit any interested gene.
To construct pli-dCas9-ftsZ(1-2), pli-dCas9-prpC(1-7) and pli-dCas9-gltA(1-4), sgRNA primers were annealed by temperature gradient PCR, the PCR product was then cut by BspQI at 50 °C for 2 h and purified. The pli-dCas9-sgRNA vector was digested by BspQI at 50 °C for 4 h and purified via electrophoresis. The inhibition plasmid was formed after ligation of the vector and PCR product. To prepare electro-competent E. coli S17-1, 3 mL volume of overnight cell culture was collected after centrifugation, the cells were then washed twice with ice-cold 10% glycerol. The ligation product was transformed into E. coli S17-1 via electroporation. Cells after transformation were placed on a LB-20 plate in the presence of 25 µg/mL chloramphenicol and cultivated overnight. Positive colonies were verified by PCR. Subsequently, the constructed plasmid was transformed into E. coli S17-1 and then conjugated into Halomonas sp. TD01.
To construct pli-dCas9-prpC6prpC7, DNA fragment containing prpC6 sgRNA, XmaI and SpeI restriction sites were amplified from PCR as the insert DNA, using pli-dCas9-prpC6 as the template. The insert DNA fragment was then cut by XmaI and SpeI. Vector pli-dCas9-prpC7 was restricted using XmaI and XbaI. After ligation of the insert DNA and the vector, pli-dCas9-prpC6prpC7 was formed, containing the reconstructed XmaI and XbaI restriction sites. The plasmid was then conjugated from E. coli S17-1 to Halomonas sp. TD01.
Designing sgRNA for repressing prpC gene
The sgRNAs were designed in the promoter region or near the ATG sequence in the targeted gene, and were right after a NGG sequence, namely, PAM sequence (protospacer adjacent motif sequence) [4]. All the sgRNAs could bind to the non-template DNA strand with sequence specificity. As shown in Fig. 4a, seven sgRNAs were designed along the prpC gene, mostly near the start site of the coding sequence or within 260 bp from the ATG sequence. In details: sgRNAs prpC1, prpC3, prpC6, prpC7, prpC4, prpC5 and PrpC2 were designed 15 bp downstream from ATG sequence, from position 16 to 38, after the PAM sequence CGG (from position 13 to 15), 35 bp downstream from ATG sequence, from position 36 to 58, after the PAM sequence CGG (from position 33 to 35), 88 bp downstream from ATG sequence, from position 89 to 111, after the PAM sequence CGG (from position 86 to 88), 127 bp downstream from ATG sequence, from position 128 to 150, after the PAM sequence CGG (from position 125 to 127), 190 bp downstream from ATG sequence, from position 191 to 213, after the PAM sequence CGG (from position 188 to 190), 237 bp downstream from ATG sequence, from position 238 to 260, after the PAM sequence GGG (from position 235 to 237) and 656 bp downstream from ATG sequence, from position 657 to 679, after the PAM sequence GGG (from position 654 to 656), respectively.
Conjugation into Halomonas sp. TD01
Plasmids were transferred from E. coli S17-1 to Halomonas sp. TD01 through conjugation. Both the donor and recipient cells were cultured overnight. Then 10 µL E. coli S17-1 and 10 µL Halomonas sp. TD01 were mixed and placed on a LB-20 plate without antibiotics and cultured in 37 °C. After four hours, the colonies from the LB-20 plate were picked up and cultured in LB-60 plates with chloramphenicol. The ingredients of LB-20 medium are (g/L): NaCl 20, tryptone 10, yeast extract 5.
Growth curve establishments
The cells were cultivated in LB-60 for 3 h, then 1 mM IPTG was added to induce the CRISPRi system. The total time of cell cultivation was 12 h. Then 200 µl bacterial fluid was inoculated into each well of a 96-well plate, with three parallel samples for each strain. Each sample in the well was diluted with LB-60 medium until OD600 reached 0.001. LB-60 medium was used as the blank control. All the samples was then cultured for 24 h under continuous rotary shaking (Thermo Scientific Varioskan Flash, Thermo Scientific, USA). OD600 was examined every half hour. After a deduction of the blank control, the average OD600 of each sample at each time point was calculated and used for growth curves.
Environmental scanning electron microscope (ESEM) analysis
The cells were harvested by centrifugation at 10,000g for 1 min, and were first fixed with 2.5% (v/v) glutaraldehyde for more than 4 h, followed by washing with 0.1 M phosphate-buffered saline (PBS) (pH 7.3) (3 times, 10 min each). Afterwards, the fixed cells were washed by ethanol in a concentration gradient (v/v) of 50, 70, 80, 90 and 100% successively, and then dehydrated by tertiary butyl alcohol mixed with ethanol in a ratio of 1:1. The cells were treated with pure tertiary butyl alcohol and used for imaging after lyophilization. The bacteria were imaged using a environmental scanning electron microscope (FEI Quanta 200, America) and analyzed utilizing XT Microscope Server imaging software.
Shake flask experiments in PHA production
After the plasmid was conjugated into Halomonas sp. TD01, the positive colonies were PCR verified. Then we obtained experimental TD strains, TD-ftsZ(1-2), TD-prpC(1-7), TD-prpC6prpC7 and TD-gltA(1-4), with each harboring a CRISPRi inhibition plasmid. Wild type Halomonas sp. TD01 and TD-sgRNA were used as the controls.
For production of PHA in Halomonas sp. TD01 and its derivate strains, the bacteria were cultivated in mineral medium (MM) containing (g/L): 60 NaCl, 30 glucose, 1 yeast extract, 1 NH4Cl, 0.2 MgSO4, 9.65 Na2HPO4-12H2O, 1.5 KH2PO4, 10 mL/L trace element solution I and 1 mL/L trace element solution II. Glucose concentration of the MM medium culturing Halomonas sp. TD01, TD-sgRNA, TD-ftsZ1 and TD-ftsZ2 in feasibility study of the constructed CRISPRi for Halomonas sp. TD01 was 20 g/L. The composition of trace element solution I was (g/L): 5 Fe(III)-NH4-citrate, 2 CaCl2, 1 M HCl. The trace element solution II contains (mg/L): 100 ZnSO4-7H2O, 30 MnCl2-4H2O, 300 H3BO3, 200 CoCl2-6H2O, 10 CuSO4-5H2O, 20 NiCl2-6H2O, 30 NaMoO4-2H2O, 1 M HCl at pH 9.0 [13].
For shake flask experiments, seed cultures were grown in 37 °C in LB-60 medium for 12 h at 200 rpm on a rotary shaker (HZQ-F160, HDL, Harbin, China). For each experimental group, three parallel samples were set. For each shake flask, 3 mL volume of the seed culture was inoculated in MM medium with 25 µg/mL chloramphenicol. The total volume of the shake flask was 50 mL. After 12 h of cultivation, IPTG was added to a final concentration at 1 mM to induce the CRISPRi inhibition system. Exceptionally, for the feasibility study of CRISPRi in Halomonas sp. TD01, TD-sgRNA, TD-ftsZ1 and TD-ftsZ2, the bacteria were cultured for 8 h before induction. The total time of cell culture was 48 h.
The bacteria were harvested, centrifuged at 10,000×g and washed once with distilled water. The cells were lyophilized and CDWs were measured. After methanolysis in chloroform at 100 °C for 4 h and cooling to room temperature, 1 mL deionized water was added. The components of the samples were then mixed by vortexing. Stratification then appeared in the sample solution, with the organic phase containing PHA. The samples were stood still for 1.5 h, and 1 mL chloroform containing PHA from the bottom layer was taken by syringe for gas chromatograph analysis. The samples were then analyzed by a gas chromatograph (GC-2014, SHIMADZU, Japan) and GCsolution software was employed to determine the PHA content [5]. Analytically pure PHB and PHBV copolymer (Sigma-Aldrich) were used as the standard samples to investigate 3HB and 3HV monomer quantities, respectively.
Real-time PCR
All the TD strains were cultivated in LB-60 medium for 3 h, then 1 mM IPTG was added to induce the CRISPRi system. The total time of cell cultivation was 12 h. The total RNA was isolated from Halomonas sp. TD01 and recombinant Halomonas sp. TD01 strains by using the RNA prep pure Cell/Bacteria Kit (Tiangen, Beijing, China). The Fastquant RT Kit (Tiangen, Beijing, China) was used to synthesize the cDNA for mRNA analysis. 16S rRNA was used as the inner standard, real-time PCR (RT-PCR) was carried out for mRNA analysis with SuperReal PreMix (SYBR Green) (Tiangen, Beijing, China).
The total extracted RNA concentration was measured by Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, USA) to design a concentration gradient for cDNA synthesis (using random primers following standard procedures described in the manufacturer’s product specification). The cDNA was used immediately in the RT-PCR analysis. The linear interval of total RNA was analyzed as a standard for the following experiments to adjust the quantity of the template within its linear range, so that the fluorescence quantitative results could be designed within a rational range. All samples were prepared with three parallel groups to obtain results of ΔCt values from the outputs of RT-PCR.
Authors’ contributions
TW and LL designed and carried out the experiments, analyzed the data and drafted the manuscript. GQC draft the basic idea and supervised the study. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to the Center of Biomedical Analysis, Tsinghua University for the ESEM analysis. Plasmid pSEVA-321 was kindly donated by Professor Víctor de Lorenzo of Centro Nacional de Biotecnolog ´ıa in Spain.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Gene sequences used in this project are from Genbank (http://www.ncbi.nlm.nih.gov/) and the restriction enzymes are from NEB.
Funding
Financial supports for this research were provided by the National Natural Science Foundation of China (Grant Nos. 31430003 and 31270146) and a special Tsinghua President Grant dedicated to this project (Grant No. 2015THZ10).
Additional file
Additional file 1: Table S1. Strains and plasmids used in this study. Table S2. Primers used for genetic manipulations. Fig. S1. Growth of recombinant Halomonas sp. TD01 harboring CRISPRi system targeting ftsZ gene in LB-60 medium. Fig. S2. Growth of recombinant Halomonas sp. TD01 with controllable prpC gene transcription via CRISPRi in LB-60 medium. Fig. S3. Growth of recombinant Halomonas sp. TD01 harboring CRISPRi system targeting gltA gene in LB-60 medium. Fig. S4. RT-PCR tests of gltA transcription levels in recombinant Halomonas sp. TD01.
Footnotes
Wei Tao and Li Lv contributed equally to this work
Contributor Information
Wei Tao, Email: taowei@phalab.org.
Li Lv, Email: lvli@phalab.org.
Guo-Qiang Chen, Phone: +86-10-62773844, Email: chengq@mail.tsinghua.edu.cn.
References
- 1.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 2.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 3.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lv L, Ren YL, Chen JC, Wu Q, Chen GQ. Application of CRISPRi for prokaryotic metabolic engineering involving multiple genes, a case study: controllable P(3HB-co-4HB) biosynthesis. Metab Eng. 2015;29:160–168. doi: 10.1016/j.ymben.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 6.Anderson AJ, Dawes EA. Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol Rev. 1990;54(4):450–472. doi: 10.1128/mr.54.4.450-472.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin DP, Williams SF. Medical applications of poly-4-hydroxybutyrate: a strong flexible absorbable biomaterial. Biochem Eng J. 2003;16:97–105. doi: 10.1016/S1369-703X(03)00040-8. [DOI] [Google Scholar]
- 8.Park SJ, Choi JI, Lee SY. Short-chain-length polyhydroxyalkanoates: synthesis in metabolically engineered Escherichia coli and medical applications. J Microbiol Biotechnol. 2005;15(1):206–215. [Google Scholar]
- 9.Chen GQ. A microbial polyhydroxyalkanoates (PHA) based bio- and materials industry. Chem Soc Rev. 2009;38:2434–2446. doi: 10.1039/b812677c. [DOI] [PubMed] [Google Scholar]
- 10.Witholt B, Kessler B. Perspectives of medium chain length poly(hydro-xyalkanoates), a versatile set of bacterial bioplastics. Curr Opin Biotechnol. 1999;10:279–285. doi: 10.1016/S0958-1669(99)80049-4. [DOI] [PubMed] [Google Scholar]
- 11.Lee SY. Bacterial polyhydroxyalkanoates. Biotechnol Bioeng. 1996;49(1):1–14. doi: 10.1002/(SICI)1097-0290(19960105)49:1<1::AID-BIT1>3.3.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 12.Cai L, Tan D, Aibaidula G, Dong XR, Chen JC, Tian WX, Chen GQ. Comparative genomics study of polyhydroxyalkanoates (PHA) and ectoine relevant genes from Halomonas sp. TD01 revealed extensive horizontal gene transfer events and co-evolutionary relationships. Microb Cell Fact. 2011;10:88. doi: 10.1186/1475-2859-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan D, Xue YS, Aibaidula G, Chen GQ. Unsterile and continuous production of polyhydroxybutyrate by Halomonas TD01. Bioresour Technol. 2011;102:8130–8136. doi: 10.1016/j.biortech.2011.05.068. [DOI] [PubMed] [Google Scholar]
- 14.Fu XZ, Tan D, Aibaidula G, Wu Q, Chen JC, Chen GQ. Development of Halomonas TD01 as a host for open production of chemicals. Metab Eng. 2014;23:78–91. doi: 10.1016/j.ymben.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 15.Bi E, Lutkenhaus J. Cell division inhibitors SulA and MinCD prevent formation of the FtsZ ring. J Bacteriol. 1993;175(4):1118–1125. doi: 10.1128/jb.175.4.1118-1125.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loose M, Mitchison TJ. The bacterial cell division proteins FtsA and FtsZ self-organize into dynamic cytoskeletal patterns. Nat Cell Biol. 2014;16:38–46. doi: 10.1038/ncb2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Margolin W. FtsZ and the division of prokaryotic cells and organelles. Nat Rev Mol Cell Biol. 2005;6:862–871. doi: 10.1038/nrm1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang XR, Wang H, Shen R, Chen GQ. Engineering the bacterial shapes for enhanced inclusion bodies accumulation. Metab Eng. 2015;29:227–237. doi: 10.1016/j.ymben.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 19.Ewering C, Heuser F, Benölken JK, Brämer CO, Steinbüchel A. Metabolic engineering of strains of Ralstonia eutropha and Pseudomonas putida for biotechnological production of 2-methylcitric acid. Metab Eng. 2006;8:587–602. doi: 10.1016/j.ymben.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Silva LF, Gomez JGC, Oliveira MS, Torres BB. Propionic acid metabolism and poly-3-hydroxybutyrate-co-3-hydroxyvalerate (P3HB-co-3HV) production by Burkholderia sp. J Biotechnol. 2000;76:165–174. doi: 10.1016/S0168-1656(99)00184-4. [DOI] [PubMed] [Google Scholar]
- 21.Tan D, Wu Q, Chen JC, Chen GQ. Engineering Halomonas TD01 for low cost production of polyhydroxyalkanoates. Metab Eng. 2014;26:34–47. doi: 10.1016/j.ymben.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 22.Gest H. Evolution of the citric acid cycle and respiratory energy conversion in prokaryotes. FEMS Microbiol Lett. 1981;12:209–215. doi: 10.1111/j.1574-6968.1981.tb07643.x. [DOI] [Google Scholar]
- 23.Zhang FY, Li JJ, Liu HW, Liang QF, Qi QS. ATP-based ratio regulation of glucose and xylose improved succinate production. PLoS ONE. 2016;11(6):e0157775. doi: 10.1371/journal.pone.0157775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao XW, Zheng WT, Hou X, Liang J, Li ZJ. Metabolic engineering of Escherichia coli for poly(3-hydroxybutyrate) production under microaerobic condition. Biomed Res Int. 2015;2015:789315. doi: 10.1155/2015/789315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bäckström BT, Brockelbank JA, Rehm BHA. Recombinant Escherichia coli produces tailor-made biopolyester granules for applications in fluorescence activated cell sorting: functional display of the mouse interleukin-2 and myelin oligodendrocyte glycoprotein. BMC Biotechnol. 2007;7:3. doi: 10.1186/1472-6750-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cress BF, Linhardt RJ, Koffas MAG, et al. CRISPathBrick: modular combinatorial assembly of type II-A CRISPR arrays for dCas9-mediated multiplex transcriptional repression in E. coli. ACS Synth Biol. 2015;4:987–1000. doi: 10.1021/acssynbio.5b00012. [DOI] [PubMed] [Google Scholar]
- 27.Wendt KE, Ungerer J, Cobb RE, Zhao H, Pakrasi HB. CRISPR/Cas9 mediated targeted mutagenesis of the fast growing cyanobacterium Synechococcus elongatus UTEX 2973. Microb Cell Fact. 2016;15:115. doi: 10.1186/s12934-016-0514-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cress BF, Linhardt RJ, Koffas MAG, et al. Rapid generation of CRISPR/dCas9-regulated, orthogonally repressible hybrid T7-lac promoters for modular, tuneable control of metabolic pathway fluxes in Escherichia coli. Nucleic Acids Res. 2016;44:4472–4485. doi: 10.1093/nar/gkw231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martínezgarcía E, Aparicio T, et al. SEVA 2.0: an update of the Standard European Vector Architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res. 2015;43:D1183–D1189. doi: 10.1093/nar/gku1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Gene sequences used in this project are from Genbank (http://www.ncbi.nlm.nih.gov/) and the restriction enzymes are from NEB.