

Abstract
The tree shrew (Tupaia belangeri) is a small mammal with a close relationship to primates and it has been proposed as an alternative experimental animal to primates in biomedical research. The recent release of a high-quality Chinese tree shrew genome enables more researchers to use this species as the model animal in their studies. With the aim to making the access to an extensively annotated genome database straightforward and easy, we have created the Tree shrew Database (TreeshrewDB). This is a web-based platform that integrates the currently available data from the tree shrew genome, including an updated gene set, with a systematic functional annotation and a mRNA expression pattern. In addition, to assist with automatic gene sequence analysis, we have integrated the common programs Blast, Muscle, GBrowse, GeneWise and codeml, into TreeshrewDB. We have also developed a pipeline for the analysis of positive selection. The user-friendly interface of TreeshrewDB, which is available at http://www.treeshrewdb.org, will undoubtedly help in many areas of biological research into the tree shrew.
The tree shrew (Tupaia belangeri), currently placed in Order Scandentia, has a wide distribution in South Asia, Southeast Asia and Southwest China1. Because of several characteristics including small adult body size, easy and low-cost of maintenance, high brain-to-body mass ratio, a short reproductive cycle, and a close relationship to primates, the tree shrew has been increasingly used as a viable model animal, and as an alternative to rodents and primates, in biomedical research2. For example, in studies into hepatitis virus infection3,4, myopia5, social stress and depression6,7,8, aging research9 and learning behaviors10.
Genome analysis of the tree shrew has received wide attention in recent years, because of the unresolved debate regarding the phylogenetic relationship of tree shrews to primates and their phylogenetic position in the Euarchontoglires11,12. Genome analysis is also an important aid to improving our understanding of the basic biology of this increasingly popular animal. The Broad Institute provided the first (yet rough) assembly of a northern tree shrew (Tupaia belangeri) genome, with annotations for 15,414 genes (most of them are partial genes) in 2008 (http://www.ensembl.org/Tupaia_belangeri/Info/Index). However, the low coverage (2X) of the assembly limited its wide usage in tree shrew related research. So as to improve on this situation, we recently completed a high-quality draft genome of the Chinese tree shrew (Tupaia belangeri chinensis), with a high depth coverage (79X) and a scaffold N50 size of 3.7 Mb13. Based on this high-quality genome information, we have shown that tree shrews have a relatively close relationship to non-human primates, thereby settling a long-running debate regarding the phylogenetic position of the tree shrew within eutherian mammals. By extensively analyzing the key classes of molecules relevant to the nervous and immune systems of the Chinese tree shrew, we have described the genetic basis of the related disease models using this animal13.
However, the retrieval of information regarding a particular gene of interest from the raw tree shrew genomic data is not easy for many researchers. Therefore, to make the access to the extensively annotated genome data easier, we have developed the tree shrew database (TreeshrewDB) (http://www.treeshrewdb.org/) as a highly integrated information platform for data storage, retrieval, visualization and analysis of the Chinese tree shrew genome. The user-friendly interface and the major functions of the web-based platform will undoubtedly make it easier to use the tree shrew as a model animal.
Results
Updates to the gene set and annotations of Chinese tree shrew genome
So as to identify the orthologous genes between the reported northern tree shrew genome from the Broad Institute and the Chinese tree shrew genome determined in our previous study13, we subjected the northern tree shrew genome (Ensembl release 64) and the Chinese tree shrew proteins to BlastP14 analysis with a similar cutoff threshold of e = 1e−5. We defined the reciprocal best-match pairs as 1:1 orthologs with the criteria that more than 70% of the aligned sequence showed an identity above 80%. In total, we detected 12,851 of these 1:1 orthologs between the northern tree shrew and the Chinese tree shrew. We followed similar methods to identify orthologous gene between the human and the Chinese tree shrew, but with a lower cut-off of identity (70%). Additional orthologous genes among other species, including human, chimpanzee, gorilla, rhesus monkey, mouse, rat, rabbit, dog and cow, were identified using the Ensembl ortholog_one2one gene database (version 64)15 for each pair of genomes.
Due to algorithm limitations in the gene prediction programs, we found some errors regarding the predicted gene structure in the early version of annotated sequences of Chinese tree shrew as reported in our previous study13 during the compilation of related gene information for the TreeshrewDB. To eliminate these potential errors, we next compared the length variance between the Chinese tree shrew protein sequences and their orthologous gene protein sequences in human and mouse genomes following the rationale below. If the length difference (which is defined as (A-B)/A, A is length of the longer one and B is length of the shorter one) of orthologous gene is above 0.3, the gene is regarded as under loose prediction. A total of 3,212 genes showed signs of a potential prediction error and were checked for accuracy again. We first aligned the genes retrieved from the Chinese tree shrew genome with their corresponding human orthologous protein sequences by tblastn14. Best hit regions of each gene together with a 5 Kb flanking region sequence were cut down and re-aligned using GeneWise16, which helped to define the detailed exon-intron structure of each gene. We further filtered out the genes not in gene synteny blocks and checked the remaining gene sequences manually. Finally, 2,729 genes were manually confirmed as harboring annotation errors, and we fixed all these errors in the updated gene sets. In addition, we gathered 553 genes mentioned in our previous paper13 and added them to the gene set. We also excluded 495 genes in the old version of the gene set13 that had no functional annotations and mRNA expression in 7 tissues (see below). By using these methods we produced a gene set with 22,121 protein-coding genes.
The individual genes of the Chinese tree shrew have been annotated based on the best match derived from the alignments of protein sequences as described in the SwissProt and TrEMBL17 databases using Blastp (with default parameters)14. We annotated motifs and domains using InterPro18 by searching against publicly available databases, including Pfam (http://pfam.sanger.ac.uk/), PRINTS (http://www.bioinf.manchester.ac.uk/dbbrowser/PRINTS/index.php), PROSITE (http://prosite.expasy.org/), ProDom (http://prodom.prabi.fr/prodom/current/html/home.php), and SMART (http://smart.embl-heidelberg.de/). Descriptions of the gene products included Gene Ontology19; this information was retrieved from InterPro18. We also annotated pathway information by mapping KEGG database20 reference genes and finding the best hit for each gene. The functional annotation is summarized in Table 1. Finally, 20,225 genes had been functionally annotated and stored in our database.
Table 1. Statistics of gene functional annotation and non-coding RNA annotation in the Chinese tree shrew.
| (a) Functional annotation of Chinese tree shrew genes by various methods. | |||
|---|---|---|---|
| Methods | Number | Percent (%) | |
| Annotated | SwissProt and TrEMBL | 20,225 | 91.4 |
| InterPro | 17,420 | 78.7 | |
| KEGG | 16,593 | 75.0 | |
| GO | 14,758 | 66.7 | |
| Unannotated | - | 1,896 | 8.6 |
| Total | - | 22,121 | - |
We used RNA-seq data published in our previous study13 to optimize the gene structure by extending the 3′- or 5′-sequences and calculated the mRNA expression levels in the 7 tissues, brain, liver, heart, kidney, pancreas, ovary and testis. In brief, the raw RNA-seq data were trimmed to remove sequencing adapters and reads containing one or more Ns > 5%, or of low quality (more than 20% of the base's qualities were less than 10). We then aligned these filtered reads to the Chinese tree shrew genome using Tophat 1.4.121. The gene structure was optimized according to the distribution of the reads, information of paired-end and the annotation of reference gene. We followed the previously described procedure to define transcriptionally active regions (TAR)22. A contiguous expression region with each base being supported by at least four reads in the 7 tissues was considered as a TAR. The TARs joined by at least one set of paired-end reads were connected into a putative gene. A total of 7,777 5′-UTRs and 10,222 3′-UTRs of 11,522 transcripts were extended.
For calculating the mRNA expression levels of the Chinese tree shrew gene, we used the RPKM measure (Reads Per kb per Million reads)23 with the following settings:
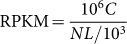 |
RPKM (A) refers to mRNA expression of gene A, C is number of reads that uniquely aligned to gene A, N is the total number of reads that were uniquely aligned to all genes, and L is the base number in the CDS of gene A. The RPKM method is able to eliminate the influence of different gene lengths and sequencing discrepancy on the calculation of gene expression. In total, mRNA expression levels of 19,448 genes were detected in at least one of the 7 tissues. We further performed a principal component analysis (PCA) to show the enrichments and relationships among these tissues according to mRNA expression pattern (Figure 1).
Figure 1. Principal component analysis plot of the global gene expression pattern for tissues from the Chinese tree shrew, including brain, liver, heart, kidney, pancreas, ovary and testis.

Non-coding RNAs (ncRNAs) play various roles in cells. For instance, rRNAs and tRNAs participated in mRNA translation, small nuclear RNAs (snRNAs) are involved in splicing, and small nucleolar RNAs (snoRNAs) are involved in the modification of rRNAs24. By searching databases using the SINE pre-masked genome sequence, we detected four types of non-coding small RNA in the Chinese tree shrew genome. We looked for snRNA and miRNA sequences by using the Rfam database (Release 9.1)25 as the reference. For rRNA sequences, we used human full-length rRNA as the reference. Blastn14 with a similar cutoff threshold of e = 1e−5 was employed in the prediction. Next, tRNAscan-SE (version 1.23)26 was used to search for tRNAs. In total, we have identified 2,794 non-coding small RNAs in the Chinese tree shrew genome (Table 1).
Database organization
We built the TreeshrewDB with three functional modules: a retrieval module, an analysis module and a data transfer module (Figure 2).
Figure 2. Overall structure of the retrieval module, analysis module and data transfer module of the TreeshrewDB.

(a) Retrieval module
For the retrieval module, we provide various retrieval methods. A user can retrieve gene information by gene symbol, ID, full name, blast search, GO term, domain information or pathway information. The retrieval module is incorporated into the home, search and gene information pages. At the home page, we provide a search tool to get gene information quickly by using the gene symbol. At the search page, we provide a “one-stop” retrieval system for viewing gene information by gene symbol, gene ID or gene full name (Figure 3). When jumping to the gene information page, a user sees 5 tables:
Basic information table. In this table, there is basic information on the particular gene queried, including the gene symbol, reference sequence ID, location, and the genomic map. In the location region, a user can obtain the genomic DNA sequence and/or the flanking region sequence for a particular gene. In the genomic map region, we have embedded a graphic to show the gene structure and gene mapping by using Gbrowse software27. The graphic was also linked to genome browser page.
Sequence table. This table provides the corresponding CDS sequence, UTR sequence, exon and intron sequence, as well as the deduced protein sequence.
Orthologous gene table. This table displays all the identified orthologous genes by using the above-mentioned methods, with links to Ensembl database (http://www.ensembl.org). In order to provide an automatic alignment and comparison of the orthologous gene sequences from different species we provide a button for the rapid alignment of the selected sequences.
Function annotation table. This table displays gene functional annotation information. A user can obtain the gene function classification from the GO annotation, pathway information from the KEGG annotation, and domain information from the InterPro annotation.
mRNA expression table. This table contains RPKM values and a histogram showing the mRNA expression levels of a particular gene in 7 tissues from the Chinese tree shrew.
Figure 3. Gene searching and browsing interface and pipeline of the TreeshrewDB.

Users can retrieve genes via the four methods listed in the lefthand boxes and get more information by clicking the gene information page.
The gene information page also provides links to other databases so as to allow the user to get more detailed information. A user can select the link to wiki (http://en.wikipedia.org) or Genecards (http://www.genecards.org) to view more information about a particular gene, and all the annotation information is linked to the referenced database. For example, the GO term is linked to Gene Ontology database (http://www.geneontology.org/), domain annotation is linked to the InterPro database (http://www.ebi.ac.uk/interpro/), and pathway information is linked to the KEGG database (http://www.genome.jp/kegg/).
In addition, the retrieval module provides for function classification retrieval and small RNA retrieval. A user can search genes by GO term, domain information and pathway information taxonomy and download gene sequences by function classification in batch mode. The ncRNA retrieval option is provided to view 4 types of small RNAs information in the Chinese tree shrew genome.
(b) Analysis module
For the analysis module, we have integrated the common programs Blast14, Muscle28, Gbrowse27, and codeml from the PAML package29, developed three sequence processing tools (ExtractSeq, ReverseSeq and TranslateSeq) and included a positive selection analysis pipeline (PSAP) accessible by a set of perl scripts, which are available online.
GBrowse27 is a popular genome browser for displaying annotations from genomes by combining the database and interactive web pages, it has been extensively applied in many genome databases, such as Flybase30, SilkDB31 and BGD32. In the TreeshrewDB, we have integrated Gbrowse to display the Chinese tree shrew genes and annotations. We also combine Gbrowse and gene retrieval module, and display the gene information by the links in GBrowse.
The TreeshrewDB had several sequence alignment tools, such as Blast14, Genewise16 and Muscle28. The Blast14 tool can find regions of local similarity between sequences. A user can compare a particular nucleotide or protein sequence of a gene to the Chinese tree shrew genome sequence, CDS sequence or protein sequence, and calculate the statistical significance of the matching. The blast results can be downloaded as needed. Genewise16 is a pairwise sequence alignment tool. It can provide highly accurate and sensitive prediction of gene structure by comparing a protein sequence to a genomic DNA sequence. Muscle28 is an accurate multiple sequences alignment tool and can be used to align and compare orthologous genes from multiple species.
Among the three sequence processing tools developed by ourselves, ExtractSeq can be used to extract a sequence from the Chinese tree shrew genome by scaffold position information; ReverseSeq can convert a DNA sequence into its reverse, complement, or reverse-complement counterpart; and TranslateSeq can translate a coding DNA sequence to a protein sequence.
In the TreeshrewDB, we provide two methods to perform positive selection analysis by using codeml from the PAML package29. The first method applies to all species. A user can upload an aligned sequence of the species that they want to analysis using the infile style for PHYLIP (http://evolution.genetics.washington.edu/phylip.html) and the species tree; and then select the model to finish the computation by codeml. The second method only applies for 10 species, human, chimpanzee, gorilla, rhesus monkey, Chinese tree shrew, mouse, rat, rabbit, dog and cow. This tool is named the positive selection analysis pipeline (PSAP), and has been developed using perl scripts, muscle28 and codeml from the PAML package29, and provides positive selection information. The PSAP has the following data as its core: (i) the 1:1 ortholog relationship between the 10 species; (ii) the longest coding region sequences and protein sequences of these species from the Ensembl database, except for the Chinese tree shrew; (iii) the phylogenetic tree list for these species with reference to Murphy et al.33. When the gene name or ID, species and model have been selected, the pipeline will automatically extract the related sequences according to the orthologous relationship list and choose the phylogenetic tree from the tree list. The extracted CDS sequences will be aligned by Muscle 3.728 with the guidance of the aligned protein sequences, and the positive selection analysis was performed with the program codeml from PAML package29. Compared with the first method, PSAP has many advantages, including a saving in time on aligning the sequences, or by just selecting the species without providing the phylogenetic tree. However, each method has its own advantages and disadvantages, and users should select the method according to their needs.
(c) Data transfer module
In the data transfer module, we provide services for batch data downloading and sending to e-mail. A user can download all the raw reads of Chinese tree shrew genome and transcriptome, and the processed data including the Chinese tree shrew genome data, gene sequences, gene function information, pseudogene sequences and information, etc. All the results can be downloaded and/or sent to other people by e-mail.
Discussion
As a mammal with a close relationship to primates, the tree shrew has got wide attention and has been considered to be an alternative experimental animal to primates in biomedical research1,34,35. Despite the fact that we had completed a high-quality draft genome of Chinese tree shrew, there were many obstacles for researchers in the accessing of the data. Firstly, it is very inconvenient to use the large raw data files of the Chinese tree shrew. Secondly, there has been much debate over the annotation of the genes. Even the best gene predictors and genome annotation pipelines hardly exceed accuracies of 80% at the exon level36, and for this reason we found some structure errors in the early version of gene set. Finally, most genes lacked sufficient annotation information. To solve these problems, we have built TreeshrewDB to improve access to the Chinese tree shrew genome data. This database provides a dynamic platform allowing us to correct errors of gene prediction, and to update the comprehensive gene set and gene structure, as well as to offer relative mRNA expression pattern based on the RNA-seq data.
In addition, TreeshrewDB provides some useful tools for sequence data analysis and processing. Considering the unique evolution position of the tree shrew and the poor level of the functional annotation of the tree shrew genome, the tools we provide are focused on molecular evolution analysis and sequence retrieval. For example, a user can use Muscle28 to compare the conservation among species and use codeml29 or PSAP to detect selection signal and also retrieve and extract sequences by combining several tools, such as Blast14, GeneWise16 and ExtractSeq. Each tool offers simplified operation of these programs via the user-friendly interface. A user can obtain the related information by choosing the species of interest and following a few simple steps.
In the future, we will update the gene sequences regularly and release the latest annotated version. Also, we will continue to collect materials regarding the tree shrew, such as biological data, live videos, images and the latest findings for this species. We hope that this publicly available annotated genome of the Chinese tree shrew will offer an opportunity to extend our knowledge of the genetic basis of the tree shrew and attract more and more researchers to work with this increasingly popular animal model. Undoubtedly, TreeshrewDB will offer a powerful platform for sharing information about the tree shrew.
Methods
Datasets
The original data that we used as the foundation of the TreeshewDB were taken from our previous study13, and were composed of Chinese tree shrew genome data and RNA-seq data from 7 tissues (brain, liver, heart, kidney, pancreas, ovary and testis). We included an updated gene set (see above) with a systematic functional annotation and demonstrated the mRNA expression pattern in each tissue.
Database implementation
The Tree shrew Database (TreeshrewDB) runs on a dual-processor server with Ubuntu operating system and is implemented under the LAMP (Linux-Apache-MySQL-Perl) software stack. The application of mod_perl increases the security and efficiency of our Apache server. The entire Chinese tree shrew genome, gene set, gene annotation and other reference information are stored in the MySQL that are administrated with the help of phpMyAdmin. Web interfaces are developed by various languages such as HTML, CSS, JavaScript and Perl.
Acknowledgments
We are grateful to Ian Logan and Fan Long for their helpful suggestions and for editing the language. And thanks to the members in Yao's laboratory for helpful discussions. This assembly of the Chinese tree shrew genome was performed at the HPC Platform, Large-scale Instrument Regional Center of Biodiversity, Kunming Institute of Zoology. This study was supported by the National 863 Project of China (2012AA021801) and grants from Chinese Academy of Sciences (KSCX2-EW-R-11 and KSCX2-EW-J23) and Yunnan Province (2013FB071).
Footnotes
The authors declare no competing financial interests.
Author Contributions Y.-G.Y. managed the project, Y.F. performed data analysis and the construction of the website. Y.F. and Y.-G.Y. wrote the manuscript, with input from D.Y.
References
- Peng Y. Z. et al. Biology of Chinese Tree Shrews (Tupaia belangeri chinensis). (Yunnan Science and Technology Press, Kunming, China, 1991). [Google Scholar]
- Xu L. et al. Tree shrews under the spot light: emerging model of human diseases. Zool Res 34, 59–69 (2013). [DOI] [PubMed] [Google Scholar]
- Zhao X. et al. Primary hepatocytes of Tupaia belangeri as a potential model for hepatitis C virus infection. J Clin Invest 109, 221–232 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R. Q. et al. Human hepatitis B virus and hepatocellular carcinoma. I. Experimental infection of tree shrews with hepatitis B virus. J Cancer Res Clin Oncol 122, 283–288 (1996). [DOI] [PubMed] [Google Scholar]
- Norton T. T., Amedo A. O. & Siegwart J. T. Jr Darkness causes myopia in visually experienced tree shrews. Invest Ophthalmol Vis Sci 47, 4700–4707 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U., van Kampen M., Isovich E., Flugge G. & Fuchs E. Chronic psychosocial stress regulates the expression of both GR and MR mRNA in the hippocampal formation of tree shrews. Hippocampus 11, 329–336 (2001). [DOI] [PubMed] [Google Scholar]
- Fuchs E. Social stress in tree shrews as an animal model of depression: an example of a behavioral model of a CNS disorder. CNS Spectr 10, 182–190 (2005). [DOI] [PubMed] [Google Scholar]
- Wang J., Zhou Q. X., Tian M., Yang Y. X. & Xu L. Tree shrew models: a chronic social defeat model of depression and a one-trial captive conditioning model of learning and memory. Zool Res 32, 24–30 (2011). [DOI] [PubMed] [Google Scholar]
- Yamashita A., Fuchs E., Taira M., Yamamoto T. & Hayashi M. Somatostatin-immunoreactive senile plaque-like structures in the frontal cortex and nucleus accumbens of aged tree shrews and Japanese macaques. J Med Primatol 41, 147–157 (2012). [DOI] [PubMed] [Google Scholar]
- Bartolomucci A., de Biurrun G., Czeh B., van Kampen M. & Fuchs E. Selective enhancement of spatial learning under chronic psychosocial stress. Eur J Neurosci 15, 1863–1866 (2002). [DOI] [PubMed] [Google Scholar]
- Xu L., Fan Y., Jiang X. L. & Yao Y. G. Molecular evidence on the phylogenetic position of tree shrews. Zool Res 34, 70–76 (2013). [DOI] [PubMed] [Google Scholar]
- Xu L., Chen S. Y., Nie W. H., Jiang X. L. & Yao Y. G. Evaluating the phylogenetic position of Chinese tree shrew (Tupaia belangeri chinensis) based on complete mitochondrial genome: implication for using tree shrew as an alternative experimental animal to primates in biomedical research. J Genet Genomics 39, 131–137 (2012). [DOI] [PubMed] [Google Scholar]
- Fan Y. et al. Genome of the Chinese tree shrew. Nat Commun 4, 1426 (2013). [DOI] [PubMed] [Google Scholar]
- Altschul S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella A. J. et al. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 19, 327–335 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E., Clamp M. & Durbin R. GeneWise and Genomewise. Genome Res 14, 988–995 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan C. et al. High-quality protein knowledge resource: SWISS-PROT and TrEMBL. Brief Bioinform 3, 275–284 (2002). [DOI] [PubMed] [Google Scholar]
- Mulder N. & Apweiler R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol Biol 396, 59–70 (2007). [DOI] [PubMed] [Google Scholar]
- Ashburner M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. & Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Pachter L. & Salzberg S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G. et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res 20, 646–654 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., Williams B. A., McCue K., Schaeffer L. & Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5, 621–628 (2008). [DOI] [PubMed] [Google Scholar]
- Mattick J. S. & Makunin I. V. Non-coding RNA. Hum Mol Genet 15 Spec No 1, R17–29 (2006). [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–124 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe T. M. & Eddy S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlin M. J. Using the Generic Genome Browser (GBrowse). Curr Protoc Bioinformatics Chapter 9, Unit 9 9 (2007). [DOI] [PubMed] [Google Scholar]
- Edgar R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24, 1586–1591 (2007). [DOI] [PubMed] [Google Scholar]
- McQuilton P., St Pierre S. E., Thurmond J. & FlyBase C. FlyBase 101--the basics of navigating FlyBase. Nucleic Acids Res 40, D706–714 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J. et al. SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res 38, D453–456 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese J. T. et al. Bovine Genome Database: supporting community annotation and analysis of the Bos taurus genome. BMC Genomics 11, 645 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy W. J., Pringle T. H., Crider T. A., Springer M. S. & Miller W. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res 17, 413–421 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J., Yang E. B., Su J. J., Li Y. & Chow P. The tree shrews: adjuncts and alternatives to primates as models for biomedical research. J Med Primatol 32, 123–130 (2003). [DOI] [PubMed] [Google Scholar]
- Fuchs E. & Corbach-Söhle S. The UFAW Handbook on the Care and Management of Laboratory and Other Research Animals. 262–275 (Wiley-Blackwell, Oxford, 2010). [Google Scholar]
- Yandell M. & Ence D. A beginner's guide to eukaryotic genome annotation. Nat Rev Genet 13, 329–342 (2012). [DOI] [PubMed] [Google Scholar]