

Abstract
Background
Modeling of microbial metabolism is a topic of growing importance in biotechnology. Mathematical modeling helps provide a mechanistic understanding for the studied process, separating the main drivers from the circumstantial ones, bounding the outcomes of experiments and guiding engineering approaches. Among different modeling schemes, the quantification of intracellular metabolic fluxes (i.e. the rate of each reaction in cellular metabolism) is of particular interest for metabolic engineering because it describes how carbon and energy flow throughout the cell. In addition to flux analysis, new methods for the effective use of the ever more readily available and abundant -omics data (i.e. transcriptomics, proteomics and metabolomics) are urgently needed.
Results
The jQMM library presented here provides an open-source, Python-based framework for modeling internal metabolic fluxes and leveraging other -omics data for the scientific study of cellular metabolism and bioengineering purposes. Firstly, it presents a complete toolbox for simultaneously performing two different types of flux analysis that are typically disjoint: Flux Balance Analysis and 13C Metabolic Flux Analysis. Moreover, it introduces the capability to use 13C labeling experimental data to constrain comprehensive genome-scale models through a technique called two-scale 13C Metabolic Flux Analysis (2S-13C MFA). In addition, the library includes a demonstration of a method that uses proteomics data to produce actionable insights to increase biofuel production. Finally, the use of the jQMM library is illustrated through the addition of several Jupyter notebook demonstration files that enhance reproducibility and provide the capability to be adapted to the user’s specific needs.
Conclusions
jQMM will facilitate the design and metabolic engineering of organisms for biofuels and other chemicals, as well as investigations of cellular metabolism and leveraging -omics data. As an open source software project, we hope it will attract additions from the community and grow with the rapidly changing field of metabolic engineering.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-017-1615-y) contains supplementary material, which is available to authorized users.
Keywords: Flux analysis, 13 C Metabolic Flux Analysis, -omics data, Predictive biology
Background
Metabolism is the set of physical and chemical processes through which living organisms extract energy and mass from their environment in order to sustain life [1]. Hence, an understanding of metabolism is fundamental in order to set the stage for, and understand the physical limitations of, every biological process. For example, metabolic disorders lie at the heart of a variety of human illnesses such as diabetes [2], obesity [3] and cancer [4].
Understanding of metabolism is also of central importance to the microbial production of drugs, bioproducts and biofuels through metabolic engineering [5]. This application of microbial bioengineering to produce biofuels and other commodity products has attracted significant attention due to its potential to reduce the CO2 emissions which cause climate change, reduce society’s reliance on fossil fuels for energy and chemicals, and provide for enhanced energy security. The production of commodity chemicals from lignocellulosic biomass is critical for achieving these goals, particularly in view of expected future population growth in emergent economies coupled with a marked improvement in their living standards [6].
Metabolic engineering rarely succeeds at the first attempt to introduce and activate a heterologous pathway in a host organism. Rather, most of the time in this process is spent troubleshooting the pathways for acceptable production [7]. Very often, the metabolic engineer diagnoses the problem in the bioengineered microbe by relying on a variety of -omics data (transcriptomics, proteomics, metabolomics) that are expected to provide an explanation of why the system does not behave as expected [8–10]. However, this diagnosing through -omics data is a non trivial process: often the amount of data is much larger than what metabolic engineers are typically trained to analyze, and it is non-trivial to extract actionable items (e.g. overexpress this specific gene or change that promoter) that will reliably improve the system’s behavior [11, 12]. To complicate matters, the reason for the underperformance of a given pathway may not rely on the pathway itself but on other parts of the host metabolism that affect the pathway’s performance in intricate and non-direct ways (e.g. cofactor availability or gene regulation) [13].
Mathematical modeling helps contextualize experimental data into a model that provides an explanation of the observed phenomena and provides predictions for changes in the system under study. Metabolic modeling, in particular, has been successfully used to e.g. increase product yield [14] or identify bottlenecks and competing pathways [15]. Moreover, data analysis and modeling is expected to increase in scope and importance as the creation of increasingly larger sets of -omics data sets are enabled by the rapid drop of DNA sequencing costs [16] and the availability of high-throughput mass spectrometry workflows [17, 18]. Hence, research institutes such as the Joint BioEnergy Institute (JBEI), tasked to produce the scientific and technological underpinnings that will enable lignocellulosic biofuels [19], have devoted research effort not only to develop drop-in biofuels [20] but also tools to speed up bioengineering through mathematical modeling.
Some of the mathematical tools created at JBEI to enable better prediction of bacterial and eukaryotic metabolism are presented here in the form of the JBEI Quantitative Metabolic Modeling library (jQMM). The jQMM library includes algorithms to measure and predict internal metabolic fluxes using three different techniques: 13C Metabolic Flux Analysis (13C MFA [21]), Flux Balance Analysis (FBA [22]) and two-scale 13 C Metabolic Flux Analysis (2S-13C MFA [23]). It also includes methods to produce actionable insights from -omics data to improve pathway yield [24], and methodologies for the flux analysis of microbial communities [25]. We hope to add to the jQMM library in the future tools and algorithms currently being developed at JBEI, as well other institutions who may consider contributing to this library.
Methods
Key capabilities
The JBEI QMM library includes code to perform the following techniques: FBA [22], 13C MFA [21], 2S-13C MFA [23], Principal Component Analysis of Proteomics (PCAP [24]), and 13C MFA for microbial communities [25]. Mathematical details for each method are provided in the supplementary material (Additional file 1). FBA, 13C MFA and 2S-13C MFA are techniques used to calculate internal metabolic fluxes; detailed explanations regarding the differences between each method can be found in Garcia Martin et al. [23]. This library contains sample demonstration scripts to, for example, replicate all the figures from Garcia Martin et al. in the form of Jupyter notebooks (see Table 1). The code in the jQMM library focuses on 13C MFA, 2S-13C MFA, since there is already an open source python-based library [26] for FBA. For 13C MFA there exists some closed-source packages (e.g. METRAN [27], INCA [28], 13CFLUX2 [29]), a few open-source packages (OPENFLUX [30] and OPENFLUX2 [31]) but no open source python-based libraries. For 2S-13C MFA, jQMM is the first public library.
Table 1.
Table of iPython Notebooks
| iPython Notebook number | Topic |
|---|---|
| A0 | module tests |
| A1 | Core demo |
| A2 | Enhanced lists demo |
| A3 | ReactionNetworks demo |
| A4 | FluxModels demo |
| A5 | GAMSclasses demo |
| A6 | Predictions demo |
| A7 | Labeling and DB demo |
| B1 | FBA demo |
| B2 | TCA 13 C MFA demo |
| B3 | Toya data 13 C MFA demo |
| B4 | Toya data 2S- 13 C MFA demo |
| B5 | PCAP example |
| B6 | 13C MFA for microbial communities |
Implementation
jQMM is implemented in Python 2.7, and we have aimed to provide a code base that is as modular as possible, in order to facilitate understanding and reusability. The code is divided into a set of modules that incorporate classes and methods which will typically be used together. Figure 1 details the different modules and how they interact with each other for flux-based analysis. The following subsections provide a brief explanation of the purpose of each module and Figs. 2, 3 and 4 (and Additional file 1: Figures S1-S3) provide graphical depictions of each module together with their main classes and methods. Tutorials are provided for each module, along with unit tests.
Fig. 1.
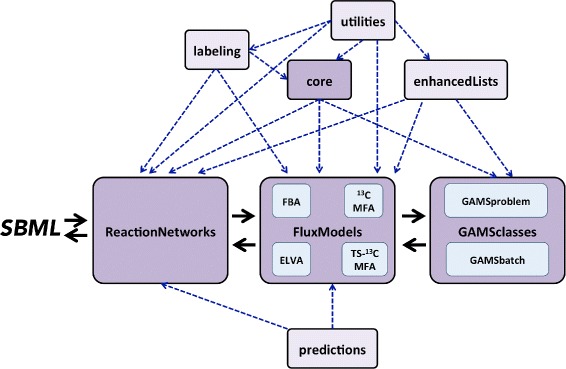
Diagram of jQMM library module relationships and typical flow for flux analysis. Flux calculations typically start with information stored in an SBML (Systems Biology Markup Language) file and translated into a reaction network. That reaction network is enclosed in a flux model of the appropriate type for the desired method (FBA, 13C MFA, 2S-13C MFA, ELVA). The flux model instance uses the GAMSclasses module in order to solve the appropriate optimization problem, and turns it back to the flux model instance, which stores the information as a reaction network or an SBML file. The core, labeling, utilities and enhancedLists modules are used by these “main workflow” modules (in darker shade). Predictions use information from reaction networks and flux models to make flux predictions for genetic modifications
Fig. 2.
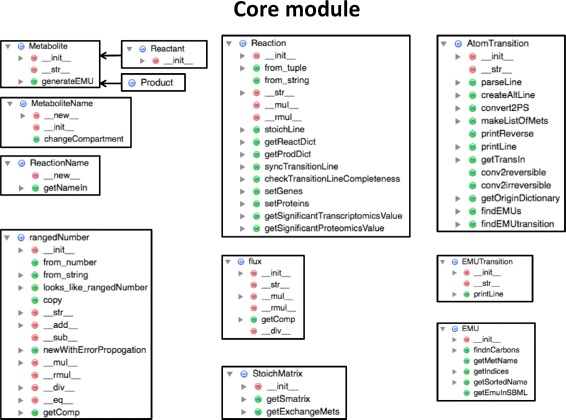
Core module class diagram. The core module contains the core classes for the rest of the library: classes for metabolite, reaction, flux, elementary metabolite units (EMU), atom transitions, emu transitions and stoichiometry matrices
Fig. 3.
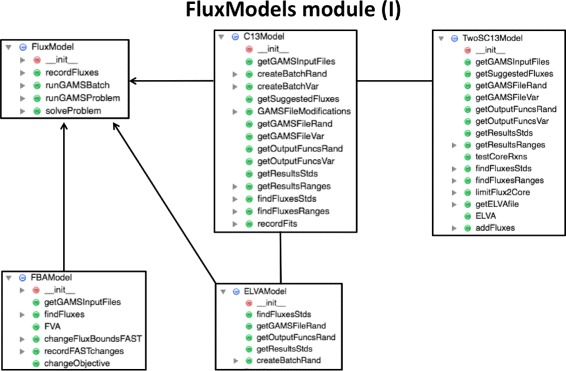
FluxModels module class diagram (part I). The FluxModels module contains classes for the different types of models used for each flux analysis type: FBA, 13C MFA, 2S-13C MFA, ELVA, etc. Arrows indicate derived classes
Fig. 4.
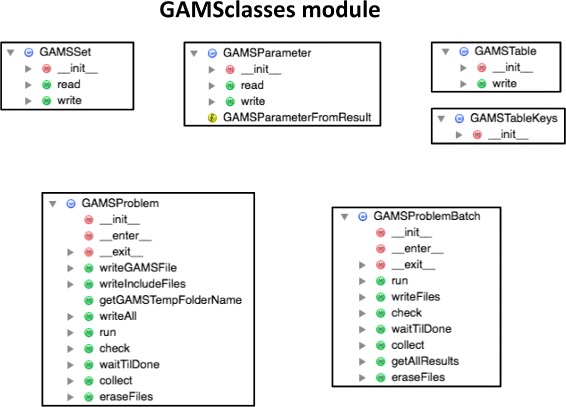
GAMSclasses module class diagram. The GAMSclasses module contains the classes needed to solve the optimization problems through the GAMS algebraic modeling system. Each of the optimization problems is described through a GAMS file, GAMS sets, GAMS parameters or GAMS tables, which are used as input for the GAMSproblem instance (see Fig. 1). GAMS batches are sets of GAMS problems, to be solved in series or parallel
In order to facilitate reproducibility, the lack of which represents one of the main problems afflicting biological research [32], all results and tutorials are provided in the form of Jupyter notebooks [33, 34] (see Table 1 and jupyter.org). This approach is meant to facilitate the reproduction of published results and the creation of new results based on these. The Jupyter notebooks are divided into two types: type A demonstrates the library’s capabilities and type B reproduces published results using code from the jQMM library (Table 1). To further enhance reproducibility we have provided a docker container in the github repository that includes all dependencies. Docker (http://www.docker.com) containers wrap a piece of software in a complete filesystem containing everything needed to run: code, systems libraries, systems tools and anything else that can be installed in a server. This guarantees that it will always run correctly and in the same way, regardless of the system environment it is running in. The jQMM docker container can be run on any computer and any cloud computing service such as Amazon Web Services (AWS), Google Cloud Platform or Microsoft Azure. The container does not include GAMS, CPLEX or CONOPT licences, which must be provided by the user.
Modules
This section contains a succint explanation for each module and some of its classes and capabilities. A more detailed description of each class containing all possible functionality can be found in the Jupyter notebooks (Table 1) and the jQMM code on the github website (see availability below).
Core
The core module contains the classes that constitute the base for all other functionality (Fig. 2). These basic classes include the Reaction and Metabolite classes, along with the derived Product and Reactant classes. These classes are defined as required to describe labeling experiments and, as such, are different from equivalently named classes in COBRApy [26] (defined for FBA purposes). However, we expect in the future to derive these classes from the corresponding COBRApy classes. Other basic classes include the flux class, which is used to describe fluxes for a given reaction (See Jupyter notebook A1).
Core classes that describe concepts exclusively used in 13 C labeling experiments include the atomTransition, emu and emuTransition classes. The atomTransition class is used to hold carbon transition rules (information on the fate for each carbon atom in a reaction [23]), e.g.:
![]()
Note that this is different from the Reaction class, which would only hold information on the reaction name, products and reactants for a reaction:
![]()
By using two classes to differentiate transitions and reactions, we minimize confusion between two related but different concepts. The emu class describes elementary metabolite units (emus, [35]), which are used to efficiently calculate labeling patterns corresponding to a flux profile. Emus represent moieties comprising any distinct subset of a compound’s atoms: for example, if pyruvate consists of 3 carbons, an emu is a subset of any number of these 3 carbons: e.g. pyr_1_2, pyr_1_2_3 or pyr_2_3 are emus. The EMUTransition class describes how emus transition into each other for each reaction, for example for citrate synthase:
![]()
indicates that emus accoa_c_1_2 and oac_c_2 condense into emu cit_c_3_4_5. This initial decomposition of atom transitions into emus and emu transitions is fundamental to solving the forward problem (i.e. determine labeling from fluxes) in 13C MFA, and is provided as part of the core module, as shown in Jupyter notebook A2.
A final core class is the rangedNumber class, which describes floating point numbers with a lower and upper limit, reflecting confidence intervals. These type of numbers are used, for example, to hold information for a flux obtained from 2S- 13 C MFA, where we have a best fit value (0.5), plus upper (0.6) and lower limits (0.3) which represent the upper and lower flux values compatible with the experimental data (see Equation 23 in [23]):
![]()
ReactionNetworks
The ReactionNetworks module (see Additional file 1: Figure S1) contains the classes needed to store three types of reaction networks: stoichiometric (reactionNetwork, used for FBA), carbon transitions (C13reactionNetwork, for 13C MFA) and stochiometric with carbon transitions only for a defined core (TSreactionNetwork, for 2S-13C MFA). The first class contains the methods for manipulating purely stoichiometric reaction networks, such as those used in FBA, the second one contains the methods for dealing with carbon transition networks typically used in 13C MFA and the final class contains the methods used to manipulate networks where genome-scale stoichiometric information is mixed with carbon transitions information (used in 2S-13C MFA). Examples of these types of networks along with tutorials on how to use these classes to manipulate them can be found in Jupyter notebook A3.
FluxModels
The FluxModels module (Fig. 3, and Additional file 1: Figure S2) contains the classes used to solve each of the various types of flux problems solved in this library: FBA (FBAModel), 13C MFA (C13Model), 2S-13C MFA (TwoSC13Model) and ELVA (External Labeling Variability Analysis, ELVAModel, a subproblem of 2S-13C MFA, [23]). Flux Variability Analysis (FVA [36]) and 13C Flux Variability Analysis (13C FVA [23]) can be solved from the same model as for FBA and 13C MFA respectively, since they require the same information. Aditionally, classes for the results obtained from each of these types of models are included, which are used to plot, analyze and manipulate results (FBAResults, FVAResults, C13Results, TSResults, ELVAResults, C13FVAResults and TSFVAResults). More details can be found in Jupyter notebook A4.
EnhancedLists
The enhancedLists module (Additional file 1: Figure S3) holds lists that have been enhanced with methods useful for the objects contained. In this way, there is e.g. a ReactionList class that holds reactions and e.g. has a carbonTransitionsOK() method that tests if carbon transition and reaction information are compatible (same reactants, same products, etc). The types of lists included are ReactionList, MetaboliteList, EMUList, EMUTransitionList and AtomTransitionList. Their use is demonstrated in Jupyter notebook A2.
GAMSclasses
The GAMSclasses module contains the classes used to solve the optimization problems for each model (see below) through the GAMS modeling system (GAMS Development Corporation), either in series or in parallel (Fig. 4). As depicted in Fig. 1, all flux techniques require solving an optimization problem using a different type of solver (either CPLEX or CONOPT in GAMS), which are accessed through the GAMS framework. At this moment, this interaction is encoded in this class, but we expect to replace it soon by use of the GAMS python API. More information and tests can be found in Jupyter notebook A5.
Predictions
This module provides the classes needed to make flux predictions using the Minimization of Metabolic Adjustment (MoMA [36]) and Regulatory on/off minimization (ROOM [37]) methods that were used in the original 2S-13C MFA paper [23]. A demonstration can be found in Jupyter notebook A6.
Labeling
The Labeling module contains all classes needed to analyze, manipulate and plot labeling patterns (a.k.a Mass Distribution Vector, MDV, the fraction of molecules with n =1, 2, 3... labeled carbons) and fragments. The base class labelingData is used to derive LCMSLabelData, CEMSLabelData and GCMSLabelData, which are classes for each type of mass spectroscopy data. The fragment class contains all information for the MS fragment harboring the MDV and is used to derive classes GCMSfragment, LCMSfragment and CEMSfragment. More details can be found in Jupyter notebook A7.
DB
The DB module houses a database of all hard-coded information, conveniently located in a single module. This includes, for example, a dictionary of fragments with all their detailed information: name, abbreviation, formula, number of carbons, formula without carbon backbone, etc.
Utilities
The utilities module stores small functions that are useful in a variety of modules: e.g. a function to extract the atoms numbers from an elemental composition string (′ H6NO2Si ′⇒ [ (′ H ′,6),(′ N ′,1),(′ O ′,2),(′ Si ′,1)]).
Results and discussion
We will now demonstrate the use of the jQMM library for each of the techniques included in the package using data from the literature.
13C Metabolic Flux Analysis for TCA toy model
The first demonstration (Fig. 5) involves the toy model of the TCA cycle originally used by Antoniewicz et al. [35] to showcase the Elementary Metabolite Unit (EMU) method (Fig. 12 in [35]). The toy model is a simplification of the tricarboxylic acid (TCA) cycle that still retains its main features. Acetyl coenzyme A (AcCoA) and aspartate are the two substrates, and glutamate and carbon dioxide are the products. The labeling for glutamate is calculated assuming a mixture of 25% [2-13C]AcCoA and 25% [1,2-13C]AcCoA as the tracer input (with everything else unlabeled), while aspartate is considered unlabeled. Fluxes are fixed to predetermined values, since the goal in this case is to showcase the calculation of labeling patterns from fluxes. Jupyter Notebook B2 demonstrates how, using jQMM, EMU transitions are generated, and solved through GAMS and CONOPT without the need to decompose the network into decoupled EMU reaction networks. The computed labeling pattern is the same as obtained through 13CFLUX2 [29](see Fig. 5, and Jupyter Notebook B2 for more details).
Fig. 5.
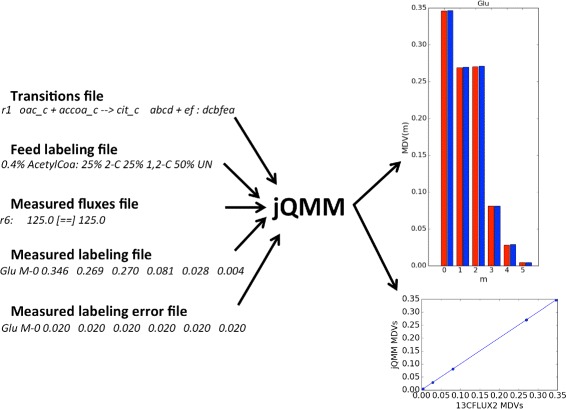
Example jQMM inputs and outputs for TCA toy model. The TCA toy model represents a symplification of the TCA cycle that still retains its main features and is used to test 13C MFA tools [35]. In this case, the input consists of the carbon transitions (the fate of each carbon for each the reaction), the feed labeling information (AcetylCoA 50% unlabeled, 25% labeled in the second carbon, 25% labeled in the first two carbons), the measured labeling patterns (glutamate MDVs), the error in these measurements, and the fluxes which are known (e.g. r6). File line examples are given in cursive below. In this case, all fluxes are assumed to be measured and the goal is to find the corresponding MDV for glutamate, which is given in the top right part of the figure (in red experimentally measured MDV, in blue theoretically calculated MDV). The lower right part of the figure provides a comparison with 13CFLUX2 [29], a well known 13C MFA package (See Jupyter Notebook B2)
13C Metabolic Flux Analysis for E. coli data
The next demonstration of the jQMM library involves a traditional use of 13 C MFA to calculate fluxes using the data from Toya et al. [38], in order to reproduce supplementary figure S18 from Garcia Martin et al. [23]. Fluxes are calculated for wild type E. coli fitting labeling data for 9 different intracellular metabolites. This calculation uses the original small-scale model for central carbon metabolism that lumps several reactions together and displays only an approximate account of fluxes to biomass. For example, as discussed in Garcia Martin et al. [23], the large fluxes draining acetyl-CoA into biomass and to the exterior of the cell as acetate (each a third of the pyruvate dehydrogenase flux, PDH) imposed in the original publication have been overestimated. The typical biomass function used in these minimal 13 C MFA models with reactions lumped together involves a specific stoichiometry of central carbon intermediates converted to biomass. However, this represents an approximation since some of the metabolites required for biomass growth are not present in the minimal network model and need to be substituted by their requirements in terms of intermediates considered in the minimal network (acetyl-CoA, in this case). These effects require significant effort to be accurately incorporated into a small-scale model, but they are properly handled by the genome-scale model, as demonstrated in the next example. Jupyter Notebook B3 shows how to calculate fluxes through 13 C MFA for this case.
2S- 13C Metabolic Flux Analysis for E. coli data
This example calculates fluxes for a full comprehensive genome-scale model [23] constrained by 13C labeling data through the 2S-13C MFA algorithm (Fig. 6). The input is the same as for the 13C MFA case in the previous section, with the addition of the iJR904 genome-scale model [39]. As discussed in depth in Garcia Martin et al. [23], the solution displays nearly the same values as 13C MFA for central metabolism (see e.g. Figure S4 and S18 in [23]). This similarity is expected since 2S-13C MFA is designed to mimic 13C MFA for this part of metabolism (see “Limiting flux to core reactions” section in [23]). The only difference for the Toya et al. data set can be found in the TCA cycle flux. These differences appear because genome-scale models account for fluxes to biomass in a more realistic manner and because they do not rule out unexpected metabolic routes compatible with the available data.
Fig. 6.
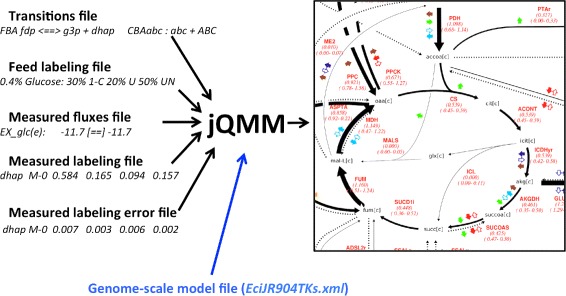
Example jQMM inputs and outputs for E. coli 2S-13C MFA. 2S-13C MFA allows for the calculation of fluxes for genome-scale models constrained by 13C labeling data [23]. The input is the same as for 13C MFA(Fig.5), but with the addition of a genome-scale model. The output consists of fluxes for the genome-scale model, which are visualized in a metabolic map (see Jupyter Notebook B4)
Furthermore, the use of genome-scale models allows for some information that can not be obtained from smaller, non-comprehensive models, such as an account of all reactions producing and consuming e.g. NADPH as constrained by 13C labeling data (Fig. 6 in [23]). Other advantages of using genome-scale models include the prediction of non measured fluxes (Fig. S13 in [23]) and quantitative full predictions of directly measurable data (Fig. 10 in [23]). Jupyter Notebook B4 shows how to reproduce all figures from Garcia Martin et al. [23].
13C Metabolic Flux Analysis for a soil microbial community
In order to show the general capabilities of the jQMM library for non-standard 13C MFA we will show how one can use this library to calculate fluxes for microbial communities as shown in Hagerty et al. [25]. The problem presented and solved in this paper is related to 13C MFA but, at the same time, the technical details differ notably. In this and other experiments [40, 41], Dijkstra et al. collected several soil samples and incubated them with two different carbon sources (glucose and pyruvate), where each source was chosen to have a different initial labeling (1-13C or fully labeled for glucose and 1-13C or 2,3-13C for pyruvate). For each carbon source and labeling type, the labeling fraction of CO2 emmited from the soil was measured. For each carbon source, the two relative CO2 labeling profiles provided enough information to fit a small model of carbon metabolism that represented the combination of reactions for all microbial entities in the soil. The good fits supported the assumption that all members of the community display a similar metabolic flux state, and the results are used to derive the carbon utilization efficiency (i.e. how much carbon is diverted into biomass and how much is lost as CO2, a critical parameter for climate models [42]). Jupyter notebook B6 shows how to do these calculations using modular combinations of the jQMM library without recourse to complicated excel data sheets [25].
Flux balance analysis
The jQMM library provides the means to perform FBA (Jupyter notebook B1) and other COBRA methods for the sake of comparison with 13C-based methods [23]. However, the main purpose of jQMM is not to provide a COBRA library, since actively developed open source python libraries such as COBRApy [26] and cameo (http://cameo.bio/) are available for this purpose. Methods for FBA, FVA, Minimization of Metabolic Adjustment (MoMA [36]) and Regulatory on/off minimization (ROOM [37]) are provided. In order to showcase these capabilities we replicated part of the tutorial from the COBRA v 1.0 package in Becker et al. [43], which can be found in Jupyter notebook B1. Future versions of the library will likely be changed to use cameo for this functionality.
Principal Component Analysis of Proteomics (PCAP)
While most of the jQMM library is focused on flux analysis, we have found it useful to statistically model -omics data without the recourse to mechanistic models [24], an approach which we expect to be increasingly popular in the coming years given the increasing availability of -omics data. In Jupyter notebook B5, we show how to use the jQMM library and quantitative proteomics data to increase biofuel yields by replicating the PCAP (Principal Component Analysis for Proteomics) analysis with a recreation of previously published figures [24] (Fig. 7).
Fig. 7.

Example jQMM inputs and outputs for Principal Component Analysis of Proteomics (PCAP). PCAP is a method developed to leverage proteomics data in order to improve bioproduct yield. The inputs are measured proteomic profiles and bioproduct production levels and the output consists of a plot that can be used to increase yields as shown in Alonso-Gutierrez et al. [24]. (see Jupyter Notebook B5). File line examples are provided in cursive below
This case represents a prototypical example of pathway engineering, in which a series of exogenous genes from a variety of organisms (E. coli, S. cerevisiae, S. aureus, and plants such as M. spicata) were assembled together in a pathway able to produce limonene [44], a jet-fuel [45]. However, when assembling this pathway together, there was no easy way to figure out the design parameters for optimal function: what promoter strength to use, when to induce? how much? In this case, a combinatorial approach was used, using promoters of different strength (weak, medium, strong), different induction strengths (low, medium, high) and induction times (low, medium, high). For each of these scenarios, limonene production and protein expression for all genes in the pathway were measured. The raw proteomics data did not provide clear trends but the use of a basic data analysis tool such as a Principal Component Analysis (PCA) produced actionable results which allowed for the improvement of limonene production by 40% and bisabolene production by 200% leading to the highest producing strain of this biodiesel replacement to that date [24]. Jupyter notebook B5 shows how to perform this analysis.
Conclusion
We have presented here the jQMM library, an open-source modular library for modeling metabolism and the first open-source library for 13C MFA in Python. The jQMM library is also the first one to provide a method to calculate fluxes for genome-scale models constrained by 13C labeling data through 2S-13C MFA, and is the first to unify the FBA, 13C MFA and 2S-13C MFA in a single package. Unlike METRAN [27], INCA [28] and 13CFLUX2 [29]), it is open source and, unlike OPENFLUX [30] and OPENFLUX2 [31], it is written in Python. Furthermore, it includes an example on how to calculate fluxes for microbial communities through 13C MFA, and the tools to leverage other types of -omics data (e.g. proteomics for biofuel production).
While the examples here are mainly focused on various types of flux analysis (FBA, 13C MFA, 2S-13C MFA, etc) and biofuels, the tools are of general aplicability to other fields (e.g. pharma, biomedicine, environmental microbiology) and include statistical methods as well (e.g. PCAP). jQMM presents not only methods for modeling and generating actionable items form -omics data, but also libraries for data manipulation and visualization (fluxes and labeling patterns see Figs. 6 and 5), including classes for most common concepts in metabolism (e.g. reactions, metabolites, GC-MS fragments, reaction networks) in order to facilitate their manipulation, and which are of utility beyond the examples presented here. Every module in jQMM is explained in detail in Jupyter notebooks that allow for immediate reproducibility. Python unit tests are included for all modules for ease of testing when adding functionality.
jQMM will continue growing as the methods developed and used at JBEI for biofuel production expand and mature, while other authors contribute to it. At the moment jQMM is heavily dependent on the closed-source commercial platform GAMS and the CPLEX and CONOPT solvers. Our first future improvement to the jQMM library is planned to involve providing an interface to open-source and free version of solvers equivalent to GAMS. Further future improvements will likely include integration with COBRApy [26], cameo (cameo.bio) or KBase (kbase.us).
Availability of data and materials
Project name: jQMM library
Project home page: https://github.com/JBEI/jqmm
Operating systems: Platform independent
Programming language: Python, GAMS
Other requirements: CONOPT large scale nonlinear solver
All data used in this paper can be found in the github library itself. Additional file 1 contains a mathematical description of the algorithms, and Figures S1-S3.
Acknowledgements
Not applicable.
Funding
This work was part of the DOE Joint BioEnergy Institute (http://www.jbei.org) supported by the U. S. Department of Energy, Office of Science, Office of Biological and Environmental Research, and was part of the Agile BioFoundry (http://agilebiofoundry.org) supported by the U.S. Department of Energy, Energy Efficiency and Renewable Energy, Bioenergy Technologies Office, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the U. S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. This research is also supported by the Basque Government through the BERC 2014-2017 program and by Spanish Ministry of Economy and Competitiveness MINECO: BCAM Severo Ochoa excellence accreditation SEV-2013-0323.
Authors’ contributions
HGM, GWB, AG, VSK, DW and DA wrote the code and analyzed the data. HGM, GWB, DA, AA and JDK wrote the paper. TWHB provided the docker containers. All authors have read and approve of the final version of the manuscript.
Competing interests
JDK has financial interests in Amyris and Lygos. All other authors have no competing interests to declare.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- 13C MFA
13C Metabolic Flux Analysis
- 2S-13C MFA
Two-scale 13C Metabolic Flux Analysis
- AcCoa
Acetyl-CoA
- API
Application Program Interface
- COBRA
COnstraint Based Reconstruction and Analysis
- ELVA
External Labeling Variability Analysis
- EMU
Elementary Metabolite Unit
- FBA
Flux Balance Analysis
- FVA
Flux Variability Analysis
- JBEI
Joint BioEnergy Institute
- jQMM
JBEI Quantitative Metabolic Modeling
- MDV
Mass Distribution Vector
- MoMA
Minimum of Metabolic Adjustment
- NADPH
Nicotinamide adenine dinucleotide phosphate
- PDH
Pyruvate Dehydrogenase
- PCAP
Principal Component Analysis of Proteomics
- ROOM
Regulatory On/Off Minimization
- TCA
trycarboxylic acid
Additional file
Supplementary file containing the mathematical description of algorithms, and supplementary figures S1-S3. (PDF 2250 kb)
Contributor Information
Garrett W. Birkel, Email: gwbirkel@lbl.gov
Amit Ghosh, Email: amitghosh@lbl.gov.
Vinay S. Kumar, Email: satvinay@gmail.com
Daniel Weaver, Email: weaver@ai.sri.com.
David Ando, Email: david.ando@lbl.gov.
Tyler W. H. Backman, Email: TBackman@lbl.gov
Adam P. Arkin, Email: aparkin@lbl.gov
Jay D. Keasling, Email: jdkeasling@lbl.gov
Héctor García Martín, Email: hgmartin@lbl.gov.
References
- 1.Nelson DL, Lehninger AL, Cox MM. Lehninger Principles of Biochemistry. New York: W. H. Freeman and Company; 2008. [Google Scholar]
- 2.Xu F, Tavintharan S, Sum CF, Woon K, Lim SC, Ong CN. Metabolic signature shift in type 2 diabetes mellitus revealed by mass spectrometry-based metabolomics. J Clin Endocrinol Metab. 2013; 98(6). doi:10.1210/jc.2012-4132. [DOI] [PubMed]
- 3.Cummins TD, Holden CR, Sansbury BE, Gibb AA, Shah J, Zafar N, Tang Y, Hellmann J, Rai SN, Spite M, Bhatnagar A, Hill BG. Metabolic remodeling of white adipose tissue in obesity, Am J Physiol Endocrinol Metab. 2014;307(3):262–77. doi: 10.1152/ajpendo.00271.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu L, Gomes A, Sinclair D. Geroncogenesis: Metabolic changes during aging as a driver of tumorigenesis. 2014. NIHMS150003. doi:10.1016/j.ccr.2013.12.005. [DOI] [PMC free article] [PubMed]
- 5.Chubukov V, Mukhopadhyay A, Petzold CJ, Keasling JD, Martín HG. Synthetic and systems biology for microbial production of commodity chemicals. npj Syst Biol Appl. 2016;2:16009. doi: 10.1038/npjsba.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lorek S, Spangenberg JH. Sustainable consumption within a sustainable economy – beyond green growth and green economies. J Clean Prod. 2014;63:33–44. doi: 10.1016/j.jclepro.2013.08.045. [DOI] [Google Scholar]
- 7.Nielsen J, Keasling JD. Engineering cellular metabolism. Cell. 2016;164(6):1185–97. doi: 10.1016/j.cell.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 8.Lee SY, Lee DY, Kim TY. Systems biotechnology for strain improvement. Trends Biotechnol. 2005;23(7):349–58. doi: 10.1016/j.tibtech.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Han MJ, Yoon SS, Lee SY. Proteome analysis of metabolically engineeredescherichia coli producing poly (3-hydroxybutyrate) J Bacteriol. 2001;183(1):301–8. doi: 10.1128/JB.183.1.301-308.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.George KW, Chen A, Jain A, Batth TS, Baidoo EE, Wang G, Adams PD, Petzold CJ, Keasling JD, Lee TS. Correlation analysis of targeted proteins and metabolites to assess and engineer microbial isopentenol production. Biotech Bioeng. 2014;111(8):1648–58. doi: 10.1002/bit.25226. [DOI] [PubMed] [Google Scholar]
- 11.Palsson B, Zengler K. The challenges of integrating multi-omic data sets. Nat Chem Biol. 2010;6(11):787–9. doi: 10.1038/nchembio.462. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen J, Fussenegger M, Keasling J, Lee SY, Liao JC, Prather K, Palsson B. Engineering synergy in biotechnology. Nat Chem Biol. 2014;10(5):319–22. doi: 10.1038/nchembio.1519. [DOI] [PubMed] [Google Scholar]
- 13.Javidpour P, Pereira JH, Goh EB, McAndrew RP, Ma SM, Friedland GD, Keasling JD, Chhabra SR, Adams PD, Beller HR. Biochemical and structural studies of nadh-dependent fabg used to increase the bacterial production of fatty acids under anaerobic conditions. Appl Environ Microbiol. 2014;80(2):497–505. doi: 10.1128/AEM.03194-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhan N, Xu P, Khalidi O, Koffas MA. Redirecting carbon flux into malonyl-coa to improve resveratrol titers: proof of concept for genetic interventions predicted by optforce computational framework. Chem Eng Sci. 2013;103:109–14. doi: 10.1016/j.ces.2012.10.009. [DOI] [Google Scholar]
- 15.Lee KH, Park JH, Kim TY, Kim HU, Lee SY. Systems metabolic engineering of escherichia coli for l-threonine production. Mol Syst Biol. 2007;3(1):149. doi: 10.1038/msb4100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wetterstrand KA.DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). 2016. http://www.genome.gov/sequencingcosts. Accessed 05 Feb 2016.
- 17.Batth TS, Singh P, Ramakrishnan VR, Sousa MML, Chan LJG, Tran HM, Luning EG, Pan EHY, Vuu KM, Keasling JD, Adams PD, Petzold CJ. A targeted proteomics toolkit for high-throughput absolute quantification of Escherichia coli proteins. Metab Eng. 2014;26C:48–56. doi: 10.1016/j.ymben.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 18.Fuhrer T, Zamboni N. High-throughput discovery metabolomics. Curr Opin Biotechnol. 2015;31:73–8. doi: 10.1016/j.copbio.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Blanch HW, Adams PD, Andrews-Cramer KM, Frommer WB, Simmons BA, Keasling JD. Addressing the need for alternative transportation fuels: the Joint BioEnergy Institute. ACS Chem Biol. 2008;3(1):17–20. doi: 10.1021/cb700267s. [DOI] [PubMed] [Google Scholar]
- 20.Beller HR, Lee TS, Katz L. Natural products as biofuels and bio-based chemicals: fatty acids and isoprenoids. Nat Prod Rep. 2015;32.10:1508–26. doi: 10.1039/C5NP00068H. [DOI] [PubMed] [Google Scholar]
- 21.Wiechert W. 13C metabolic flux analysis. Metab Eng. 2001;3(3):195–206. doi: 10.1006/mben.2001.0187. [DOI] [PubMed] [Google Scholar]
- 22.Lewis NE, Nagarajan H, Palsson BO. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat Rev Microbiol. 2012;10(4):291–305. doi: 10.1038/nrmicro2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia Martin H, Kumar VS, Weaver D, Ghosh A, Chubukov V, Mukhopadhyay A, et al. A Method to Constrain Genome-Scale Models with 13C Labeling Data. PLoS Comput Biol. 2015;11(9):e1004363. doi: 10.1371/journal.pcbi.1004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alonso-Gutierrez J, Kim EM, Batth TS, Cho N, Hu Q, Chan LJG, Petzold CJ, Hillson NJ, Adams PD, Keasling JD, Garcia-Martin H, Soon Lee T. Principal component analysis of proteomics (PCAP) as a tool to direct metabolic engineering. Metab Eng. 2014;28:123–33. doi: 10.1016/j.ymben.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 25.Hagerty SB, van Groenigen KJ, Allison SD, Hungate BA, Schwartz E, Koch GW, Kolka RK, Dijkstra P. Accelerated microbial turnover but constant growth efficiency with warming in soil. Nat Clim Chang. 2014;4(10):903–6. doi: 10.1038/nclimate2361. [DOI] [Google Scholar]
- 26.Ebrahim A, Lerman JA, Palsson BO, Hyduke DR. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst Biol. 2013;7(1):74. doi: 10.1186/1752-0509-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crown SB, Ahn WS, Antoniewicz MR. Rational design of 13C-labeling experiments for metabolic flux analysis in mammalian cells. BMC Syst Biol. 2012;6(1):43. doi: 10.1186/1752-0509-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Young JD. INCA: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinforma (Oxford, England) 2014;30(9):1333–5. doi: 10.1093/bioinformatics/btu015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weitzel M, et al. 13CFLUX2Ůhigh-performance software suite for 13C-metabolic flux analysis. Bioinformatics. 2013;29.1:143–5. doi: 10.1093/bioinformatics/bts646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quek LE, Wittmann C, Nielsen LK, Krömer JO. OpenFLUX: efficient modelling software for 13C-based metabolic flux analysis. Microb Cell Factories. 2009;8(1):25. doi: 10.1186/1475-2859-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shupletsov MS, Golubeva LI, Rubina SS, Podvyaznikov DA, Iwatani S, Mashko SV. OpenFLUX2: 13 C-MFA modeling software package adjusted for the comprehensive analysis of single and parallel labeling experiments. Microb Cell Factories. 2014;13(1):152. doi: 10.1186/s12934-014-0152-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plant AL, Locascio LE, May WE, Gallagher PD. Improved reproducibility by assuring confidence in measurements in biomedical research. Nat Methods. 2014;11(9):895–8. doi: 10.1038/nmeth.3076. [DOI] [PubMed] [Google Scholar]
- 33.Wilson G, Aruliah DA, Brown CT, Chue Hong NP, Davis M, Guy RT, Haddock SHD, Huff KD, Mitchell IM, Plumbley MD, Waugh B, White EP, Wilson P. Best Practices for Scientific Computing. PLoS Bio. 2014; 12(1). http://arxiv.org/abs/arXiv:1210.0530v3. [DOI] [PMC free article] [PubMed]
- 34.Pérez F, Granger BE. IPython: a system for interactive scientific computing. Comput Sci Eng. 2007;9(3):21–9. doi: 10.1109/MCSE.2007.53. [DOI] [Google Scholar]
- 35.Antoniewicz MR, Kelleher JK, Stephanopoulos G. Elementary metabolite units (EMU): A novel framework for modeling isotopic distributions. Metab Eng. 2007;9(1):68–86. doi: 10.1016/j.ymben.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segrè D, Vitkup D, Church GM. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci U S A. 2002;99(23):15112–7. doi: 10.1073/pnas.232349399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shlomi T, Berkman O, Ruppin E. Regulatory on/off minimization of metabolic flux changes after genetic perturbations, Proc Natl Acad Sci U S A. 2005;102(21):7695–00. doi: 10.1073/pnas.0406346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toya Y, Ishii N, Nakahigashi K, Hirasawa T, Soga T, Tomita M, Shimizu K. 13C-Metabolic flux analysis for batch culture of Escherichia coli and its pyk and pgi gene knockout mutants based on mass isotopomer distribution of intracellular metabolites. Biotechnol Prog. 2010;26(4):975–92. doi: 10.1002/btpr.420. [DOI] [PubMed] [Google Scholar]
- 39.Reed JL, Vo TD, Schilling CH, Palsson BO. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR) Genome Biol. 2003;4(9):54. doi: 10.1186/gb-2003-4-9-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dijkstra P, Dalder JJ, Selmants PC, Hart SC, Koch GW, Schwartz E, Hungate BA. Modeling soil metabolic processes using isotopologue pairs of position-specific 13C-labeled glucose and pyruvate. Soil Biol Biochem. 2011;43(9):1848–57. doi: 10.1016/j.soilbio.2011.05.001. [DOI] [Google Scholar]
- 41.Dijkstra P, Thomas SC, Heinrich PL, Koch GW, Schwartz E, Hungate BA. Effect of temperature on metabolic activity of intact microbial communities: Evidence for altered metabolic pathway activity but not for increased maintenance respiration and reduced carbon use efficiency. Soil Biol Biochem. 2011;43(10):2023–31. doi: 10.1016/j.soilbio.2011.05.018. [DOI] [Google Scholar]
- 42.Cox PM, Betts RA, Jones CD, Spall SA, Totterdell IJ. Acceleration of global warming due to carbon-cycle feedbacks in a coupled climate model. Nature. 2000;408(6809):184–7. doi: 10.1038/35041539. [DOI] [PubMed] [Google Scholar]
- 43.Becker SA, Feist AM, Mo ML, Hannum G, Palsson BO, Herrgard MJ. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat Protoc. 2007;2(3):727–38. doi: 10.1038/nprot.2007.99. [DOI] [PubMed] [Google Scholar]
- 44.Alonso-Gutierrez J, Chan R, Batth TS, Adams PD, Keasling JD, Petzold CJ, Lee TS. Metabolic engineering of Escherichia coli for limonene and perillyl alcohol production. Metab Eng. 2013;19:33–41. doi: 10.1016/j.ymben.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Chuck CJ, Donnelly J. The compatibility of potential bioderived fuels with Jet A-1 aviation kerosene. Appl Energy. 2014;118:83–91. doi: 10.1016/j.apenergy.2013.12.019. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Project name: jQMM library
Project home page: https://github.com/JBEI/jqmm
Operating systems: Platform independent
Programming language: Python, GAMS
Other requirements: CONOPT large scale nonlinear solver
All data used in this paper can be found in the github library itself. Additional file 1 contains a mathematical description of the algorithms, and Figures S1-S3.