

Worsening renal function (WRF) is common during the treatment of heart failure (HF) and has been associated with decreased survival, hospitalizations and disease progression.1 There are a number of hypothetical mechanisms including inflammation, oxidant stress or induction of apoptosis by uremic toxins by which a reduction in renal function could directly lead to mortality.2, 3 However, patients who experience WRF also often exhibit multiple markers of increased HF-disease severity and are less likely to respond to diuretics.4, 5 As a result, it is difficult to determine whether the frequently observed association between WRF and adverse outcomes results directly from the reduction in glomerular filtration rate (GFR) or is merely serving as a marker of greater HF disease severity.
Over the last several years, it been described that not all forms of WRF are prognostically equivalent and WRF that occurs in the setting of otherwise beneficial HF therapies, like renin angiotensin aldosterone system (RAAS) antagonists or aggressive diuresis, appears to have a negligible impact on outcomes.6–10 RAAS antagonists, a cornerstone of guideline-based medical therapy for HF with reduced ejection fraction (HFrEF), can lead to perturbations in glomerular hemodynamics, secondary to a more pronounced vasodilation of the efferent arteriole, yielding a decrease in filtration fraction and thus at times GFR.11 As a result, it is not surprising that WRF is commonly observed during treatment with these medications.7, 12 Despite the increased frequency of WRF, we and others have found that WRF in the setting of HFrEF treatment with RAAS antagonists is relatively benign compared to WRF unprovoked by RAAS antagonism.7, 9, 12 So while the evidence for prognostic subtypes of WRF is well established, questions remain as to what causes the difference in prognosis. Is unprovoked WRF simply identifying sicker patients whereas RAAS antagonist-induced WRF does not, and the association is all confounding? Or are all decreases in GFR equally and directly harmful, but WRF provoked by RAAS antagonists has this disadvantage offset by the mortality benefit of the RAAS antagonists? If the latter is true, then in a group that did not experience benefit from RAAS antagonism, we should see worsened outcomes associated with RAAS-induced WRF.
In this issue of Circulation: Heart Failure, Damman and colleagues set out to examine just that: to compare the outcomes associated with WRF in patients with HFrEF (who derive survival advantage from RAAS antagonism) to HF with preserved ejection fraction (HFpEF, where a survival advantage is absent with RAAS antagonism). Damman and coauthors undertook a meta-analysis of nearly 29,000 patients from randomized, placebo-controlled trials of RAAS antagonists.13 WRF was defined in accordance with the individual clinical trial definition using either a percent decrease in eGFR or a ≥ 0.3 increase in serum creatinine. Not surprisingly, WRF in all patients was associated with increased mortality (RR=1.35, 95% CI 1.25–1.46) as well as greater risk for HF hospitalization (RR=1.44, 95% CI 1.30–1.59). WRF occurred more frequently in patients on RAAS antagonists than placebo (13% vs. 9%) with a high background rate of WRF in the absence of RAAS. Consistent with previous reports, the magnitude of the impact of WRF on increased mortality in HFrEF was greater in patients on placebo (RR=1.48, 95% CI 1.35–1.62, p<0.001) with a more subtle risk associated with WRF in the setting of RAAS antagonists (RR=1.19, 95% CI 1.08–1.31, p<0.001; p-interaction=0.005). Conversely, in patients with HFpEF, the magnitude of the association between WRF and increased mortality was instead greater in patients randomized to ACE or ARB antagonists (RR=1.78, 95% CI 1.43–2.21, p<0.001) as compared to placebo, with WRF in the placebo group no longer demonstrating a significant relationship with mortality (RR=1.25, 95% CI 0.88–1.77, p=0.29; p-interaction 0.092). Data were not presented on aldosterone antagonists for HFpEF. There was no significant difference in the magnitude of the risk of HF hospitalization associated with WRF between those patients on RAAS antagonists compared to those on placebo, regardless of HF phenotype.13
Although the analysis by Damman et al. certainly improves our understanding of WRF in the setting of RAAS antagonism, has it advanced our understanding of the causal vs. confounding nature of the WRF-mortality association? When examining the available evidence, particularly regarding RAAS antagonists, separating the effects of the etiology of the WRF from the reduction in GFR itself is challenging. Any reduction in GFR can theoretically lead to retention of uremic toxins and a cascade of hypothetical problems such as inflammation/oxidant stress, which may have direct adverse effects.2, 3 Regardless of what caused the GFR to deteriorate, the GFR has worsened and thus we would expect the same harmful toxin buildup. However, we and others have described that the clinical context in which WRF occurs (during RAAS antagonism, a drop in blood pressure, elevated natriuretic peptide levels, or aggressive diuresis) differentially affects the magnitude of the association between WRF and mortality despite similar decrements in GFR.6–10, 14, 15 If the direct effects of WRF were entirely responsible for inferior outcomes, then the mechanism driving the decrease in GFR should be irrelevant. However, from the observational studies cited above, this does not appear to be the case.
Similarly, if the low GFR itself was the primary driver for increased mortality in HF, then improvement in renal function (IRF) should yield improved outcomes. To the contrary, we have described a strong association between IRF and poor outcomes.16–18 In a post hoc analysis of the Diuretic Strategies in Patients with Acute Decompensated Heart Failure (DOSE) trial, in which hospitalized HF patients were randomized to a high or low intensity diuretic strategy, an improvement in eGFR during the study period was associated with markedly increased risk of the composite endpoint of death, HF hospitalization or emergency department visit (Figure 1).19 This negative association persisted despite adjustment for baseline characteristics and in-hospital treatment-related factors. Additionally, in a trial of bardoxolone methyl specifically aimed at improving renal dysfunction in type 2 diabetics with concomitant stage 4 chronic kidney disease, bardoxolone resulted in significant improvement in GFR, yet the trial was terminated early for increased cardiovascular events in the bardoxolone arm.20 This growing literature on the negative associations between IRF and survival certainly calls into question an overwhelmingly causal relationship between GFR and outcomes as the primary explanation for the WRF association; however, it does not completely exclude a small causal component. In the case of IRF it is very likely that these patients were inadequately treated with neurohormonal antagonists and diuretics, thus potentially counteracting the benefit of the improved GFR. Similarly, bardoxolone may have substantial direct toxicity that outweighs a smaller positive effect of improved GFR, and it was not studied in the setting of pre-existing cardiac dysfunction where the GFR effects may be more prominent.20
Figure 1. Relationship between changes in renal function and clinical outcomes.
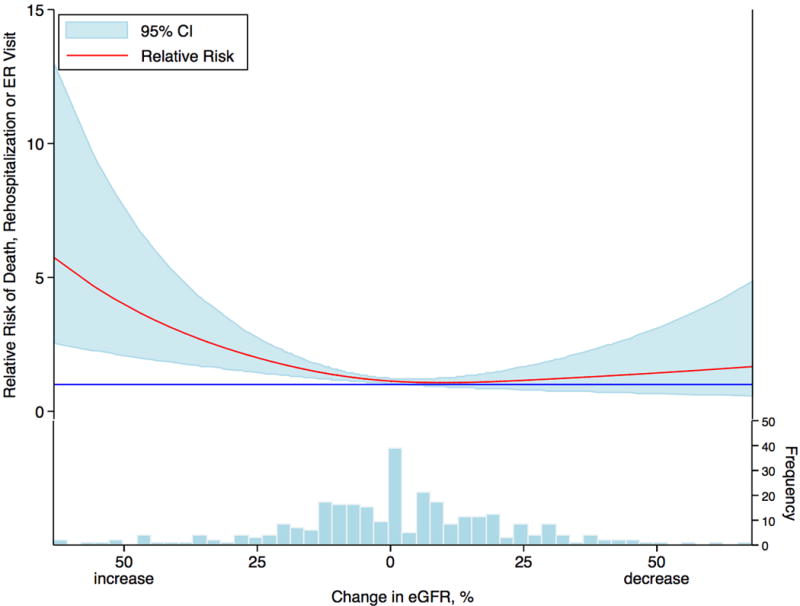
Relationship between the percent change in eGFR from baseline to 72 hours and the composite outcome of death, rehospitalization, or emergency room visit within 60 days in patients admitted with acute decompensated heart failure and enrolled in a clinical trial of diuretic strategies. The solid blue line represents a hazard ratio of 1. CI: confidence interval; eGFR: estimated glomerular filtration rate; ER: emergency room. Reproduced with permission from Brisco MA, Zile MR, Hanberg JS, Wilson FP, Parikh CR, Coca SG, Tang WH and Testani JM. Relevance of Changes in Serum Creatinine During a Heart Failure Trial of Decongestive Strategies: Insights From the DOSE Trial. J Card Fail. 2016;22:753–60.19
Does the fact that there appears to be no mortality benefit with RAAS antagonists in HFpEF to counterbalance the concomitant GFR reduction provide support that the WRF had direct detrimental effects, settling the argument of cause versus disease severity indicator once and for all? Unfortunately it does not. If the decrease in GFR itself is the cause for the inferior outcomes, then in HFpEF where RAAS antagonism has no benefit, we would expect the risk of death associated with WRF to be identical in the placebo and RAAS antagonist groups.21 To the contrary, the risk of death associated with RAAS antagonist-induced WRF was 53% greater than WRF in the placebo group. Notably, WRF in the placebo group was not actually significantly associated with worse outcomes (p=0.21).13 Unless there was a tremendous asymmetry of the magnitude of loss in GFR in the RAAS-antagonist WRF group (which was not reported here, but has not been observed in other analyses), this proves substantial confounding was driving the differential risk.7 The most likely explanation is that the hemodynamic and neurohormonal challenge of the RAAS antagonist served as a cardio-renal “stress test” and simply identified greater disease severity.12 So really the decrease in GFR was just a marker of a sick patient rather than something that directly made the patient sick. For example, if we see a hypotensive response to exercise on a treadmill stress test, we know that is associated with a poor prognosis.22 However, we would never believe that the brief period of mild hypotension caused the poor prognosis. Rather the underlying pathology of severe three vessel coronary disease or critical aortic stenosis that provoked the abnormal response is the cause for the poor prognosis. Ultimately, what we do know for certain, as supported in this meta-analysis, is that WRF in HF is epidemiologically complex, with mechanistically and prognostically distinct forms that appear to differ by HF phenotype. We also now know from this analysis that WRF after starting a RAAS antagonist identifies high-risk patients, but it does not inform the question if we start a HFpEF patient on a RAAS antagonist for a valid clinical reason, and they have WRF, that the drug should or should not be discontinued.13 Only through prospective randomized trials will we be able to move past these perplexing epidemiologic signals and learn how to use changes in renal function to help us guide therapy.
Supplementary Material
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Damman K, Valente MAE, Voors AA, O’Connor CM, van Veldhuisen DJ, Hillege HL. Renal impairment, worsening renal function, and outcome in patients with heart failure: an updated meta-analysis. Eur Heart J. 2014;35:455–469. doi: 10.1093/eurheartj/eht386. [DOI] [PubMed] [Google Scholar]
- 2.Vanholder R, Baurmeister U, Brunet P, Cohen G, Glorieux G, Jankowski J, European Uremic Toxin Work G A bench to bedside view of uremic toxins. J Am Soc Nephrol. 2008;19:863–870. doi: 10.1681/ASN.2007121377. [DOI] [PubMed] [Google Scholar]
- 3.Rosner MH, Ronco C, Okusa MD. The role of inflammation in the cardio-renal syndrome: a focus on cytokines and inflammatory mediators. Semin Nephrol. 2012;32:70–78. doi: 10.1016/j.semnephrol.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Testani J, Cappola T, Mccauley B, Chen J, Shen J, Shannon R, Kimmel S. Impact of worsening renal function during the treatment of decompensated heart failure on changes in renal function during subsequent hospitalization. Am Heart J. 2011;161:944–949. doi: 10.1016/j.ahj.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Testani J, Mccauley B, Kimmel S, Shannon R. Characteristics of patients with improvement or worsening in renal function during treatment of acute decompensated heart failure. Am J Cardiol. 2010;106:1763–1769. doi: 10.1016/j.amjcard.2010.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Testani JM, Coca SG, McCauley BD, Shannon RP, Kimmel SE. Impact of changes in blood pressure during the treatment of acute decompensated heart failure on renal and clinical outcomes. Eur J Heart Fail. 2011;13:877–884. doi: 10.1093/eurjhf/hfr070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Testani JM, Kimmel SE, Dries DL, Coca SG. Prognostic Importance of Early Worsening Renal Function After Initiation of Angiotensin-Converting Enzyme Inhibitor Therapy in Patients With Cardiac Dysfunction. Circ Heart Fail. 2011;4:685–691. doi: 10.1161/CIRCHEARTFAILURE.111.963256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Testani JM, Chen J, McCauley BD, Kimmel SE, Shannon RP. Potential effects of aggressive decongestion during the treatment of decompensated heart failure on renal function and survival. Circulation. 2010;122:265–272. doi: 10.1161/CIRCULATIONAHA.109.933275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lesogor A, Cohn JN, Latini R, Tognoni G, Krum H, Massie B, Zalewski A, Kandra A, Hua TA, Gimpelewicz C. Interaction between baseline and early worsening of renal function and efficacy of renin-angiotensin-aldosterone system blockade in patients with heart failure: insights from the Val-HeFT study. Eur J Heart Fail. 2013;15:1236–1244. doi: 10.1093/eurjhf/hft089. [DOI] [PubMed] [Google Scholar]
- 10.van Kimmenade RRJ, Januzzi J, James L, Baggish AL, Lainchbury JG, Bayes-Genis A, Richards AM, Pinto YM. Amino-terminal pro-brain natriuretic Peptide, renal function, and outcomes in acute heart failure: redefining the cardiorenal interaction? J Am Coll Cardiol. 2006;48:1621–1627. doi: 10.1016/j.jacc.2006.06.056. [DOI] [PubMed] [Google Scholar]
- 11.Schoolwerth AC, Sica DA, Ballermann BJ, Wilcox CS, Council on the Kidney in Cardiovascular D and the Council for High Blood Pressure Research of the American Heart A Renal considerations in angiotensin converting enzyme inhibitor therapy: a statement for healthcare professionals from the Council on the Kidney in Cardiovascular Disease and the Council for High Blood Pressure Research of the American Heart Association. Circulation. 2001;104:1985–1991. doi: 10.1161/hc4101.096153. [DOI] [PubMed] [Google Scholar]
- 12.Clark H, Krum H, Hopper I. Worsening renal function during renin-angiotensin-aldosterone system inhibitor initiation and long-term outcomes in patients with left ventricular systolic dysfunction. Eur J Heart Fail. 2014;16:41–48. doi: 10.1002/ejhf.13. [DOI] [PubMed] [Google Scholar]
- 13.Beldhuis I, Streng KW, Maaten JMT, Voors AA, Meer Pvd, Rossignol P, McMurray JJV, Damman K. Renin agniotensin system inhibition, worsening renal function and outcome in heart failure patients with reduced and preserved ejection fraction. Circ Heart Fail. 2017;10:e003588. doi: 10.1161/CIRCHEARTFAILURE.116.003588. [DOI] [PubMed] [Google Scholar]
- 14.Testani JM, Damman K, Brisco MA, Chen S, Laur O, Kula AJ, Tang WH, Parikh C. A combined-biomarker approach to clinical phenotyping renal dysfunction in heart failure. J Card Fail. 2014;20:912–919. doi: 10.1016/j.cardfail.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Testani JM, Coca SG, Shannon RP, Kimmel SE, Cappola TP. Influence of renal dysfunction phenotype on mortality in the setting of cardiac dysfunction: analysis of three randomized controlled trials. Eur J Heart Fail. 2011;13:1224–1230. doi: 10.1093/eurjhf/hfr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Testani JM, McCauley BD, Chen J, Coca SG, Cappola TP, Kimmel SE. Clinical Characteristics and Outcomes of Patients With Improvement in Renal Function During the Treatment of Decompensated Heart Failure. J Card Fail. 2011;17:993–1000. doi: 10.1016/j.cardfail.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brisco MA, Coca SG, Chen J, Owens AT, McCauley BD, Kimmel SE, Testani JM. Blood urea nitrogen/creatinine ratio identifies a high-risk but potentially reversible form of renal dysfunction in patients with decompensated heart failure. Circ Heart Fail. 2013;6:233–239. doi: 10.1161/CIRCHEARTFAILURE.112.968230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brisco MA, Kimmel SE, Coca SG, Putt ME, Jessup M, Tang WW, Parikh CR, Testani JM. Prevalence and prognostic importance of changes in renal function after mechanical circulatory support. Circ Heart Fail. 2014;7:68–75. doi: 10.1161/CIRCHEARTFAILURE.113.000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brisco MA, Zile MR, Hanberg JS, Wilson FP, Parikh CR, Coca SG, Tang WH, Testani JM. Relevance of Changes in Serum Creatinine During a Heart Failure Trial of Decongestive Strategies: Insights From the DOSE Trial. J Card Fail. 2016;22:753–760. doi: 10.1016/j.cardfail.2016.06.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ, McMurray JJ, Meyer CJ, Parving HH, Remuzzi G, Toto RD, Vaziri ND, Wanner C, Wittes J, Wrolstad D, Chertow GM, Investigators BT Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013;369:2492–2503. doi: 10.1056/NEJMoa1306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Damman K, Perez AC, Anand IS, Komajda M, McKelvie RS, Zile MR, Massie B, Carson PE, McMurray JJ. Worsening renal function and outcome in heart failure patients with preserved ejection fraction and the impact of angiotensin receptor blocker treatment. J Am Coll Cardiol. 2014;64:1106–1113. doi: 10.1016/j.jacc.2014.01.087. [DOI] [PubMed] [Google Scholar]
- 22.Le VV, Mitiku T, Sungar G, Myers J, Froelicher V. The blood pressure response to dynamic exercise testing: a systematic review. Prog Cardiovasc Dis. 2008;51:135–160. doi: 10.1016/j.pcad.2008.07.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.