

Abstract
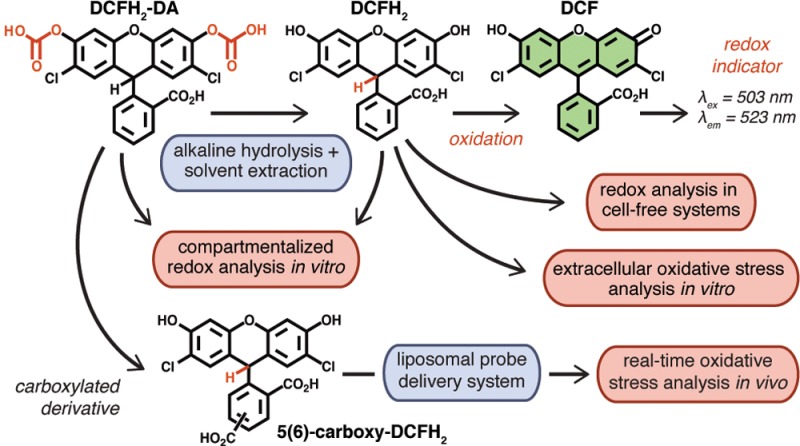
Oxidative stress, a state in which intra- or extracellular oxidant production outweighs the antioxidative capacity, lies at the basis of many diseases. DCFH2-DA (2′,7′-dichlorodihydrofluorescein diacetate) is the most widely used fluorogenic probe for the detection of general oxidative stress. However, the use of DCFH2-DA, as many other fluorogenic redox probes, is mainly confined to the detection of intracellular oxidative stress in vitro. To expand the applicability of the probe, an alkaline hydrolysis and solvent extraction procedure was developed to generate high-purity DCFH2 (2′,7′-dichlorodihydrofluorescein) from DCFH2-DA using basic laboratory equipment. Next, the utility of DCFH2 was exemplified in a variety of cell-free and in vitro redox assay systems, including oxidant production by transition metals, photodynamic therapy, activated macrophages, and platelets, as well as the antioxidative capacity of different antioxidants. In cells, the concomitant use of DCFH2-DA and DCFH2 enabled the measurement and compartmentalized analysis of intra- and extracellularly produced oxidants, respectively, using a single read-out parameter. Furthermore, hepatocyte-targeted liposomes were developed to deliver the carboxylated derivative, 5(6)-carboxy-DCFH2, to hepatocytes in vivo. Liposome-delivered 5(6)-carboxy-DCFH2 enabled real-time visualization and measurement of hepatocellular oxidant production during liver ischemia-reperfusion. The liposomal 5(6)-carboxy-DCFH2 can be targeted to other tissues where oxidative stress is important, including cancer.
Oxidants in the form of reactive oxygen or nitrogen species (ROS or RNS), transition metals, and peroxidases are extensively studied because of their involvement in numerous health and disease states. To this end, redox-sensitive fluorogenic probes are commonly employed because of their low cost, relative nontoxicity, and easy application.1,2 Of the commercially available probes, 2′,7′-dichlorodihydrofluorescein diacetate (DCFH2-DA) is the most widely used. Following diffusion across the plasma membrane, esterases cleave the acetate groups to generate the more hydrophilic 2′,7′-dichlorodihydrofluorescein (DCFH2) that is retained in the cytosol. Oxidation of DCFH2 yields the highly fluorescent 2′,7′-dichlorofluorescein (DCF, λex = 503 nm, λem = 523 nm) that can be measured spectrofluorometrically (Figure 1A). DCFH2 reacts with a wide array of oxidants, albeit at different reaction constants.2,3 The probe is therefore an ideal indicator of redox shifts and general oxidative stress.
Figure 1.

(A) Structure of 2′,7′-dichlorodihydrofluorescein diacetate (DCFH2-DA, nonfluorescent), its deacetylated derivative 2′,7′-dichlorodihydrofluorescein (DCFH2, nonfluorescent), and its oxidized end-product 2′,7′-dichlorofluorescein (DCF, fluorescent). (B) Standard operating procedure for the preparation and purification of DCFH2 (detailed in the Supporting Information, section S–VIII).
Because of its hydrophilicity and consequent membrane impermeability, DCFH2 holds unique benefits over DCFH2-DA in terms of detecting extracellularly formed oxidants as well as its potential for targeted delivery, for example, through encapsulation in liposomes. DCFH2 can be prepared from its parent compound DCFH2-DA though alkaline hydrolysis. Methods for the preparation of DCFH2 from DCFH2-DA have been described by others,3−8 but the reaction conditions have never been optimized and the end-product has never been completely characterized. Moreover, DCFH2 prepared through these methods needed to be used immediately due to its limited stability in aqueous solvent and contained residual salts from the hydrolysis step, rendering it unsuitable for some assays. To overcome these limitations, a simple and fast alkaline hydrolysis and liquid two-phase extraction method was developed and optimized (Figure 1B).
The method presented here yields high purity, stable DCFH2 that can be dissolved in different organic solvents depending on its intended use. As a result, the probe has a long shelf life and can be employed in multifarious assays, as is exemplified below. The preparation procedure takes less than 2 h and can be performed in any laboratory with standard reagents and equipment at minimal cost. Moreover, the method can be applied to prepare the more hydrophilic 5(6)-carboxy-DCFH2 (CDCFH2) from its commercially available diacetylated precursor. CDCFH2 can be used in liposomal formulations for selective delivery of the probe to target tissues, such as the liver or solid tumors, to intravitally measure oxidative stress in vivo. For these purposes, a selectively hepatotargeted liposomal probe delivery system was developed to visualize and quantify hepatocellular oxidant formation. The proof-of-concept is provided here for hepatic ischemia-reperfusion.
Experimental Section
A concise description of the preparation of DCFH2 from DCFH2-DA is provided here. The standard operating procedure as well as all other materials and methods are detailed in the Supporting Information, indicated by the prefix “S–”. The standard operating procedure is described in section S–VIII.
Step 1: Deacetylation of DCFH2-DA
DCFH2 was prepared from DCFH2-DA by alkaline hydrolysis of the acetate groups (Figure 1A). The deacetylation of DCFH2-DA at increasing concentrations of NaOH was assessed by absorption spectroscopy (S–II) and thin layer chromatography (S–III). From these experiments, it was concluded that the optimal NaOH concentration for DCFH2-DA deacetylation was 0.1 M.
Step 2: Purification of DCFH2
Following DCFH2-DA deacetylation in aqueous NaOH, DCFH2 was purified by liquid phase extraction (Figure 1B). An equivalent volume of 0.2 M aqueous HCl was added under continuous vortexing to precipitate DCFH2. The precipitated DCFH2 was subsequently washed thrice (2000 × g, 15 min, 4 °C) in ice-cold acidified Milli-Q (pH = 1) to remove excess salt. Chloroform was then added under continuous vortexing to extract DCFH2 from the aqueous phase. Following phase separation, the aqueous phase was removed and all chloroform was evaporated under a continuous stream of nitrogen gas. The DCFH2 pellet was dissolved in dimethyl sulfoxide or methanol and stored under a nitrogen atmosphere at −20 °C in the dark. It is imperative that DCFH2 is shielded from light during all steps, as DCFH2 auto-oxidizes upon light exposure.
Results and Discussion
The most important features of DCFH2 prepared through the method presented here are, first, a purity of ≥95% (S–V) at a 54.2% yield (S–IV), which can be increased to ∼90% by omitting the washing steps. No chloride from the washing step was detectable in the methanol stock (S–VII), implying that other ions are likely also absent when precipitated DCFH2 is directly extracted into chloroform. Second, the mean ± SEM molar extinction coefficient of DCFH2 in methanol, which can be used to spectrophotometrically determine stock concentrations, is 7.6 ± 1.5 × 103 M–1·cm–1 at the absorption maximum of 287 nm (S–IX). The absorption maximum is comparable in nonbuffered water and neutral HEPES buffer, but undergoes a bathochromic shift in aqueous alkaline solvent (S–X). Third, the DCFH2 stock solution is stable in methanol and dimethyl sulfoxide for at least 4 wk in the dark (S–XI), but stock solutions stored up to 20 months have been used without indication of decay (i.e., formation of DCF). Finally, DCFH2 is oxidized to DCF in the presence of oxidants, which was demonstrated in a variety of cell-free, cell-based, and in vivo assays.
In the cell-free assays, DCFH2 was employed to spectrofluorometrically measure the redox catalytic activity of the transition metals Mn2+, Fe2+, Cu+, and Zn2+ in neutral HEPES buffer (Figure 2A) and pH-neutralized Milli-Q (Figure S11) at ambient O2 tension. Iron and copper are clinically relevant since both play a role in oxidative stress-mediated diseases such as hemochromatosis9 and Alzheimer’s disease.10 Fe2+, for instance, is presumed to bridge between organic molecules (e.g., DNA, proteins, or DCFH2) and O2, and facilitate O2-mediated oxidation of the organic compound involved.11 Fe2+ can moreover reduce O2 to O2•–, thereby further enhancing oxidant formation.11 DCFH2 oxidation by Fe2+ occurred at a faster rate than by Cu+ and was slow and absent in the presence of Mn2+ and Zn2+, respectively.
Figure 2.
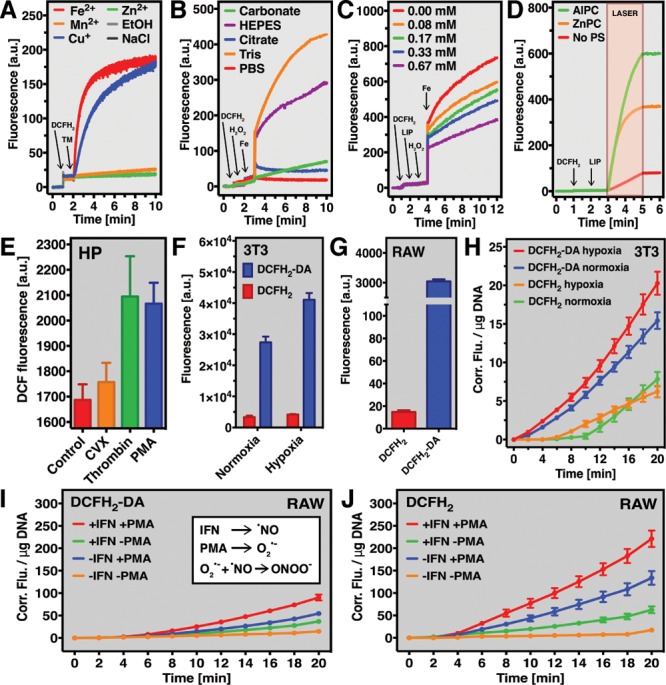
DCFH2 in cell-free and cell-based redox assays. (A) Transition metal (TM) ion-catalyzed DCFH2 oxidation in HEPES buffer over time. (B) Fenton reaction-mediated oxidation of DCFH2 in different buffer solutions. The oxidant-scavenging potential of antioxidant liposomes (LIP) under Fenton reaction conditions measured using DCFH2 is detailed in (C). (D) Oxidant-generating potential of photosensitizer-encapsulating (AlPC or ZnPC) liposomes (LIP) upon laser-light irradiation, which was analyzed using DCFH2. Generation of DCF by resting (control) and activated human platelets (HP) is presented in (E). The extracellular localization of DCFH2 was confirmed in oxidant-producing 3T3 fibroblasts (F) and RAW 264.7 macrophages (G) using flow cytometry. (H) Generation of DCF from DCFH2 or DCFH2-DA by 3T3 fibroblasts subjected to hypoxia/reoxygenation. DCF formation from DCFH2-DA (I) or DCFH2 (J) by RAW 264.7 macrophages stimulated with IFN-γ and/or PMA. The inset in Panel I shows which oxidants are likely involved.
Redox reactions are affected by pH and are therefore frequently assayed in buffered systems to mimic physiological conditions. However, the type of buffer used in redox assays is critical for experimental outcome. In a standardized Fenton system (Fe2+ + H2O2 → Fe3+ + •OH + OH–), which is part of the Haber-Weiss reaction,12 redox reactions proceed best in Tris- and HEPES-buffered solutions, indicating that these are preferred buffers for similar experiments. DCFH2 oxidation is impaired in solutions buffered with carbonate, citrate, and PBS (Figure 2B). These findings are in accordance with data on the rate of Fe2+ auto-oxidation in different buffer systems.13
The probe can also be used to determine the antioxidative and pro-oxidative properties of pharmaceutical preparations. In case of the former, PEGylated DSPC liposomes containing the antioxidants γ-tocopherol, α-tocopherol, curcumin, melatonin, and α-lipoic acid effectively scavenged oxidants in a DCFH2-probed Fenton system (Figure 2C). DCFH2 was further instrumental in measuring the oxidant-generating potential of illuminated photosensitizer-containing liposomes used for photodynamic therapy of cancer.14 Both assays allow comparative analysis of outcome parameters to, for example, determine which antioxidant or pro-oxidant formulation is optimal for its intended purpose.
The formation of oxidants in cells is part of regular metabolism and biochemical signaling that, under pathological conditions, can become disproportional and lead to oxidative stress and ultimately cell death. Platelets, for example, execute specific functions through ROS (O2•–, H2O2, •OH) and RNS (•NO).15 DCFH2 was used to analyze extracellular oxidant formation platelets activated by agonists that bind different platelet receptors (Figure 2E). Incubation of (activated) platelets in the presence of DCFH2 revealed that the native platelet agonist thrombin induced extracellular oxidant formation at an equal propensity as the intracellular O2•– generator PMA (phorbol 12-myristate 13-acetate), suggesting that the PMA-induced O2•– (or derivate oxidants) diffused out of the platelets. Snake venom-derived convulxin triggered minimal DCFH2 oxidation, altogether in support of previous reports.16
The proposition that hydrophilic DCFH2 does not transgress the plasma membrane was confirmed in resting as well as oxidant-producing 3T3 fibroblasts (Figure 2F) and RAW 264.7 macrophages (Figure 2G) using flow cytometry. Consequently, DCFH2 and DCFH2-DA may be used complementarily to discern between intracellular and extracellular oxidant formation in these cell types. The simultaneous application of both probes unveiled that oxidants were predominantly formed intracellularly in 3T3 fibroblasts under normoxic conditions (Figure 2H, blue vs green line). Whereas intracellular oxidant formation increased when cells were subjected to hypoxia/reoxygenation (red vs blue line), extracellular oxidant formation remained unchanged under these conditions (orange vs green line). These data indicate that cytosolic oxidative stress occurs during hypoxia/reoxygenation, presumably as a secondary effect of mitochondrial oxidant formation.17
In RAW 264.7 macrophages, oxidant formation was most abundant in cells stimulated with interferon gamma (IFN-γ) and PMA as a result of the formation of •NO (Figure S12) and O2•–, respectively (Figure 2I,J). Oxidative stress was more pronounced in the extracellular compartment, most likely due to the localization of NOX2-dependent O2•– formation (i.e., at the plasma membrane) in combination with the long half-life and membrane-transgressing properties of iNOS-derived •NO. Accordingly, inhibition of iNOS and NOX2 by L-NAME (Nω-nitro-l-arginine methyl ester hydrochloride) and diphenyleneiodonium (DPI), respectively, significantly hampered DCFH2 oxidation (Figure S13). Reduced iNOS activity resulted in a ∼50% decrease in DCF formation compared to •NO- and O2•–-producing activated cells (Figure S13), whereas NOX2 inactivation reduced DCFH2 oxidation to a level that was comparable to resting cells (i.e., ∼97% decrease). These findings pinpoint cellular O2•– formation as a crucial factor for the extracellular formation of oxidants by activated RAW 264.7 macrophages, either through its reaction with •NO (Figure 2I, inset) or via its dismutation into H2O2. Nevertheless, since DPI inhibits mitochondrial O2•– formation as well,18 sources other than NOX2 could also contribute to the extracellular formation of oxidants by activated RAW 264.7 macrophages.
Lastly, intracellular oxidative stress was measured in vivo using liposome-encapsulated CDCFH2, a more hydrophilic form of DCFH2 that is oxidized to the highly fluorescent CDCF (λex = 495 nm, λem = 525 nm) by oxidants.19 The alkaline hydrolysis and two-phase liquid extraction method was used to prepare CDCFH2 from CDCFH2-DA. Next, CDCFH2 was encapsulated in hepatotargeted liposomes. The liposomes, composed of DPPC, cholesterol, lactosyl-phosphatidylethanolamine (LPE) and monosialotetrahexosylganglioside (GM1) in varying molar ratios, were optimized in vitro (S–XVI through S–XIX) and investigated in vivo in terms of intrahepatic accumulation (S–XX and S–XXI) and distribution to hepatocytes and nonparenchymal cells (Figure 3A). CDCFH2 was subsequently encapsulated into the optimal formulation for hepatocellular targeting (i.e., DPPC:cholesterol:GM1 in a 55:40:5 molar ratio) to visualize oxidative stress in vivo. Hepatocellular oxidative stress was induced using a standardized mouse model of hepatic ischemia-reperfusion injury,20 which is associated with extensive oxidative stress in the reperfusion phase. Accordingly, hepatocellular CDCF formation was observed in the ischemia-reperfusion group but not in sham-operated control animals (Figure 3B–D). These data are the first to show selective hepatocellular oxidant formation during early reperfusion. Proof-of-concept was provided only in the liver, where redox perturbations lie at the basis of numerous disorders. Nevertheless, the applicability of the method is expandable to other redox-pertinent tissues to which liposomes can be targeted, such as solid tumors21 and atherosclerotic plaques.22
Figure 3.
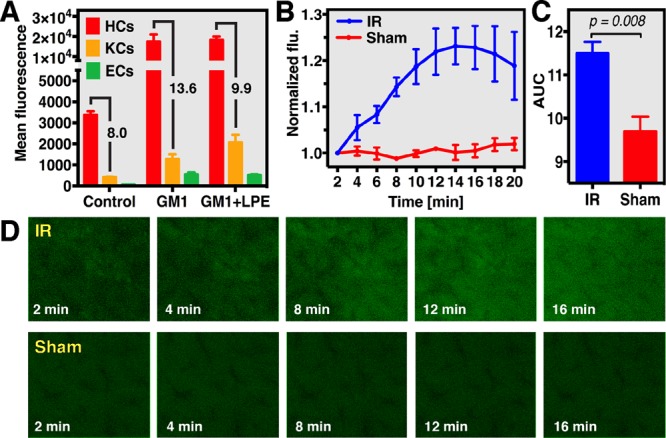
Real-time analysis of hepatocellular oxidative stress using liposome-delivered CDCFH2 during hepatic ischemia-reperfusion (IR) and sham operation in mouse livers. (A) Uptake of NBD-labeled GM1 and GM1 + lactosyl-PE (LPE) liposomes by hepatocytes (HCs), Kupffer cells (KCs), and endothelial cells (ECs), which was analyzed using flow cytometry. Oxidant formation during IR was analyzed by intravital fluorescence (flu) microscopy (D) and spectroscopy (B) in a standardized mouse model of liver IR (60 min ischemia) using CDCFH2-encapsulating GM1 liposomes. (C) Cumulative fluorescence formation during 20 min reperfusion, which was significantly higher in the IR group compared to sham controls.
As with all experimental methods for the detection of oxidative stress, there are aspects that exact attention when implementing (C)DCFH2 in any experimental protocol. Formation of O2•– as a byproduct of DCFH2 oxidation as well as the presumed pH-dependence of DCFH2 reactivity should always be taken into consideration when interpreting data obtained with DCFH2(-DA).2 Furthermore, the possibility of intercellular differences in esterase activity on DCFH2-DA deacetylation and/or in basal redox state implies that care should be taken with directly comparing different cell types. Transporter-mediated DCF(H2) uptake and efflux trough organic anion-transporting polypeptides 1B1 or 3 (OATP1B1/3)23,24 and MDR-associated ABC transporters (e.g., multidrug-resistance protein-1; MRP-1),23,25 respectively, are expected to influence assay outcomes. Consequently, it is not recommended to use DCFH2 in assays involving cells that express OATP1B1/3, such as hepatocyte-derived HepG2 or HepaRG cells.26 The presence of efflux proteins, for example, MRP-1 on RAW 264.7 macrophages,25 is likely less troublesome insofar as its effects will be limited to an underestimation of intracellular DCF levels. Furthermore, assay interference by efflux proteins is not specific for DCF(H2) since affinity toward MDR-associated ABC transporters has also been reported for a variety of redox indicators (i.e., dihydrorhodamine, MitoSOX, and dihydroethidium) as well as other frequently used dyes (e.g., JC1 and Hoechst 33342).25 Underestimation of intracellular DCF levels should therefore always be considered when DCFH2-DA is used on MDR-associated ABC transporter-expressing cells, regardless of whether the assay is coconducted with DCFH2.
Nevertheless, general oxidative stress can be easily measured using DCFH2 in tailored settings and conditions. Aside from the relatively low cost of DCFH2-DA versus other redox probes, the use of DCFH2 holds several important benefits. The membrane-impermeability and consequent extracellular localization of DCFH2 does not apply to most commercially available fluorogenic redox probes such as membrane-transgressing dihydroethidium and dihydrorhodamine.1 This is particularly useful in light of having an intracellularly localizing, structurally identical counterpart (i.e., DCFH2-DA). Although compartmentalized analysis of redox processes has been reported,16,27 these studies used different probes for the measurement of intra- and extracellular oxidant formation. Such approaches significantly reduce comparability of the data while underscoring the significant advantage of using DCFH2 in concert with DCFH2-DA. Moreover, DCFH2 does not require cofactors, as is the case with, for example, Amplex Red1 and lucigenin,2 which may interfere with probe reaction(s). Lastly, the high fluorescence quantum yield of DCF (0.96 in PBS28 compared to, for example, 0.0091 for ethidium in H2O29) allows detection in the nanomolar range.
Conclusions
DCFH2 is an easy-to-prepare DCFH2-DA derivative that enables a variety of innovative probe applications. These include various cell-free assays in which DCFH2 can be employed to analyze specific redox reactions. The simultaneous use of DCFH2-DA and DCFH2 moreover allows for the concomitant detection of intra- and extracellular oxidative stress in vitro. Lastly, the same method can be used to prepare CDCFH2 from CDCFH2-DA that, when encapsulated into a liposomal probe delivery system, can be used for the detection of organ- or tissue-specific oxidant formation in vivo.
Acknowledgments
DCFH2 and liposomal CDCFH2 are available to interested parties upon request to the corresponding author.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b00043.
The authors declare no competing financial interest.
Supplementary Material
References
- Gomes A.; Fernandes E.; Lima J. L. J. Biochem. Biophys. Methods 2005, 65, 45–80. 10.1016/j.jbbm.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Wardman P. Free Radical Biol. Med. 2007, 43, 995–1022. 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Wrona M.; Patel K.; Wardman P. Free Radical Biol. Med. 2005, 38, 262–70. 10.1016/j.freeradbiomed.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Royall J. A.; Ischiropoulos H. Arch. Biochem. Biophys. 1993, 302, 348–55. 10.1006/abbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- LeBel C. P.; Ischiropoulos H.; Bondy S. C. Chem. Res. Toxicol. 1992, 5, 227–231. 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- Burkitt M. J.; Wardman P. Biochem. Biophys. Res. Commun. 2001, 282, 329–33. 10.1006/bbrc.2001.4578. [DOI] [PubMed] [Google Scholar]
- Cathcart R.; Schwiers E.; Ames B. N. Anal. Biochem. 1983, 134, 111–6. 10.1016/0003-2697(83)90270-1. [DOI] [PubMed] [Google Scholar]
- Wrona M.; Wardman P. Free Radical Biol. Med. 2006, 41, 657–67. 10.1016/j.freeradbiomed.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Pietrangelo A. Hepatology 2007, 46, 1291–301. 10.1002/hep.21886. [DOI] [PubMed] [Google Scholar]
- Hung Y. H.; Bush A. I.; Cherny R. A. JBIC, J. Biol. Inorg. Chem. 2010, 15, 61–76. 10.1007/s00775-009-0600-y. [DOI] [PubMed] [Google Scholar]
- Welch K. D.; Davis T. Z.; Eden M. E. V.; Aust S. D. Free Radical Biol. Med. 2002, 32, 577–583. 10.1016/S0891-5849(02)00760-8. [DOI] [PubMed] [Google Scholar]
- Wardman P.; Candeias L. P. Radiat. Res. 1996, 145, 523–531. 10.2307/3579270. [DOI] [PubMed] [Google Scholar]
- Welch K. D.; Davis T. Z.; Aust S. D. Arch. Biochem. Biophys. 2002, 397, 360–369. 10.1006/abbi.2001.2694. [DOI] [PubMed] [Google Scholar]
- Weijer R.; Broekgaarden M.; Krekorian M.; Alles L. K.; van Wijk A. C.; Mackaaij C.; Verheij J.; van der Wal A. C.; van Gulik T. M.; Storm G.; Heger M. Oncotarget. 2015, 7, 3341–3356. 10.18632/oncotarget.6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krötz F.; Sohn H. Y.; Pohl U. Arterioscler., Thromb., Vasc. Biol. 2004, 24, 1988–96. 10.1161/01.ATV.0000145574.90840.7d. [DOI] [PubMed] [Google Scholar]
- Bakdash N.; Williams M. S. Free Radical Biol. Med. 2008, 45, 158–166. 10.1016/j.freeradbiomed.2008.03.021. [DOI] [PubMed] [Google Scholar]
- Chouchani E. T.; Pell V. R.; Gaude E.; Aksentijević D.; Sundier S. Y.; Robb E. L.; Logan A.; Nadtochiy S. M.; Ord E. N.; Smith A. C.; Eyassu F.; Shirley R.; Hu C. H.; Dare A. J.; James A. M.; Rogatti S.; Hartley R. C.; Eaton S.; Costa A. S.; Brookes P. S.; Davidson S. M.; Duchen M. R.; Saeb-Parsy K.; Shattock M. J.; Robinson A. J.; Work L. M.; Frezza C.; Krieg T.; Murphy M. P. Nature 2014, 515, 431–435. 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Trush M. A. Biochem. Biophys. Res. Commun. 1998, 253, 295–299. 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- Hempel S. L.; Buettner G. R.; O’Malley Y. Q.; Wessels D. A.; Flaherty D. M. Free Radical Biol. Med. 1999, 27, 146–159. 10.1016/S0891-5849(99)00061-1. [DOI] [PubMed] [Google Scholar]
- van Golen R. F.; Reiniers M. J.; Heger M.; Verheij J. J. Hepatol. 2015, 62, 975–977. 10.1016/j.jhep.2014.12.014. [DOI] [PubMed] [Google Scholar]
- Allen T. M. Nat. Rev. Cancer 2002, 2, 750–763. 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- Joner M.; Morimoto K.; Kasukawa H.; Steigerwald K.; Merl S.; Nakazawa G.; John M. C.; Finn A. V.; Acampado E.; Kolodgie F. D.; Gold H. K.; Virmani R. Arterioscler., Thromb., Vasc. Biol. 2008, 28, 1960–1966. 10.1161/ATVBAHA.108.170662. [DOI] [PubMed] [Google Scholar]
- Zamek-Gliszczynski M. J.; Xiong H.; Patel N. J.; Turncliff R. Z.; Pollack G. M.; Brouwer K. L. R. J. Pharmacol. Exp. Ther. 2003, 304, 801–809. 10.1124/jpet.102.044107. [DOI] [PubMed] [Google Scholar]
- De Bruyn T.; Fattah S.; Stieger B.; Augustijns P.; Annaert P. J. Pharm. Sci. 2011, 100, 5018–5030. 10.1002/jps.22694. [DOI] [PubMed] [Google Scholar]
- Procházková J.; Kubala L.; Kotasová H.; Gudernová I.; Šrámková Z.; Pekarová M.; Sarkadi B.; Pacherník J. Free Radical Res. 2011, 45, 779–787. 10.3109/10715762.2011.579120. [DOI] [PubMed] [Google Scholar]
- Hart S. N.; Li Y.; Nakamoto K.; Subileau E. A.; Steen D.; Zhong X. B. Drug Metab. Dispos. 2010, 38, 988–994. 10.1124/dmd.109.031831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begonja A. J.; Gambaryan S.; Geiger J.; Aktas B.; Pozgajova M.; Nieswandt B.; Walter U. Blood 2005, 106, 2757–2760. 10.1182/blood-2005-03-1047. [DOI] [PubMed] [Google Scholar]
- Zhang X. F.; Zhang J.; Liu L. J. Fluoresc. 2014, 24, 819–826. 10.1007/s10895-014-1356-5. [DOI] [PubMed] [Google Scholar]
- Brand L.; Gohlke J. R. Annu. Rev. Biochem. 1972, 41, 843–868. 10.1146/annurev.bi.41.070172.004211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.