

Crystal structure, thermal behaviour and phase transitions of formamidinium iodide were studied by DTG, DSC, powder diffraction and X-ray crystallography.
Keywords: crystal structure, formamidinium iodide, phase transitions, powder diffraction, synchrotron radiation
Abstract
At a temperature of 100 K, CH5N2 +·I− (I), crystallizes in the monoclinic space group P21/c. The formamidinium cation adopts a planar symmetrical structure [the r.m.s. deviation is 0.002 Å, and the C—N bond lengths are 1.301 (7) and 1.309 (8) Å]. The iodide anion does not lie within the cation plane, but deviates from it by 0.643 (10) Å. The cation and anion of I form a tight ionic pair by a strong N—H⋯I hydrogen bond. In the crystal of I, the tight ionic pairs form hydrogen-bonded zigzag-like chains propagating toward [20-1] via strong N—H⋯I hydrogen bonds. The hydrogen-bonded chains are further packed in stacks along [100]. The thermal behaviour of I was studied by different physicochemical methods (thermogravimetry, differential scanning calorimetry and powder diffraction). Differential scanning calorimetry revealed three narrow endothermic peaks at 346, 387 and 525 K, and one broad endothermic peak at ∼605 K. The first and second peaks are related to solid–solid phase transitions, while the third and fourth peaks are attributed to the melting and decomposition of I. The enthalpies of the phase transitions at 346 and 387 K are estimated as 2.60 and 2.75 kJ mol−1, respectively. The X-ray powder diffraction data collected at different temperatures indicate the existence of I as the monoclinic (100–346 K), orthorhombic (346–387 K) and cubic (387–525 K) polymorphic modifications.
Chemical context
Compounds with the general formula ABX 3 [where A denotes an organic cation e.g. methylammonium (MA, CH3NH3 +) or formamidinium [FA = CH(NH2)2, CH3NH3]; B = Pb, Sn; X = I, Br, Cl] belong to a class of hybrid organic–inorganic perovskites and perform as outstanding light harvesters. These compounds gave birth to a new field of photovoltaics – perovskite solar cells – when Kojima and co-authors used (MA)PbI3 as a light sensitizer for the first time in dye-sensitized solar cells (DSSCs) in 2009 and showed 3.8% efficiency (Kojima et al., 2009 ▸). Since then, a revolutionary breakthrough has occured in this area and the highest efficiency now has reached 22.1%.
In 2014, the formamidinium cation was proposed to replace methylammonium and the further investigation of (FA)PbI3 disclosed its superiority to (MA)PbI3 (Koh et al., 2014 ▸; Pang et al., 2014 ▸). In particular, it was found that (FA)PbI3 exhibits higher thermal and moisture stability and has a lower bandgap than (MA)PbI3 which gives a greater capacity for sunlight absorption (Koh et al., 2014 ▸; Han et al., 2016 ▸). Recently, it was shown that the properties of the compounds may be further optimized by tuning the MA/FA ratio and an efficiency of 20.5% has been reached for a mixed compound (Li et al., 2016 ▸; Jeon et al., 2015 ▸).
The main precursors to obtain (FA)PbI3 are PbI2 and formamidinium iodide (FA)I. Several methods of perovskite synthesis include steps where it can be obtained directly from (FA)I in a crystalline form (Zhou et al., 2015 ▸; Leyden et al., 2015 ▸). It also appears in a crystalline form and leads to a formation of low-dimensional phases when an excess of it is taken (Xi et al., 2016 ▸; Ma et al., 2017 ▸). Thus, the understanding of the (FA)I crystal structure gives valuable information for understanding the crystallization of formamidinium-based lead halide perovskites. Knowledge of the (FA)I crystal structure may also be of particular interest for computer simulations of the processes related to the crystallization of these perovskites. Surprisingly, despite the hundreds of papers published over the last several years that have mentioned (FA)I as a major precursor for hybrid lead halide perovskites, its crystal structure has remained unknown so far.
In this work, we investigated the structure of (FA)I (I) and its thermal behaviour by different physico-chemical methods.
Structural commentary
At a temperature of 100 K, compound I crystallizes in the monoclinic space group P21/c. The formamidinium cation adopts a planar symmetrical structure [r.m.s. deviation is 0.002 Å, and the C—N bond lengths are 1.301 (7) and 1.309 (8) Å; Fig. 1 ▸]. The iodide anion does not lie within the cation plane, but deviates from it by 0.643 (10) Å. The cation and anion in I form a tight ionic pair by the strong N1—H1A⋯I1 hydrogen bond (Table 1 ▸ and Fig. 1 ▸).
Figure 1.

The molecular structure of salt I. Displacement ellipsoids are shown at the 50% probability level. H atoms are presented as small spheres of arbitrary radius. Dashed line indicates the intermolecular N—H⋯I hydrogen bond.
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1A⋯I1 | 0.90 | 2.77 | 3.612 (5) | 156 |
| N2—H2A⋯I1i | 0.90 | 2.74 | 3.622 (4) | 166 |
Symmetry code: (i) 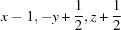 .
.
In order to understand the thermal behaviour of I at elevated temperatures, the sample was investigated by TG and DSC methods in the temperature region from 293 to 750 K at a rate of 5 K min−1. The mass loss started from ∼520 K (Fig. 2 ▸). Differential scanning calorimetry revealed three narrow endothermic peaks at 346, 387 and 525 K, and one broad endothermic peak at ∼605 K. The first and the second peaks are related to solid–solid phase transitions, while the third and the fourth peaks are attributed to the melting and decomposition of I. Enthalpy of the phase transitions at 346 and 387 K are estimated as 2.24 and 2.87 kJ mol−1, respectively.
Figure 2.

Thermogravimetry and differential scanning calorimetry analyses for I.
The X-ray powder diffraction data collected at different temperatures confirm the existence of different phases (Fig. 3 ▸). At low temperatures, salt I exists in a monoclinic phase and exhibits a significant change of the parameters with a rise in temperature (100 → 195 → 293 K, Fig. 3 ▸). A phase existing at 358 K is indexed in an orthorhombic crystal system [a = 7.3915 (8) Å, b = 6.3358 (8) Å, c = 5.2391 (9) Å; M(20) = 25, F(20) = 45]. Another high-temperature phase is cubic, exhibiting only a few reflections at 400 K [a = 5.0571 (5) Å; M(13) = 126, F(13) = 109]. It seems to be a plastic phase similar to a plastic phase for methylammonium iodide (Ishida et al., 1995 ▸; Yamamuro et al., 1992 ▸).
Figure 3.

X-ray powder diffraction data for I at different temperatures.
Supramolecular features
In the crystal of I, the tight ionic pairs form hydrogen-bonded zigzag-like chains propagating toward [20 ] by the strong intermolecular N2—H2A⋯I1i hydrogen bonds (Table 1 ▸ and Fig. 4 ▸). The hydrogen-bonded chains are further packed in stacks along [100] (Fig. 4 ▸) [symmetry code: (i) x − 1, −y +
] by the strong intermolecular N2—H2A⋯I1i hydrogen bonds (Table 1 ▸ and Fig. 4 ▸). The hydrogen-bonded chains are further packed in stacks along [100] (Fig. 4 ▸) [symmetry code: (i) x − 1, −y +  , z + ].
, z + ].
Figure 4.

The crystal structure of I demonstrating the hydrogen-bonded zigzag-like chains propagating toward [20]. Dashed lines indicate the intermolecular N—H⋯I hydrogen bonds.
Synthesis and crystallization
Polycrystalline powder of I was purchased from Dyesol and used without further purification. Single crystals suitable for X-ray structural study were obtained by recrystallization from an anhydrous ethanol solution by slow cooling.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. X-ray diffraction study of I was carried out on the ‘Belok’ beamline of the National Research Center ‘Kurchatov Institute’ (Moscow, Russian Federation) using a Rayonix SX165 CCD detector. Reflection intensities measured were corrected for absorption using the Scala (Evans, 2006 ▸) program.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | CH5N2 +·I− |
| M r | 171.97 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 100 |
| a, b, c (Å) | 4.8211 (6), 13.776 (3), 7.0113 (10) |
| β (°) | 98.06 (3) |
| V (Å3) | 461.06 (14) |
| Z | 4 |
| Radiation type | Synchrotron, λ = 0.96990 Å |
| μ (mm−1) | 15.38 |
| Crystal size (mm) | 0.06 × 0.05 × 0.03 |
| Data collection | |
| Diffractometer | Rayonix SX165 CCD |
| Absorption correction | Multi-scan (SCALA; Evans, 2006 ▸) |
| T min, T max | 0.400, 0.600 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 5111, 949, 894 |
| R int | 0.070 |
| (sin θ/λ)max (Å−1) | 0.642 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.039, 0.093, 1.06 |
| No. of reflections | 949 |
| No. of parameters | 38 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.87, −0.91 |
The H atoms of the NH2 groups were localized in the difference Fourier map and refined with fixed positional and isotropic displacement parameters [U iso(H) = 1.2U eq(N)]. The CH hydrogen was placed in a calculated position, with C—H = 0.95 Å, and refined in the riding model with a fixed isotropic displacement parameter [U iso(H) = 1.2U eq(C)].
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S205698901700425X/lh5840sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S205698901700425X/lh5840Isup2.hkl
Supporting information file. DOI: 10.1107/S205698901700425X/lh5840Isup3.cml
CCDC reference: 1538402
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
This work was supported financially by the Ministry of Education and Science of the Russian Federation (the Agreement number 02.a03.21.0008). AT, AP and EG acknowledge the Russian Foundation for Basic Research for funding the reported study as part of research project No. 16-29-03291.
supplementary crystallographic information
Crystal data
| CH5N2+·I− | F(000) = 312 |
| Mr = 171.97 | Dx = 2.477 Mg m−3 |
| Monoclinic, P21/c | Synchrotron radiation, λ = 0.96990 Å |
| a = 4.8211 (6) Å | Cell parameters from 600 reflections |
| b = 13.776 (3) Å | θ = 4.0–36.0° |
| c = 7.0113 (10) Å | µ = 15.38 mm−1 |
| β = 98.06 (3)° | T = 100 K |
| V = 461.06 (14) Å3 | Prism, colourless |
| Z = 4 | 0.06 × 0.05 × 0.03 mm |
Data collection
| Rayonix SX165 CCD diffractometer | 894 reflections with I > 2σ(I) |
| φ scan | Rint = 0.070 |
| Absorption correction: multi-scan (Scala; Evans, 2006) | θmax = 38.5°, θmin = 4.0° |
| Tmin = 0.400, Tmax = 0.600 | h = −6→6 |
| 5111 measured reflections | k = −17→17 |
| 949 independent reflections | l = −8→7 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.039 | H-atom parameters constrained |
| wR(F2) = 0.093 | w = 1/[σ2(Fo2) + 0.7865P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.06 | (Δ/σ)max < 0.001 |
| 949 reflections | Δρmax = 0.87 e Å−3 |
| 38 parameters | Δρmin = −0.91 e Å−3 |
| 0 restraints | Extinction correction: SHELXL2014 (Sheldrick, 2015b), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: difference Fourier map | Extinction coefficient: 0.0064 (11) |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| I1 | 0.86658 (6) | 0.38577 (2) | 0.20022 (5) | 0.0218 (2) | |
| N1 | 0.6120 (9) | 0.4171 (3) | 0.6569 (7) | 0.0250 (10) | |
| H1A | 0.7250 | 0.4174 | 0.5650 | 0.030* | |
| H1B | 0.6251 | 0.4648 | 0.7454 | 0.030* | |
| N2 | 0.2540 (9) | 0.3389 (3) | 0.7860 (7) | 0.0254 (11) | |
| H2A | 0.1352 | 0.2882 | 0.7776 | 0.030* | |
| H2B | 0.2515 | 0.3832 | 0.8801 | 0.030* | |
| C1 | 0.4296 (11) | 0.3478 (4) | 0.6606 (8) | 0.0227 (11) | |
| H1 | 0.4233 | 0.2994 | 0.5637 | 0.027* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| I1 | 0.0230 (3) | 0.0215 (3) | 0.0217 (4) | 0.00012 (9) | 0.0054 (2) | −0.00041 (10) |
| N1 | 0.022 (2) | 0.030 (2) | 0.023 (3) | −0.0030 (18) | 0.0030 (19) | 0.000 (2) |
| N2 | 0.021 (2) | 0.022 (2) | 0.034 (3) | −0.0020 (17) | 0.008 (2) | 0.0011 (19) |
| C1 | 0.023 (2) | 0.026 (3) | 0.019 (3) | 0.002 (2) | 0.001 (2) | 0.000 (2) |
Geometric parameters (Å, º)
| N1—C1 | 1.301 (7) | N2—H2A | 0.8999 |
| N1—H1A | 0.9001 | N2—H2B | 0.9000 |
| N1—H1B | 0.9001 | C1—H1 | 0.9500 |
| N2—C1 | 1.309 (8) | ||
| C1—N1—H1A | 119.7 | H2A—N2—H2B | 120.0 |
| C1—N1—H1B | 120.3 | N1—C1—N2 | 125.8 (5) |
| H1A—N1—H1B | 120.0 | N1—C1—H1 | 117.1 |
| C1—N2—H2A | 119.7 | N2—C1—H1 | 117.1 |
| C1—N2—H2B | 120.3 |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···I1 | 0.90 | 2.77 | 3.612 (5) | 156 |
| N2—H2A···I1i | 0.90 | 2.74 | 3.622 (4) | 166 |
Symmetry code: (i) x−1, −y+1/2, z+1/2.
References
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Doyle, R. A. (2011). Marccd software manual. Rayonix L. L. C., Evanston, IL 60201, USA.
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Han, Q., Bae, S.-H., Sun, P., Hsieh, Y.-T., Yang, Y. M., Rim, Y. S., Zhao, H., Chen, Q., Shi, W., Li, G. & Yang, Y. (2016). Adv. Mater. 28, 2253–2258. [DOI] [PubMed]
- Ishida, H., Maeda, H., Hirano, A., Fujimoto, T., Kubozono, Y., Kashino, S. & Emura, S. (1995). Z. Naturforsch. Teil A, 50, 14–18.
- Jeon, N. J., Noh, J. H., Yang, W. S., Kim, Y. C., Ryu, S., Seo, J. & Seok, S. I. (2015). Nature, 517, 476–480. [DOI] [PubMed]
- Koh, T. M., Fu, K., Fang, Y., Chen, S., Sum, T. C., Mathews, N., Mhaisalkar, S. G., Boix, P. P. & Baikie, T. (2014). J. Phys. Chem. C, 118, 16458–16462.
- Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. (2009). J. Am. Chem. Soc. 131, 6050–6051. [DOI] [PubMed]
- Leyden, M. R., Lee, M. V., Raga, S. R. & Qi, Y. (2015). J. Mater. Chem. A3, 16097–16103.
- Li, X., Bi, D., Yi, C., Decoppet, J.-D., Luo, J., Zakeeruddin, S. M., Hagfeldt, A. & Gratzel, M. (2016). Science, 353, 58–62. [DOI] [PubMed]
- Ma, F., Li, J., Li, W., Lin, N., Wang, L. & Qiao, J. (2017). Chem. Sci. 8, 800–805. [DOI] [PMC free article] [PubMed]
- Pang, S., Hu, H., Zhang, J., Lv, S., Yu, Y., Wei, F., Qin, T., Xu, H., Liu, Z. & Cui, G. (2014). Chem. Mater. 26, 1485–1491.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Xi, J., Wu, Z., Xi, K., Dong, H., Xia, B., Lei, T., Yuan, F., Wu, W., Jiao, B. & Hou, X. (2016). Nano Energy, 26, 438–445.
- Yamamuro, O., Matsuo, T., Suga, H., David, W. I. F., Ibberson, R. M. & Leadbetter, A. J. (1992). Acta Cryst. B48, 329–336.
- Zhou, Y., Yang, M., Vasiliev, A. L., Garces, H. F., Zhao, Y., Wang, D., Pang, S., Zhu, K. & Padture, N. P. (2015). J. Mater. Chem. A, 3, 9249–9256.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S205698901700425X/lh5840sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S205698901700425X/lh5840Isup2.hkl
Supporting information file. DOI: 10.1107/S205698901700425X/lh5840Isup3.cml
CCDC reference: 1538402
Additional supporting information: crystallographic information; 3D view; checkCIF report