

Summary
Learning depends on experience-dependent modification of synaptic efficacy and neuronal connectivity in the brain. We provide direct evidence for physiological roles of the recycling endosome protein GRASP1 in glutamatergic synapse function and animal behavior. Mice lacking GRASP1 showed abnormal excitatory synapse number, synaptic plasticity and hippocampal-dependent learning and memory due to a failure in learning-induced synaptic AMPAR incorporation. We identified two GRASP1 point mutations from intellectual disability (ID) patients that showed convergent disruptive effects on AMPAR recycling and glutamate uncaging-induced structural and functional plasticity. Wild-type GRASP1, but not ID mutants, rescues spine loss in hippocampal CA1 neurons of Grasp1 knockout mice. Together, these results demonstrate a requirement for normal recycling endosome function in AMPAR-dependent synaptic function and neuronal connectivity in vivo, and suggest a potential role for GRASP1 in the pathophysiology of human cognitive disorders.
Keywords: glutamate receptor, long-term potentiation, recycling endosomes, structural plasticity, neuronal connectivity, intellectual disability, neurodevelopmental disorder, GRIP1, syntaxin13
Introduction
The ability to learn and adapt to the changing environment is crucial for human cognition and has long been thought to depend on experience-dependent modification of synaptic efficacy and circuit connectivity in the brain (Bailey and Kandel, 1993; Bliss and Collingridge, 1993; Kessels and Malinow, 2009). Dysregulated synaptic structure and function are associated with brain disorders ranging from neurodevelopmental to neurodegenerative diseases such as intellectual disability (ID), autism and Alzheimer’s disease (Selkoe, 2002; Volk et al., 2015). Synaptic plasticity, one of the most promising cellular models for learning and memory, often refers to a functional adaptation that transforms patterned neuronal activity into long-lasting changes in synaptic efficacy; long-term potentiation (LTP) or long-term depression (LTD). At glutamatergic synapses, LTP/LTD require the addition or removal of synaptic AMPA-type glutamate receptors (AMPARs). Therefore, the mechanisms that regulate AMPAR trafficking are critical for synaptic plasticity and human cognitive behaviors (Huganir and Nicoll, 2013; Kessels and Malinow, 2009).
AMPARs conduct the majority of fast excitatory synaptic transmission in the central nervous system and are tetrameric receptors composed of subunits GluA1–4 (Shepherd and Huganir, 2007). To adjust synaptic efficacy, AMPARs are dynamically regulated through continual endocytosis, sorting and recycling to the cell surface or lysosomal degradation (Anggono and Huganir, 2012; van der Sluijs and Hoogenraad, 2011). AMPAR-interacting proteins are key players in these processes. For example, PICK1 (protein interacting with C kinase1) is involved in the retention of endocytosed AMPARs (Perez et al., 2001; Xia et al., 1999) whereas GRIP1 (glutamate receptor interacting protein1) regulates reinsertion of AMPARs into dendrites and synapses (Dong et al., 1999; Mao et al., 2010; Steiner et al., 2005). Synapse function is also regulated by Rab family GTPases, molecular motors, or SNARE membrane fusion proteins via regulation of endocytic AMPAR trafficking under basal conditions and in response to neuronal activity (Esteban, 2008; Gu et al., 2016; Jurado et al., 2013; Petrini et al., 2009; Wang et al., 2008).
Recycling endosomes regulate the balance of lipids and membrane proteins on the plasma membrane and are important for many cellular processes including cell adhesion, migration and morphogenesis (Grant and Donaldson, 2009; Maxfield and McGraw, 2004). In cultured hippocampal neurons, recycling endosomes have been reported to be a key source to supply AMPARs and membrane per se at glutamatergic synapses. A general block of recycling endosomes with dominant negative Rab11 or Syntaxin13 (Stx13) interferes with endosome fusion and impairs activity-induced synaptic AMPAR targeting and synaptic potentiation (Park et al., 2004; Petrini et al., 2009). Similarly, blocking recycling endosome trafficking abolishes activity-dependent membrane trafficking and subsequent expansion of existing spines as well as formation of new spines (Park et al., 2006; Wang et al., 2008). While these results strongly support an activity-dependent role for recycling endosomes in coordinating synaptic structure and function in neurons, the underlying molecular mechanisms involved and their physiological roles in vivo remain unexplored.
GRASP1 (GRIP associated protein1) is a neuron-specific endosomal protein, identified as part of an AMPAR complex through a direct interaction with the AMPAR scaffolding protein GRIP1 (Ye et al., 2000). Recycling endosomes are formed through a series of membrane fusions of Rab5-positive early endosomes to Rab4-positive early/recycling endosomes, and Rab4-positive endosomes to Rab11-positive mature recycling endosomes destined for exocytosis to the plasma membrane (Sonnichsen et al., 2000). Recently, GRASP1 was reported to play an important role promoting recycling endosome maturation by facilitating the transition from Rab4 to Rab11-positive recycling endosomes (Hoogenraad et al., 2010). As a recycling endosome protein that also interacts with the AMPAR complex, we hypothesize that GRASP1 may be a key molecule essential for experience-dependent remodeling of synaptic transmission and structure necessary for proper learning and memory.
Here, we report that Grasp1 knockout (KO) mice exhibit decreased glutamatergic synaptic connectivity and defective synaptic plasticity in the hippocampus. Moreover, Grasp1 KO mice display significantly impaired associative and spatial learning and memory, likely due to a failure in learning-induced synaptic AMPAR targeting. Furthermore, we identified two GRASP1 mutations that segregated with severe ID. GRASP1 ID mutations impaired activity-dependent AMPAR recycling by trapping internalized AMPAR in recycling endosomes and altering their interactions with key endosomal proteins, GRIP1 and Stx13. Molecular replacement with GRASP1 ID mutants severely impaired glutamate uncaging-induced AMAPR incorporation and spine enlargement. Expression of wild-type GRASP1, but not ID-associated mutants, rescues spine loss in hippocampal CA1 neurons in Grasp1 KO mice, demonstrating the impacts of ID mutations in vivo. Together, our study provides insights for the physiological and pathological roles of GRASP1 in regulating experience-dependent synaptic strengthening and cognitive functions.
Results
Grasp1 KO mice exhibit decreased glutamatergic synapses
To investigate physiological functions of GRASP1 in vivo, we generated Grasp1 KO mice (Figure S1) and tested whether GRASP1 regulates basal AMPAR-mediated synaptic transmission. Spontaneous miniature excitatory postsynaptic currents (mEPSCs) were isolated and recorded from hippocampal CA1 neurons of juvenile (P21–23) Grasp1 animals. A change in mEPSC amplitude indicates a change in the number of AMPARs at postsynaptic sites, while a change in mEPSC frequency suggests alteration of synapse number or presynaptic vesicle release probability. We found no change in the mEPSC amplitude between WT and KO mice, suggesting GRASP1 does not regulate steady state postsynaptic AMPAR number (Figures 1 B&D). However, mEPSC frequency in KO mice was significantly reduced compared to WT littermates (Figures 1A&C) suggesting that GRASP1 mediates neuronal connectivity and/or presynaptic glutamate release. To test whether the reduced mEPSC frequency reflects a decrease in glutamatergic synapse number, we crossed Grasp1 mice with Thy1-GFP transgenic mice to sparsely label CA1 neurons with a GFP-fill (Figure 1E). We found that the glutamatergic synapse density, quantified as the number of spines on secondary dendrites, was significantly lower in juvenile KO than WT littermates (Figures 1 E&F), suggesting that GRASP1 regulates neuronal connectivity in the developing hippocampus.
Figure 1. Grasp1 KO mice have reduced AMPA mEPSC frequency and dendritic spines.
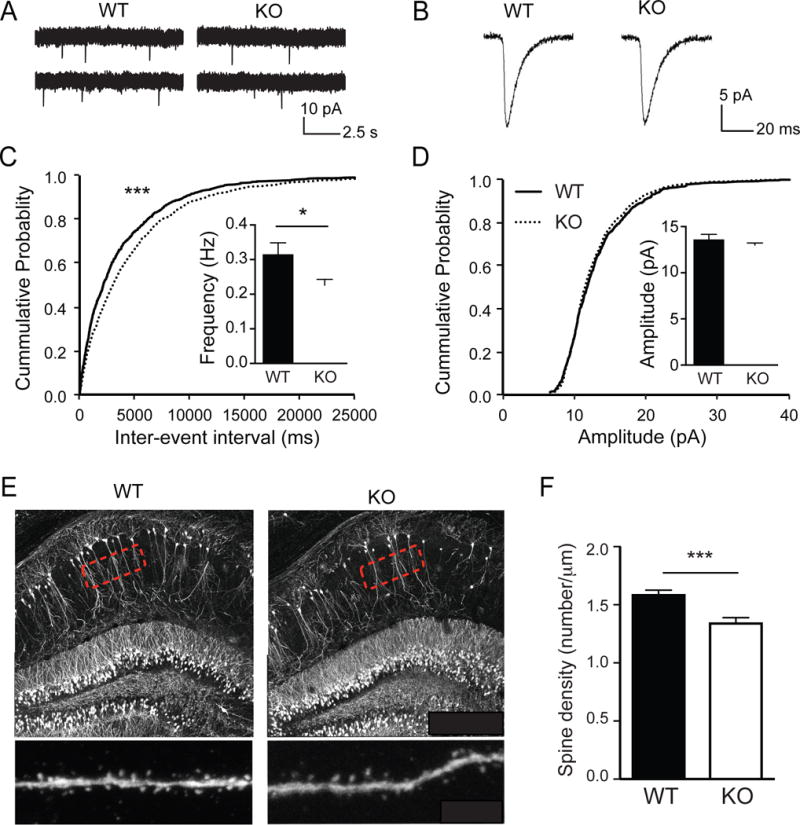
(A) Representative traces of spontaneous mEPSCs recorded from hippocampal CA1 neurons of juvenile Grasp1 mice.
(B) Representative traces of individual mEPSCs.
(C) Quantifications of mEPSC frequency and cumulative distributions of the inter-mEPSC event intervals. mEPSC frequency is lower in Grasp1 KO compared to WT littermates (WT= 0.32 ± 0.03 and KO= 0.23 ± 0.02Hz; n= 18–19 neurons/genotype; p= 0.03, Mann-Whitney test; p< 0.001, Kolmogorov-Smirnov test).
(D) Quantifications of mEPSC amplitude and cumulative distributions of the mEPSC amplitudes. mEPSC amplitudes are comparable between WT and KO littermates (WT= 13.6 ± 0.5 and KO= 13.0 ± 0.2pA, n= 180–19 neurons/genotype; p= 0.28, Mann-Whitney test; p= 0.42, Kolmogorov-Smirnov test).
(E) Representative images of CA1 neurons from Thy1-GFP × Grasp1 mice. High magnification images of secondary dendrites indicated by the highlighted regions were taken for spine number quantifications.
(F) Quantifications of spine density in (E). Spine density is lower in Grasp1 KO compared to WT littermates (WT= 1.50 ± 0.03 and KO= 1.28 ± 0.04, n= 12 neurons/genotype; p < 0.001, unpaired t-test).
All data were presented as mean ± S.E.M. *, p< 0.05; **, p< 0.01, ***, p< 0.001.
Grasp1 KO mice show deficient synaptic plasticity
To test whether GRASP1 influences synaptic plasticity, we examined NMDAR-mediated LTP induced by theta burst stimulation (TBS). LTP induction resulted in an initially less potentiated field excitatory postsynaptic potential (fEPSP) and remained reduced for at least one hour in Grasp1 KO mice (Figures 2A–C). Since this form of LTP results from an increase in postsynaptic AMPARs (Huganir and Nicoll, 2013), these results suggest that GRASP1 is required for appropriate delivery of synaptic AMPARs in response to TBS stimulation. On the other hand, NMDAR-mediated LTD was not different between KO and WT mice (Figures 2D&E), suggesting a specific role of GRASP1 in activity-dependent incorporation but not removal of synaptic AMPARs.
Figure 2. Grasp1 KO mice exhibit impaired NMDAR-dependent LTP but normal LTD and presynaptic function.
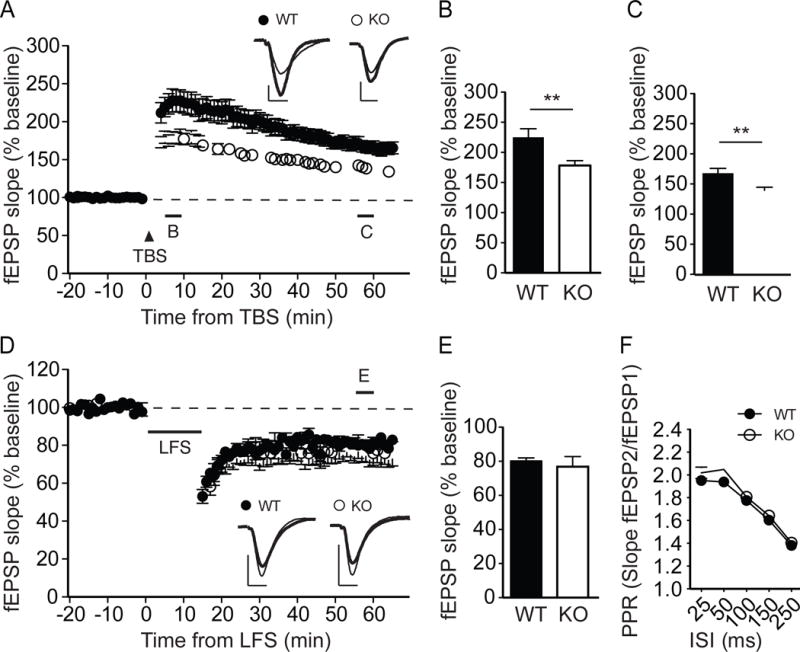
(A) TBS-induced LTP at the Schaffer collateral-CA1 synapses from juvenile Grasp1 mice. Sample traces represent fEPSPs at one min before (thin) and one hour after (thick) stimulation. Scale bars represent 0.5mV (vertical) and 5ms (horizontal).
(B) The magnitude of TBS-LTP is reduced in Grasp1 KO mice at the initial phase (5–10 min, WT= 225.3 ± 13.9 and KO= 178.3 ± 7.9%, n= 24 slices/genotype; p= 0.008, unpaired t-test).
(C) The magnitude of TBS-LTP is reduced in Grasp1 KO mice at the maintenance phase (55–60 min, WT= 167.7 ± 7.9 and KO= 139.4 ± 5.0%, n= 24 slices/genotype; p= 0.005, unpaired t-test).
(D) LFS-induced LTD. Sample traces represent fEPSPs at one min before (thin) and one hour after (thick) stimulation.
(E) LFS-LTD is comparable between Grasp1 WT and KO mice (55–60 min, WT= 80.1 ± 1.9 and KO= 76.8 ± 5.9%, n=11–14 slices/genotype; p=0.60, unpaired t-test).
(H) Paired-pulse ratios (PPRs) with different inter-stimulus intervals (ISIs) are comparable between Grasp1 WT and KO littermates (25ms: WT= 2.0 ± 0.04 and KO= 2.0 ± 0.05; 50ms: WT= 1.9 ± 0.03 and KO= 2.0 ± 0.04; 100ms: WT= 1.8 ± 0.03 and KO=1.8 ± 0.03; 150ms: WT= 1.6 ± 0.02 and KO = 1.6 ± 0.03; 250ms: WT= 1.4 ± 0.01 and KO= 1.4 ± 0.02; n= 32–34 slices/genotype; p> 0.05, two-way ANOVA).
We further tested whether GRASP1 regulates presynaptic function by comparing paired-pulse ratios (PPRs), a standard approach to assess neurotransmitter release probability. Grasp1 KO mice showed comparable PPRs in a wide range of inter-stimulus intervals suggesting that GRASP1 does not regulate presynaptic vesicle release (Figure 2F). Together with the observation that Grasp1 KO mice had fewer spines, we concluded that the deficient mEPSC frequency reflects primarily a reduction in glutamatergic inputs onto hippocampal CA1 neurons.
Grasp1 KO mice display impaired cognitive function and learning-induced synaptic AMPAR delivery
Next we tested whether GRASP1 is required for normal animal behavior using the Morris water maze (MWM) task, a hippocampal-dependent learning and memory assay (Morris, 1984). To test spatial learning, adult (2–5 months old) Grasp1 mice were trained to locate a hidden platform in association with four visual cues in a water maze. Mice were automatically tracked and their swim latencies and distances to the platform were analyzed for 8 sessions over a 4-day period. During the initial training sessions, KO and WT mice spent similar time and swam a similar distance to find the hidden platform suggesting they have comparable swim and motor capability (Figure 3A). However, WT mice learned to locate the platform rapidly and reached their plateau performance by the 5th session whereas the KO mice did not achieve similar latency until session 7 (Figure 3A). Similar learning curves were observed with swim distance analysis (Figure S2C). To test spatial memory, we conducted a probe test one day after the final training session in which mice were exposed to the same arena but the platform was removed. In contrast to WT mice, which spent significantly more time in the target quadrant, KO mice showed no spatial preference, and made fewer crossings over the prior platform location (Figures 3B&C), indicating their impaired memory retention. When exposed to the same arena with a visible platform at different quadrants, mice of either genotype showed similar latencies and distances to the platform indicating their normal visual perception and swimming capability (Figures S2D&E). Together, these data suggest that GRASP1 is critical for hippocampal-dependent spatial learning and memory.
Figure 3. Grasp1 KO mice show impaired learning and memory as well as learning-induced synaptic AMPAR incorporation.
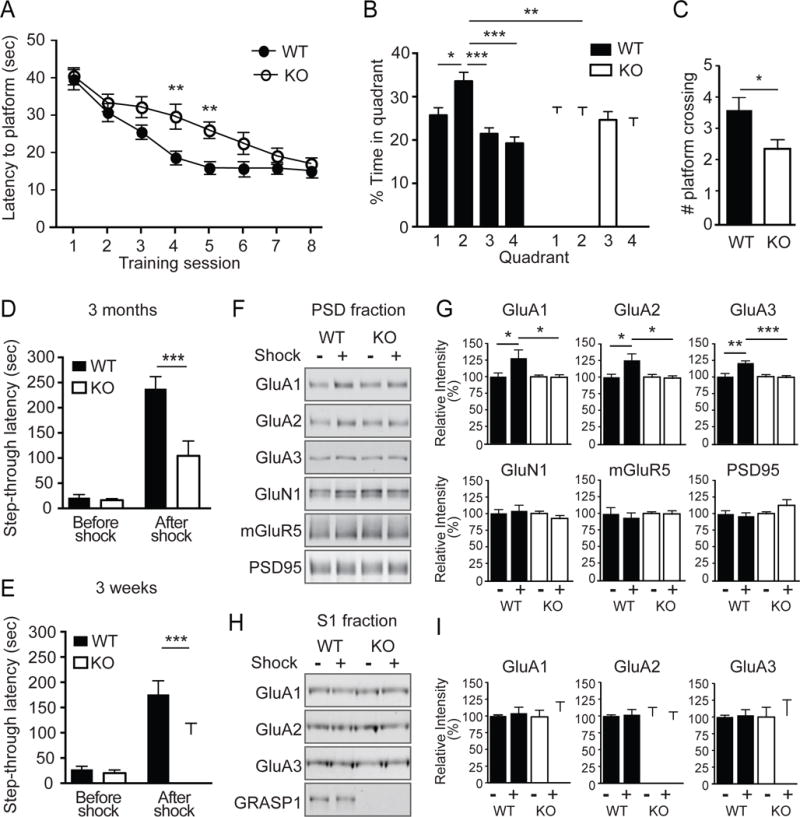
(A) Spatial learning curves measured as the latency to find the hidden platform during Morris water maze (MWM) training. Grasp1 KO mice spent a longer time to locate the hidden platform at sessions 4 and 5. (session 4: WT= 18.7 ± 1.6 and KO= 29.8 ± 3.4 sec, p= 0.003; session 5: WT= 16.0 ± 1.1 and KO= 26.1 ± 2.2 sec, p= 0.009, n=20 mice/genotype, two-way ANOVA; see Table S1 for values of all sessions).
(B) Percentage of time spent in each quadrant in probe trials. Grasp1 WT but not KO showed a significant preference for quadrant 2, the target quadrant (% time in quadrant 2: WT= 33.6 ± 2.0 and KO = 25.8 ± 1.8%; p= 0.005, two-way ANOVA; see Table S2 for values of all quadrants).
(C) Number of platform crossings in probe trials is less for Grasp1 KO mice (WT= 3.7 ± 0.4 and KO= 2.4 ± 0.3; p= 0.013, unpaired t-test).
(D) Inhibitory avoidance (IA) learning/memory measured as the tendency for the animals to avoid the dark chamber where they were shocked during IA training. Adult Grasp1 KO mice show shorter latency to crossover to the dark chamber after IA training (before shock: WT= 21.5 ± 6.2 and KO= 16.3 ± 3.3 sec; after shock: WT= 237.8 ± 24.2 and KO= 104.5 ± 29.4 sec; n= 10–13 mice/genotype; p< 0.001, two-way ANOVA).
(E) Juvenile Grasp1 KO mice showed shorter crossover latency (before shock: WT= 27.4 ± 6.2 and KO= 20.2 ± 6.1 sec; after shock: WT= 175.9 ± 27.0 and KO= 97.4 ± 21.4 sec, n= 16 mice/genotype; p< 0.001, two-way ANOVA).
(F) Representative western blots of postsynaptic density (PSD) proteins from dorsal hippocampus of Grasp1 mice with or without shock during IA training.
(G) Quantifications of (F). AMPARs are enriched in PSD fraction after shock during IA training in Grasp1 WT mice but not KO littermates (% non-shocked control; GluA1: WT= 130.3 ± 10.5 and KO= 97.0 ± 4.4, p=0.03; GluA2: WT= 125.6 ± 8.1 and KO= 98.8 ± 4.8, p=0.03; GluA3: WT= 120.8 ± 2.8 and KO= 98.0 ± 3.4, p< 0.001; GluN1: WT= 107.3 ± 7.3 and KO= 90.4 ± 5.4; mGluR5: WT= 94.1 ± 7.1 and KO= 99.2 ± 5.2; PSD95: WT= 96.8 ± 4.2 and KO mice= 114.9 ± 10.5; n=6 mice/group, p>0.05 if not stated, unpaired t-test).
(H) Representative western blots of post-nuclear supernatant1 (S1) fraction proteins.
(I) Quantifications of (H). GluA1,2 and 3 levels are comparable among all conditions (% non-shocked control; WT mice: GluA1= 104.1 ± 8.9; GluA2= 101.9 ± 7.7; GluA3= 102.5 ± 8.4; KO mice: GluA1= 108.6 ± 13.9; GluA2= 94.9 ± 11.1; GluA3= 102.3 ± 23.3, p>0.05; n=4–7 mice/group, unpaired t-test).
Learning induces LTP in the hippocampus in vivo by incorporating more AMPARs into synapses (Whitlock et al., 2006), whereas blocking synaptic delivery of AMPARs is known to impair hippocampus-dependent learning (Mitsushima et al., 2011). To test whether GRASP1 regulates experience-dependent synaptic AMPAR delivery in vivo, we used inhibitory avoidance (IA), a single trial associative learning task previously shown to induce LTP and synaptic AMPAR incorporation in the dorsal hippocampus (Cammarota et al., 1998; Whitlock et al., 2006). Adult mice were first habituated to an arena consisting of light and dark chambers separated by a guillotine-style gate. When introduced to the light side, both WT and KO littermates shortly entered the dark chamber due to a natural preference for a dark environment (Figures S2F& 3D). During IA training, a mild foot-shock was delivered to the mice upon entering the dark chamber. When tested 24 hours after shock, WT animals displayed a long step-through latency, showing clear associative memory, while Grasp1 KO mice showed significantly shorter step-though latency indicating their impairment in IA learning/memory (Figure 3D). Because impaired synaptic connectivity and LTP were observed in juvenile mice, we further tested IA on 3-week old Grasp1 mice. Similarly, juvenile KO mice showed a shorter step-through latency to the dark chamber after shock (Figure 3E), indicating the IA learning/memory impairment occurs early during development.
To test whether GRASP1 is required for learning-induced synaptic AMPAR delivery, we isolated the postsynaptic density (PSD) fraction, highly enriched in synaptic proteins, from dorsal hippocampus of adult Grasp1 mice 30 minutes following IA training. Consistent with previous findings, we found that in WT animals, IA training increased the levels of GluA1, 2 and 3 AMPAR subunits compared to littermates that did not receive a foot-shock (Figures 3F&G). The learning-induced increase in synaptic glutamate receptors is AMPAR-specific because we did not detect changes in the NMDAR subunit GluN1 or the metabotropic glutamate receptor 5 (mGluR5). Remarkably, this learning-induced increase in AMPAR is completely abolished in KO mice (Figures 3F&G). AMPARs in the post-nuclear supernatant 1 (S1) fraction remain comparable regardless the genotype and IA training, indicating that GRASP1 is specifically required for synaptic delivery of AMPARs during IA learning (Figures 3H&I). Taken together, these data demonstrate a critical role for GRASP1 in spatial (MWM) and associative (IA) learning and memory, likely via regulation of activity-dependent AMPAR trafficking and synaptic potentiation.
Neuronal activity recruits GRASP1 to synapses and increases its association with AMPARs
To understand the cellular mechanisms by which GRASP1 enhances AMPAR synaptic targeting during LTP, we used primary neurons with a well-established glycine-induced, NMDAR-dependent chemical LTP (cLTP) protocol that elicits synaptic AMPAR insertion supplied from recycling endosomes (Lu et al., 2001; Park et al., 2004). Consistent with previous reports, we observed a 20–30% increase in synaptic GluA1-3 subunits in PSD fractions upon glycine treatment (Figures 4A&B). Interestingly, we also detected drastic increases of GRASP1 and GRIP1 in the PSD, substantially larger than AMPARs (Figures 4A&B). We did not observe significant differences in the endocytosis trafficking protein, PICK1, or another GRASP1-interacting endosomal protein, Stx13 (Figures 4A&B). Notably, we saw no changes in total cellular levels of these proteins before and after cLTP indicating that the PSD enrichments of GRASP1, GRIP1 and AMPARs are as a result of trafficking events rather than an effect on total protein levels (Figures 4A&C).
Figure 4. Neuronal activity recruits GRASP1 to synapses and increases GRASP1-GluA1 interaction.
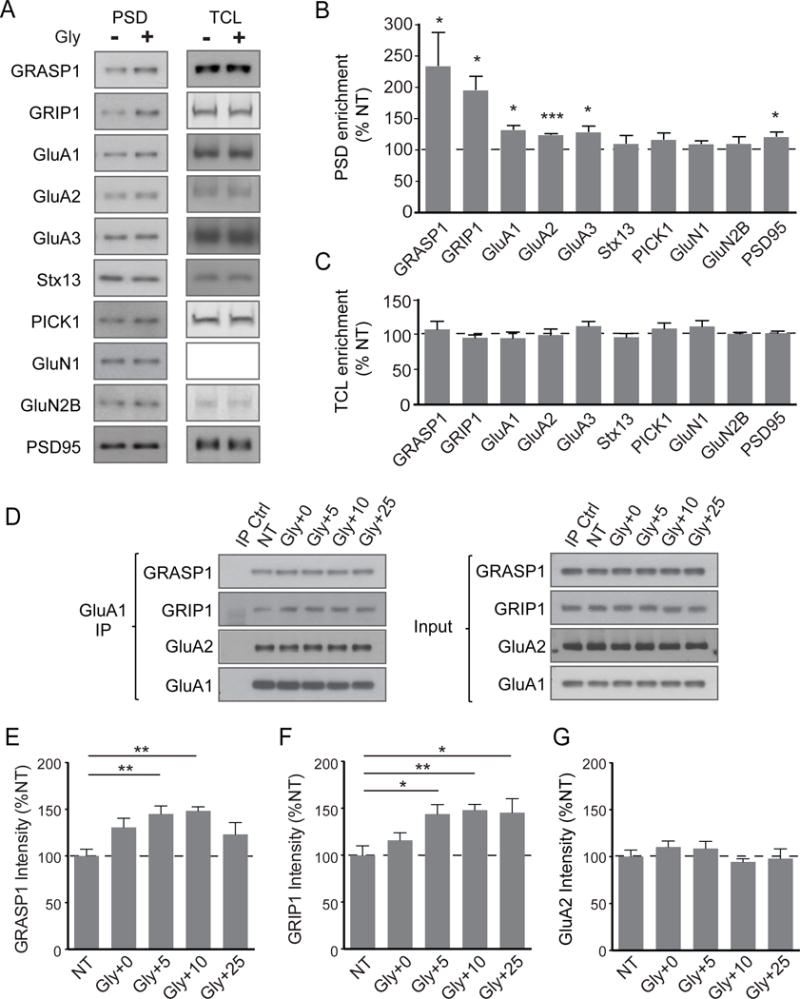
(A) Representative western blots of proteins from PSD and total cell lysate (TCL) isolated from cortical neurons either not treated (NT) or 20 minutes after a 5-minute glycine treatment.
(B) Quantifications of PSD proteins in (A). (% NT: GRASP1= 213.8 ± 32.7, p= 0.04; GRIP1 = 195.5 ± 21.0%, p= 0.02; GluA1= 131.9 ± 6.0, p= 0.013; GluA2= 123.6 ± 1.3, p< 0.001; GluA3= 128.3 ± 8.6, p= 0.046; Stx13= 109.9 ± 12.1; PICK1= 116.0 ± 10.0; NR1= 109.5 ± 3.9; NR2B= 110.0 ± 9.6; PSD95= 121.0 ± 6.4, p= 0.045; n= 4/group, one-way ANOVA).
(C) Quantifications of TCL proteins in (A). (% NT: GRASP1= 106.8 ± 11.4; GRIP1= 94.9 ± 3.9%; GluA1= 94.3 ± 8.7; GluA2= 98.7 ± 8.6; GluA3= 111.3 ± 6.6; Stx13= 95.5 ± 5.4; PICK1= 107.9 ± 7.9; NR1= 110.9 ± 8.3; NR2B= 100.4 ± 2.2; PSD95= 101.5 ± 2.8; n= 7/group, p>0.05, one-way ANOVA).
(D) Representative western blots showing co-immunoprecipitation of GluA1 with GRASP1, GRIP1 and GluA2 from neurons not treated (NT) or glycine treated for 5 minutes followed by a 0–25 minute recovery.
(E) Quantifications of GRASP1 in (D). (% NT: NT=100.0 ± 7.3; Gly+0= 130.6 ± 9.8; Gly+5= 145.0 ± 8.6, p= 0.005; Gly+10= 148.4 ± 4.3, p= 0.002; Gly+25= 123.1 ± 12.6; n= 7/group, oneway ANOVA).
(F) Quantifications of GRIP1 in (D). (% NT: NT: 99.9 ± 10.1; Gly+0= 115.9 ± 8.1; Gly+5= 144.0 ± 9.9, p= 0.019; Gly+10= 148.0 ± 6.2, p= 0.009; Gly+25= 145.4 ± 14.8, p= 0.015; n= 7/group, one-way ANOVA).
(G) Quantifications of GluA2 in (D). (% NT: NT= 100.2 ± 6.8; Gly+0= 110.4 ± 6.23; Gly+5= 108.8 ± 7.6; Gly+10= 94.4 ± 3.4; Gly+25= 98.0 ± 10.3; p> 0.05; n= 5/group, one-way ANOVA).
If GRASP1 and GRIP1 were actively recruited to synaptic fractions to facilitate AMPAR insertion during plasticity, we would expect to see an increase in their interaction with AMPARs upon glycine treatment. To test this, we immunoprecipitated AMPARs by a GluA1 antibody from cultured neurons before or after glycine treatment at various times. The presence of GRASP1 and GRIP1 in the immunoprecipitates indicated that GRASP1 is part of the GluA1/2 AMPAR protein complex (Figures 4D–G). Moreover, we observed that both GRASP1-AMPAR and GRIP1-AMPAR associations were significantly enhanced at 5 and 10 minutes following glycine stimulation (Figures 4D–F). Together, these data suggest that GRASP1 and GRIP1 function cooperatively to facilitate synaptic AMPAR delivery during activity-dependent synaptic potentiation.
Identification of GRASP1 mutations in X-linked intellectual disability patients
Given GRASP1’s function in neuronal connectivity, synaptic plasticity and learning/memory in mice, we hypothesized that GRASP1 may be important for human cognitive function. Interestingly, GRASP1 is an X-Chromosome gene located at Xp11.23, a region that has been previously associated with ID and autism (Chung et al., 2011; Edens et al., 2011; Giorda et al., 2009). We performed a screen on a cohort of well-characterized male probands (n=400) with ID and a pedigree consistent with X-Iinked (XL) inheritance (Wu et al., 2007; Zhang et al., 2007). In one XL-ID family, we identified a GRASP1 missense mutation, c.2465G>A; p.R822Q, in two affected brothers with severe ID. This mutation was segregated with the ID phenotype in this family (Figure S3) and was not found in a screen of 1,377 males with normal cognitive function. X chromosome-exome sequencing of the affected brothers identified no additional causal mutation for the ID phenotype (Niranjan et al., 2015). Further screening of this XL-ID cohort identified another missense mutation, c.218G>A; p.S73N, in a 28 years old male with severe ID and a pedigree consistent with X-linked inheritance (BH0092, Human Genetics Collection, ECACC, Salisbury, U.K.). This mutation was not found in a screen of 1,295 males with normal cognitive function. Interestingly, both R822Q and S73N mutations involve evolutionarily conserved residues suggesting that these sites may be important for GRASP1 function (Figure 5B). Two polymorphic GRASP1 missense variants, c.536C>T; p.P179L and c.1733A>C; p.E578A were identified in both ID patients and normal controls in this study.
Figure 5. GRASP1 intellectual disability (ID) mutations and their effects on basal-and activity-dependent AMPAR trafficking.
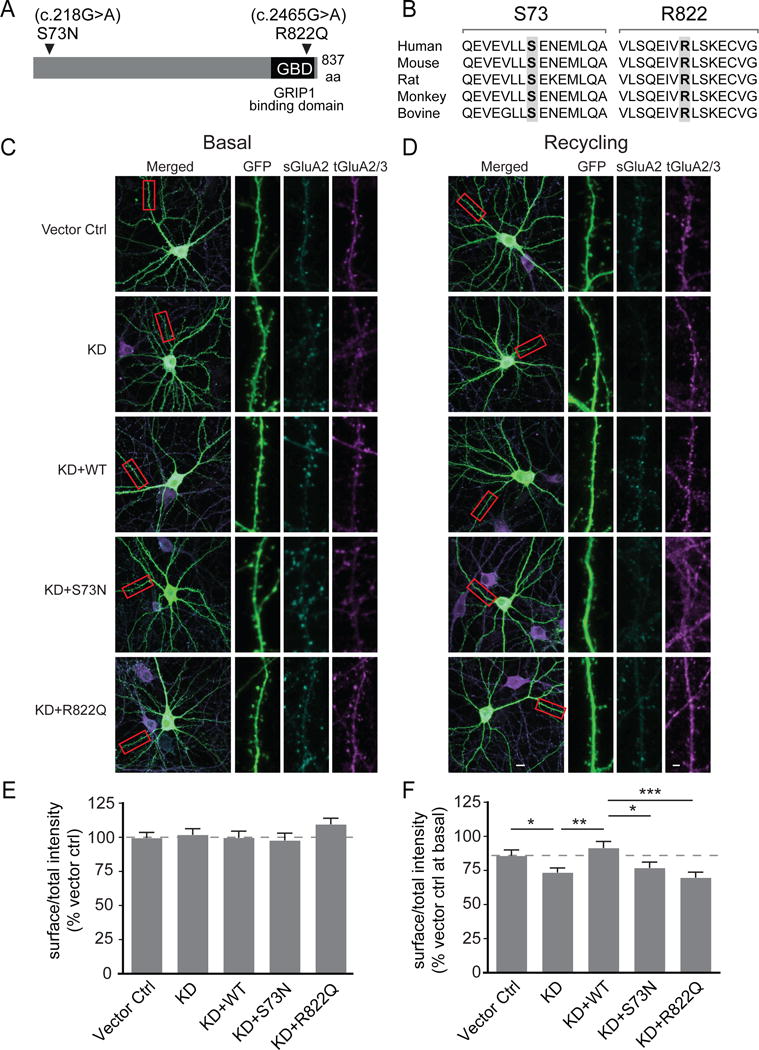
(A) Schematic of GRASP1 protein with a GRIP1 binding domain (GBD) and locations of GRASP1 mutations found in severe ID patients.
(B) Sequence alignments of GRASP1 flanking the ID mutations.
(C) Representative images of rat hippocampal neurons and dendritic segments transfected with GFP and a control vector (Vector ctrl), shRNA for GRASP1 protein knockdown (KD), or KD plus either GRASP1 WT, S73N, or R822Q with mock treatment. A highlighted dendritic region (red box) is shown at higher magnification with separate channels to show GFP, surface GluA2 (sGluA2) and total GluA2/3 (tGluA2/3) staining.
(D) Same as (C) except that neurons were AMPA-treated to induce AMPAR recycling.
(E) Quantifications of (C). Integrated intensity of sGluA2, normalized to tGluA2/3 signal, is comparable between all groups. (% Vector control: Vector ctrl= 99.9 ± 4.3; KD= 102.8 ± 4.5; KD+WT= 100.1 ± 4.8; KD+S73N= 98.2 ± 5.4; KD+R822Q= 110.0 ± 4.6; n= 84–96 dendrites from 21–24 neurons/group; p>0.05; one-way ANOVA).
(F) Quantifications of (D). Integrated intensity of s/tGluA2(3) were normalized to vector ctrl at basal state to quantify AMPA-induced AMPAR recycling. (% Vector ctrl at basal: Vector ctrl= 86.4 ± 4.2 and KD= 74.0 ± 3.3, p= 0.04; KD+WT= 92.1 ± 4.8 and KD, p=0.002; KD+WT and KD+S73N= 77.4 ± 4.2, p=0.015; KD+WT and KD+R822Q= 70.3 ± 4.1, p=0.0002; n= 88–100 dendrites from 22–25 neurons/group; one-way ANOVA).
GRASP1 ID mutants show impaired activity-dependent AMPAR trafficking
To test whether GRASP1 ID mutations impact endosomal recycling of AMPARs, we employed a molecular replacement approach to examine AMPA-induced AMPAR recycling. Hippocampal neurons were transfected with GFP alone, or together with a shRNA targeting endogenous GRASP1 for protein knockdown (KD) and shRNA-resistant GRASP1 WT, S73N or R822Q. Transfected neurons were stimulated with or without AMPA for two minutes to induce AMPAR internalization followed by a 58-minute recovery period during which internalized AMPARs recycle back to the cell surface (Hoogenraad et al., 2010). Surface levels of endogenous GluA2 (sGluA2) were used as an indicator to evaluate the effect of GRASP1 ID mutations on AMPAR recycling since we did not observe a difference in the AMPA-induced AMPAR internalization in GRASP1 ID mutant-replaced cells (Figure S4). In mock treated neurons, sGluA2 signals were comparable among all groups demonstrating that neither loss of GRASP1 nor expression of GRASP1 ID mutants affects basal AMPAR surface expression (Figures 5C&E). However, 58 min following AMPA stimulation, when most AMPARs had recycled back to surface in controls, KD neurons showed significantly less sGluA2 suggesting that GRASP1 is required for efficient AMPAR recycling (Figures 5D&F). We observed a full rescue of the sGluA2 level when neurons were co-transfected with a shRNA resistant WT GRASP1. However, neither of the ID mutations rescued the reduced sGluA2 indicating a convergent effect of these ID mutants on activity-dependent AMPAR recycling (Figures 5D&F).
GRASP1 ID mutants accumulate AMAPR in recycling endosomes and exhibited altered binding to endosomal proteins
To dissect the cellular mechanism underpinning recycling deficits in these neurons expressing mutant GRASP1, we labeled the internalized GluA2 (iGluA2) and systematically examined their colocalizations with early, recycling and late endosomes with well-known markers, early endosome antigen 1 (EEA1), Stx13 and lysosomal-associated membrane protein 1 (LAMP1), respectively. Neurons transfected with GFP, KD, and shRNA-resistant GRASP1 WT, S73N or R822Q were first live-labeled with antibodies against extracellular domain of GluA2 subunits, followed by AMPA stimulation to induce AMPAR/antibody internalization and subsequent recycling. 43-min after AMPA treatment, we blocked the surface GluA2 antibodies with non-conjugated secondary antibodies. After fixation and permeablization, iGluA2 antibodies were visualized by fluorescent-conjugated secondary antibodies to analyze their percent colocalization with each endosome marker. We found that the remaining iGluA2 in neurons replaced with either ID mutants (S73N and R822Q) showed significantly more colocalizations with Stx13 than the WT neurons (Figure 6B). The colocalizations of iGluA2 with either EEA1 or LAMP1 are, however, comparable in all neurons (Figures 6A&C) indicating that the internalized AMPARs likely accumulated at recycling endosomes but not in early or late endosomes. These results further strengthened our argument that the reduced sGluA2 following AMPA stimulation in ID mutant-replaced neurons (Figures 5D&F) are as a result of recycling effect, but not AMPAR degradation.
Figure 6. Effects of GRASP1 ID mutants on the internalized AMPAR trafficking and their interactions with endosomal proteins.
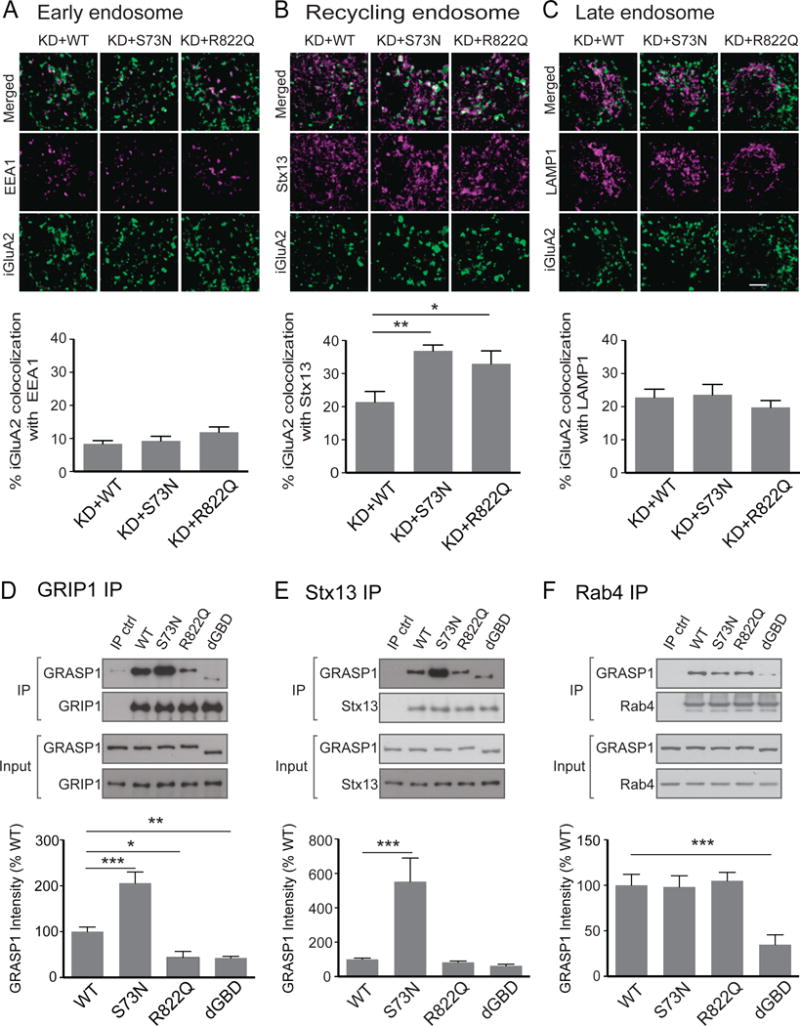
(A) Colocalization (white) of internalized GluA2 (iGluA2, green) and EEA1 (magenta) in cell bodies of hippocampal neurons transfected with a shRNA for GRASP1 knockdown (KD) and either GRASP1 WT or ID mutants (S73N, R822Q). (% iGluA2 colocalization: KD+WT= 8.4 ± 1.0; KD+S73N= 9.3 ± 1.4; KD+R822Q= 12.0 ± 1.5; n= 23–24 neurons/group, p> 0.05, one-way ANOVA).
(B) Colocalization of iGluA2 and Stx13 (magenta). (% iGluA2 colocalization: KD+WT= 21.4 ± 3.2 and KD+S73N= 36.9 ± 1.8, p= 0.002; KD+WT and KD+R822Q= 32.9 ± 3.9, p= 0.021, n= 1719 neurons/group, one-way ANOVA).
(C) Colocalization of iGluA2 and LAMP1 (magenta). (% iGluA2 colocalization: KD+WT= 22.8 ± 2.5and KD+S73N= 23.6 ± 3.1; KD+WT and KD+R822Q= 19.8 ± 2.1, n= 17 neurons/group, p> 0.05, one-way ANOVA).
(D) Co-immunoprecipitation of GRIP1 and either GRASP1 WT, ID mutants or GBD deletion mutant (dGBD) from transfected HEK cells. (% WT: WT= 100.0 ± 4.5 and S73N= 206.0 ± 24.1, p< 0.001; WT and R822Q= 43.7 ± 6.6, p= 0.011; WT and dGBD = 42.6 ± 3.6, p= 0.007, n= 4–5/group, one-way ANOVA).
(E) Co-immunoprecipitation of Stx13 and GRASP1s. (% WT: WT= 100.0 ± 7.4 and S73N= 552.6 ± 137.3, p< 0.001; R822Q= 83.2 ± 7.1; dGBD= 62.7 ± 8.9; n=7/group, one-way ANOVA).
(F) Co-immunoprecipitation of Rab4 and GRASP1s. (% WT: WT= 100.0 ± 12.0 and dGBD= 34.8± 10.2; p< 0.001; S73N= 98.2 ± 12.2; R822Q= 105.0 ± 9.2; n=5/group; one-way ANOVA).
To understand how GRASP1 ID mutations impact endosomal recycling at the molecular level, we examined the interaction of GRASP1 ID mutants with all known endosomal proteins reported to interact with GRASP1, including GRIP1, Stx13 and Rab4. Notably, R882Q is located on a previously identified GRIP1 binding domain (GBD)(Figure 5A) (Ye et al., 2000) and thus may affect GRIP1 interaction. To test the interaction of GRASP1 with GRIP1, we co-expressed GFP-tagged GRIP1 with either WT, ID mutants (S73N or R822Q) or a GBD deletion mutant (dGBD) of GRASP1 in HEK293 cells. The expression levels of mutant and WT GRASP1 are comparable suggesting that these mutations do not affect protein stability (Input, Figures 6D–F). We then immunoprecipitated GRIP1-containing protein complexes with GFP antibodies and performed western blot analysis to quantify the amount of GRASP1 in the GRIP1 protein complexes. As predicted, R822Q and dGBD mutants showed a significant decrease in GRIP1 binding (Figure 6D). Notably, the reduction of R822Q bound to GRIP1 was similar to dGBD, which deleted the major GRIP1 binding domain, demonstrating a strong disruptive effect of R822Q in GRIP1 interaction. In contrast, S73N showed a two-fold increase in GRIP1 binding (Figure 6D). To test the interaction of GRASP1 with Stx13, we immunoprecipitated endogenous Stx13-containing protein complexes with Stx13 antibodies. S73N mutant showed a dramatic increase (> 5 fold) in binding to Stx13 compared to GRASP1 WT, while R822Q and dGBD showed comparable interactions with Stx13 (Figure 6E). We did not observe any differences between Rab4 interaction with WT or ID mutants, whereas dGBD showed a significant decrease in binding to Rab4 in cells expressing GFP-tagged Rab4 and GRASP1 WT or mutants (Figure 6F). In summary, these results suggest that ID-associated GRASP1 mutations may impair brain function via altered GRASP1 interaction with GRIP1 and/or Stx13-mediated endosomal machinery. Moreover, the impact of ID mutations is likely to be independent of Rab4, or downstream of Rab4-mediated recycling endosome trafficking.
GRASP1 ID mutants impair synaptic plasticity at single spines and fail to rescue spine number deficit in Grasp1 KO animals
To examine functional impacts of GRASP1 ID mutants on synaptic delivery of AMPARs and synaptic structure, we performed live-imaging of GluA1/2 tagged with SEP, a pH-sensitive GFP that becomes fluorescent only when receptors are on the cell surface, as well as spine morphology with a cell-fill dsRed2 to monitor dynamic changes of AMPAR content and spine size during LTP. Consistent with previous findings (Matsuzaki et al., 2004), patterned activity delivered by glutamate uncaging induces LTP with a rapid and stable increase in SEP-AMPARs on uncaged spines and enlargements of the uncaged spines in control neurons (Figures 7A–F). The LTP-induced increases in AMPAR content and spine size were, however, reduced in neurons with GRASP1 KD (Figures 7A–F). Although we observed full rescue of these KD phenotypes with a hairpin resistant WT GRASP1, replacement with either S73N or R822Q failed to do so (Figures 7A–F), indicating that both ID mutants produce deficits in activity-dependent synaptic efficacy and spine structure.
Figure 7. Functional impacts of GRASP1 ID mutants on synaptic plasticity of single spines and spine number of Grasp1 animals.
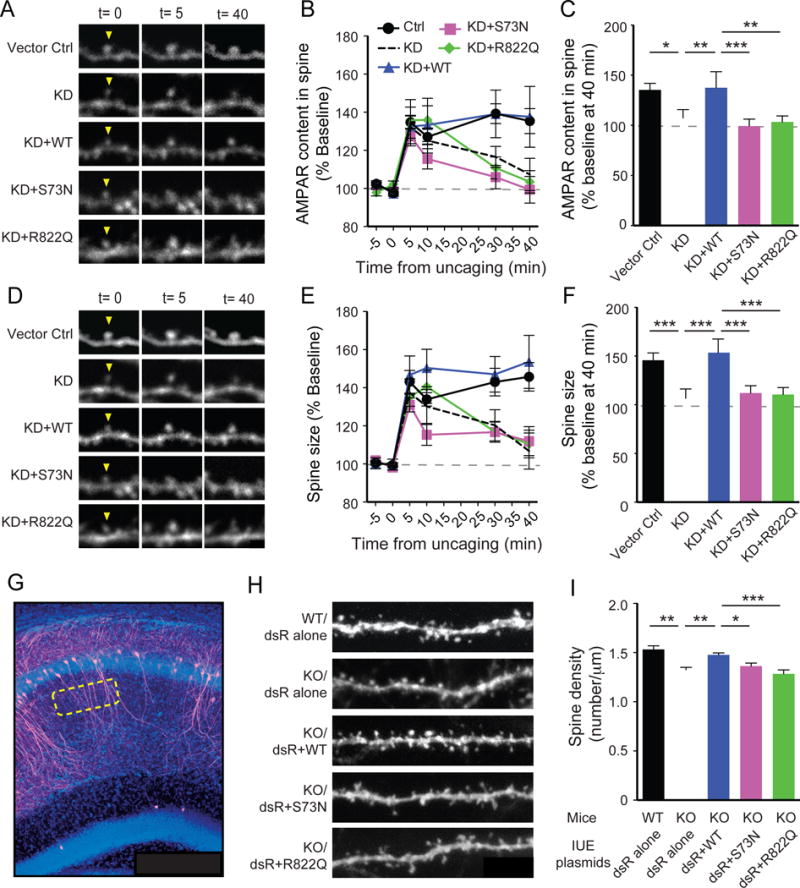
(A) Representative spine images of SEP-GluA1/2 showing the dynamic changes in spine AMPAR content before and after two-photon glutamate uncaging-induced plasticity.
(B) Quantifications of (A) at various times.
(C) Statistical analysis of AMPAR content at 40-minute in (B). (Vector ctrl = 138.5 ± 6.5 and KD = 107.4 ± 8.5, p< 0.001; KD+WT= 137.7 ± 16.0 and KD, p= 0.003; KD+WT and KD+S73N = 99.3 ± 7.0, p= 0.03; KD+WT and KD+R822Q = 103.5 ± 5.9, p< 0.001; n= 10–11 spines/group, two-way ANOVA).
(D) Representative spine images of DsRed2 (dsR) showing the dynamic changes in spine size.
(E) Quantifications of (D) at various times.
(F) Statistical analysis of spine size at 40-minute in (E). (Vector ctrl = 145.7 ± 7.5 and KD = 106.8 ± 9.4, p= 0.010; KD+WT= 153.7 ± 13.9 and KO, p= 0.006; KD+WT and KD+S73N = 112.1 ± 7.7, p< 0.001; KD+WT and KD+R822Q = 110.5 ± 7.3, p= 0.001; n= 10–11/group, two-way ANOVA).
(G) Representative image of a hippocampal slice with sparsely in utero electroporated CA1 neurons (magenta) and DAPI staining (blue) in Grasp1 mice.
(H) High magnification images of secondary dendrites from dsRed2 (dsR)-positive CA1 neurons co-electroporated with indicative constructs in Grasp1 mice.
(I) Quantifications of spine density in (H). (WT/dsR alone= 1.53 ± 0.04 and KO/dsR alone= 1.33 ± 0.02, p< 0.001; KO/WT= 1.48 ± 0.02 and KO/dsR alone, p= 0.005; KO/WT and KO/S73N= 1.36 ± 0.03, p= 0.048; KO/WT and KO/R822Q= 1.28 ± 0.04; p<0.001; n= 10 neurons/group, one-way ANOVA).
To investigate the influences of GRASP1 ID mutants in vivo we delivered GRASP1 WT or ID mutants into CA1 neurons of Grasp1 animals by in utero electroporation and examined their effects on synapse number at P21. Consistent with our findings obtained from Thy1-GFP mice, we observed that dsRed2 expressing neurons have less spines in KO mice compared to WT mice (Figures 7G–I). This deficit in Grasp1 KO mice can be rescued by expression of WT GRASP1 but not S73N or R822Q (Figures 7G–I), indicating that both ID-mutants can impact neuronal connectivity in mice.
Discussion
Endocytic membrane trafficking regulates many cellular processes by controlling lipids and membrane proteins on the cell surface. This machinery is particularly important for neurons considering the high membrane to cytoplasm ratio and the requirements for membrane receptors, channels and adhesion molecules for neuronal function. Recently, disrupted endocytic trafficking found in many brain disorders has attracted much attention. Defective endocytosis is implicated in schizophrenia and bipolar disorder (Dev and Henley, 2006; Schubert et al., 2012), impaired endosomal sorting is linked to Down’s syndrome (Wang et al., 2013) and altered endosomal recycling is associated with autism (Mejias et al., 2011). These discoveries together suggest a critical role for endocytic trafficking in human cognition, although the underlying molecular mechanisms remain largely unknown.
Here we provide direct physiological evidence for functions of the recycling endosome protein GRASP1 in neuronal connectivity and synaptic plasticity in the mammalian hippocampus. Mice lacking GRASP1 exhibited significant learning and memory impairments likely due to a failure in learning-induced synaptic AMPAR delivery. In support of a role for GRASP1 in cognition, we identified two GRASP1 mutations that segregate with severe ID phenotypes in unrelated XL-ID families. These mutations appear to drive accumulation of internalized AMPARs in the recycling endosome, likely due to altered bindings to key recycling endosomal proteins, GRIP1 and Stx13, and results in an impaired activity-dependent AMPAR recycling. Both ID mutants also showed convergent effects on blocking LTP-induced AMPAR insertion and spine enlargement at single spines as well as regulating spine density in CA1 pyramidal neurons in mice. Together, our results demonstrate critical roles of GRASP1 in regulating neuronal function and animal cognition, and link aberrant AMPAR recycling to ID etiology.
GRASP1 function in synaptic transmission
Our finding that Grasp1 KO mice have impaired LTP but normal mEPSC amplitude suggest that GRASP1 is selectively required for activity-dependent AMPAR trafficking and synaptic strengthening. Several studies in hippocampal neurons or slices have revealed the existence of multiple endocytic trafficking pathways for AMPARs. Basal synaptic transmission regulated by constitutive AMPAR trafficking pathway involves endocytosis that is clathrin/dynamin-independent, recycling that is mediated by TC10 GTPase, and exocytosis that is mediated by Rab8 and synaptobrevin2 (Esteban, 2008; Gu et al., 2016; Jurado et al., 2013; Zheng et al., 2015). The activity-dependent pathway, best studied in NMDAR-dependent LTP, involves endocytosis that is mediated by clathrin/dynamin, recycling mediated by Stx13, Rab11 and Rab8, and exocytosis that is mediated by SNAP47, Stx3 and synaptobrevin2 (Brown et al., 2007; Jurado et al., 2013; Park et al., 2004). Although there are shared proteins in both pathways, the constitutive and activity-dependent recycling are largely regulated by distinct molecular machinery and can be functionally uncoupled. Most key regulators in activity-dependent AMPAR trafficking, including Stx13, Rab11, SNAP47 and Stx3, do not affect basal synaptic transmission, similar to our findings with GRASP1 knockdown or molecular replacement of GRASP1 ID mutants. Activity-dependent AMPAR trafficking machinery therefore is engaged when certain signal transduction pathway(s) trigger their actions. Notably, GRASP1 mediates recycling endosome maturation by bridging early Rab4-positive endosomes and mature Rab11-positive recycling endosomes. Besides the well-established SNARE machinery that mediates final exocytosis step in the fusion of recycling endosomes and plasma membrane for activity-dependent AMPAR delivery, GRASP1 may serve as another gate for the regulation of the internal pools of receptors before final exocytosis to regulate the supply of AMPARs.
During LTP recycling endosomes have been observed to translocate into spines to promote synaptic AMPAR delivery (Esteves da Silva et al., 2015; Park et al., 2006; Patterson et al., 2010). Consistently, we found an ~2-fold enrichment of GRASP1 in the PSD fraction and an increased GRASP1/AMPAR association after cLTP stimulation in neurons. GRASP1-mediated recycling may therefore overcome the narrow spine neck barrier to facilitate fast and efficient AMPAR incorporation near synapses at given active spines to enhance synaptic strength. Whether GRASP1 is enriched only at active spines to perform input-specific AMPAR delivery required for proper experience-dependent synaptic strengthening and accurate memory encoding will be interesting to address in the future studies. Notably, the source for AMPAR supply is not restricted to spines. Several studies have shown that extrasynaptic plasma membrane pools of AMPARs are critical for LTP expression (Granger et al., 2013; Lin et al., 2009) and the lateral diffusion of AMPARs into spines affects synaptic AMPAR numbers during LTP (Makino and Malinow, 2009; Petrini et al., 2009). Further experiments are needed to examine whether GRASP1-mediated recycling also occurs at dendritic shafts and contributes to the lateral diffusion-dependent synaptic AMPAR incorporation. Local shaft insertion in response to activity may facilitate AMPAR delivery to neighboring, non-active synapses and underlie clustered synaptic plasticity (Harvey and Svoboda, 2007; Makino and Malinow, 2011).
Notably, the role of GRASP1 in activity-dependent synaptic delivery of AMPARs is not subunit specific. Although the traditional view of LTP is GluA1 driven, these findings rely heavily on the trafficking of ectopically overexpressed GluA1 or GluA2, which is known to preferentially forms homomers when overexpressed alone (Makino and Malinow, 2009; Shi et al., 1999). However, the majority of AMPARs at Schaffer collateral-CA1 synapses consist of GluA1/2 and GluA2/3 heteromers (Wenthold et al., 1996). Therefore, it is not clear whether the trafficking of GluA1 or GluA2 homomers represents the trafficking of endogenous GluA1 or GluA2-containing AMPARs. Our results from glycine-stimulated cultured neurons and IA-trained animals showed that the PSD enrichment of endogenous AMPARs contains GluA1, 2 and 3 subunits, likely reflecting an increase in both GluA1/2 and GluA2/3 heteromers. Our findings are consistent with reports from another group (Whitlock et al., 2006), indicating that both GluA1 and GluA2 are recruited to synapse during learning in vivo. GRASP1 knockdown in culture neurons affect both GluA1 and GluA2 recycling to the same degree (Hoogenraad et al., 2010) further suggesting that GRASP1 regulates trafficking of both GluA1 and GluA2-containing AMPARs. Although GRASP1/GRIP1 do not interact with GluA1 directly, the direct binding capability of GRIP1 to GluA2 suggests that GRASP1 can regulate GluA1-containing AMPARs in the form of GluA1/2 heterormers and GluA2 can be actively involved in AMPAR insertion during LTP. Our results from GluA1 IP during plasticity revealed a stable association of GluA1 with GluA2 subunits but rapid recruitment of GRASP1 and GRIP1 into GluA1 protein complex upon cLTP stimulation further support that GRASP1/GRIP1 can regulate the trafficking of GluA1-containing AMPAR trafficking in a form of GluA1/2 heteromers.
GRASP1 function in neuronal connectivity
Hippocampal CA1 neurons from juvenile Grasp1 KO mice exhibited decreased spine density and lower AMPA mEPSC frequency. In utero delivery of GRASP1 WT but not ID mutants rescues spine density deficits in Grasp1 KO mice indicating that mutations of GRASP1 compromise functional synapses and neuronal connectivity in the developing hippocampus. How does GRASP1 regulate synapse density? In cultured neurons, recycling endosomes positively regulate spine formation and structure. Interfering recycling endosome trafficking by manipulating Rab11 and Stx13 functions bi-directionally regulates spine density under basal states, likely through altering the supply of biological membrane (Hirling et al., 2000; Park et al., 2006). Loss of GRASP1 causes an aggregation of early and recycling endosome membranes as well as a significant decrease in spine density in cultured neurons (Hoogenraad et al., 2010). These findings suggest that recycling endosomes may serve as a more general regulator in mediating basal neuronal connectivity.
In addition to the increased synaptic AMPARs, LTP also induces robust structural remodeling, including formation of new spines and expansion of pre-existing spines (Matsuzaki et al., 2004; Park et al., 2006; Watson et al., 2015). Both types of plasticity-induced structural remodeling can be completely abolished by disrupting recycling endosome trafficking (Park et al., 2006; Wang et al., 2008). Our finding that spine enlargement at single spines after glutamate uncaging-induced LTP was blocked following knockdown of GRASP1 or molecular replacement of ID mutants provides direct evidence that GRASP1-mediated endosomal trafficking is critical for structural plasticity. Interestingly, knockdown of GRASP1 in cultured neurons appears to have no effect on cLTP-induced spine addition (Hoogenraad et al., 2010), suggesting that the GRASP1-mediated recycling endosome trafficking may be selectively required in active spines for activity-dependent spine enlargement.
GRASP1 function in cognition
Grasp1 KO mice exhibited significant impairments in learning and memory, and the identification of GRASP1 mutations from severe ID patients further supports the significance of GRASP1 in human cognitive function. The R822Q mutation of GRASP1 localized within the GBD prevents its interaction with GRIP1. GRIP1, a major scaffold protein for GluA2 and 3, is required for AMPAR recycling and exocytosis via interaction with the exocyst complex (Dong et al., 1997; Mao et al., 2010). Interestingly, GRIP1 SNPs that alter AMPAR recycling are associated with human autism, often comorbid with ID (Mejias et al., 2011). GRASP1 may thus work cooperatively with GRIP1 for selective recycling of AMPARs to maintain proper brain function. In support of this, we observed an increased association of AMPAR with GRASP1 and GRIP1 as well as a co-enrichment in the PSD fraction following cLTP. Moreover, R882Q mutation impairs GRASP1-GRIP1 interaction and accumulates AMPARs in the recycling endosomes, suggesting that AMPARs require GRASP1-GRIP1 interaction to traffic out of recycling endosomes for subsequent surface insertion. GRASP1 may thus serve as a gate to regulate internal pools of receptors before final exocytosis to regulate the supply of AMPARs and determine synaptic strength and synaptic plasticity.
Stx13 is a target-SNARE protein that can form a complex with SNAP25 and VAMP2 to mediate membrane fusion during recycling (Prekeris et al., 1998). Additionally, Stx13 is known to function together with GRASP1 to facilitate the maturation of Rab4-positive endosomes into Rab11-positive recycling endosomes (Hoogenraad et al., 2010). Disrupting Stx13 function blocks activity-dependent AMPAR insertion and synaptic strengthening (Park et al., 2004) as well as basal and cLTP-induced spine growth and formation (Park et al., 2006). These effects are very similar to the deficits we observed in Grasp1 KO mice, suggesting GRASP1 and Stx13 function collaboratively in regulating brain function. The increased binding of S73N mutant to Stx13 and GRIP1 may reflect an enhanced recycling trafficking of GRIP1 and GRIP1-scaffolded cargos such as AMPARs. Alternatively, it may reflect a failure in the dissociation of Stx13 with GRASP1 that can prevent Rab11 recycling endosome formation and an accumulation of recycling cargos. Our recycling assay showed a clear impairment of AMPAR recycling with a significant accumulation of AMPARs in the recycling endosomes in S73N neurons, strongly supporting the later possibility and suggesting that deficient recycling endosome maturation may underlie ID etiology.
In conclusion, our results indicate a critical role for GRASP1 in cognitive function. Loss of GRASP1 in mice impairs neuronal structure and function as well as learning and memory. Importantly, GRASP1 mutations identified from ID patients cause deficient AMPAR recycling as well as structural and functional plasticity, and extend the significance of GRASP1-mediated endosomal trafficking to human cognitive disorders.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard L. Huganir; email: rhuganir@jhmi.edu
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All procedures related to animal care and treatment conformed to Johns Hopkins University Animal Care and Use Committee guidelines. Grasp1 KO mice were generated with a neomycin resistance gene cassette inserted into exon 6 of GRASP1, creating a premature stop codon (Figure S1A) on a 129 background but were backcrossed to C57BL/6 background for more than 10 generations. Successful targeting was evident by southern and northern-blot analyses of the genomic DNA and RNA from Grasp1 mice (Figures S1B&C). Western-blot analysis and immunohistochemistry using a specific GRASP1 antibody verified the absence of intact GRASP1 protein in KO mice (Figures S1D&E). Homozygous KO animals are viable and have no gross developmental defects or anatomical abnormalities (Figure S1F). Grasp1 KO mice and Thy1-GFP (line-M) transgenic mice (Feng et al., 2000) were group housed and maintained in C57BL/6 background. Male Grasp1 littermates were randomly assigned to experimental groups and used at either juvenile (3–4 weeks) or adult (2–5 months) stage. Sprague Dawley rats (Harlan Laboratories) were used for hippocampal or cortical cultures at embryonic day 18 (E18) as described below. All animals were group housed in a standard 12 hours light/12 hours dark cycle.
Cell Cultures and Transfection
HEK293T cells (ATCC) were grown in DMEM supplemented with 10% FBS, 50U/mL penicillin and 50μg/mL streptomycin. Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, and processed 48 hours after transfection.
Hippocampal or cortical neurons from embryonic day-18 rat pups were plated onto poly-L-lysine coated coverslips or plates in 5% horse serum (HS)-containing Neurobasal medium with freshly added supplements (2% B27, 2 mM Glutamax, 50 U/ml penicillin and 50 mg/streptomycin). Hippocampal neurons were switched to serum-free Neurobasal medium with supplements one day post-seeding and fed once a week with same medium and supplements. Cortical neurons were switched to 1% HS containing Neurobasal medium one day post-seeding, treated with FDU (5 mM 5-Fluro-2′-deoxyuridine and 5 mM Uridine) to stop glia proliferation at day-in-vitro (DIV) 5 and fed twice a week with glia-conditioned, 1% HS containing Neurobasal medium with supplements. For staining and live imaging, hippocampal neurons were plated at a density of 80,000 and 150,000 per well into 12-well tissue culture plates. Neurons were transfected at DIV16-18 using Lipofectamine 2000 (Invitrogen) following manufacture’s manual, and the cells were used 3–4 days later. For biochemistry, cortical or hippocampal neurons were plated at a high density of 250,000 cells/well into 12-well plates or 650,000 cells/well into 6-well plates and were used at 2–3 weeks old.
X linked-lntellectual Disability (XL-ID) Human Samples
The study cohorts of XL-ID probands and normal controls were described previously (Wu et al., 2007). XL-ID patients and normal controls were recruited from Greenwood Genetic Center (Greenwood, SC), Johns Hopkins University (Baltimore, MD), Coriell Cell Repositories (Camden, NJ), and ECACC (Salisbury, U.K.). Human subject research protocols were approved by Institutional Review Board (IRB) at respective institutions. An informed consent was obtained from each study patient and/or their parents or guardians. These patients were evaluated by clinical geneticists and underwent standard laboratory evaluations for ID. All patients had a normal karyotype and a negative molecular test for fragile X syndrome, as well as a negative screen for common inborn errors of metabolism. For each patient, 5–10 ml of blood was collected to establish lymphoblast cell lines by Epstein-Barr virus transformation for preparation of genomic DNA used in the study. Variant data from the anonymous male only samples (n=525) were extracted from the master variant output of the 1,000 Genomes project (Integrated Phase 1, version 3: 20101123) as a fraction of normal controls in the study.
METHOD DETAILS
Intracellular whole-cell recordings
Pairs of Graps1 WT and KO male mice at p21-23 were recorded at one animal per day in an interleaved manner with randomized sequence (some pairs started with WT first and other pairs started with KO first). Mice were anesthetized with the inhalation anesthetic isoflurane prior to decapitation. Whole brain coronal slices of 350 μm were prepared in ice-cold, oxygenated N-methyl-D-glucamine (NMDG)-based cutting solution (135 mM NMDG, 1 mM KCl, 1.2 mM KH2PO4, 0.5 mM CaCl2, 1.5 mM MgCl2, 24.2 mM choline bicarbonate and 13 mM glucose). Slices containing dorsal hippocampus were transferred to a static submersion chamber filled with oxygenated ACSF1 (119 mM NaCl, 2.5 mM KCl, 1 mM NaH2PO4, 2.5 mM CaCl2, 1.3 mM MgSO4, 26.2 mM NaHCO3 and 11 mM glucose) at room temperature allowing recovery for at least 1 hour before recording. For spontaneous AMPA mEPSC recording, slices were perfused in ACSF1 in the presence of 1 μM TTX and 100 μM picrotoxin at a flow rate of ~2 ml/min. Whole cell recording pipettes (3–6 MΩ) were filled with internal solution (115 mM Cs-MeSO3, 0.4 mM EGTA, 5 mM TEA-Cl, 2.8 mM NaCl, 20 mM HEPES, 3 mM Mg-ATP, 0.5 mM Na2-GTP, pH 7.2, osmolality 295–300 mOsm). Hippocampal CA1 neurons were patched and held at −70 mV holding potential and recording was performed at room temperature. Upon entering whole cell mode, we allowed 5 minutes for dialysis of the intracellular solution before collecting data. Signals were measured with MultiClamp 700B amplifier and digitized using a Digidata 1440A analog-to-digital board. Data acquisition were performed with pClamp 10.2 software and digitized at 10 kHz. mEPSCs were detected with a template matching algorithm in Clampfit 10.2 software. All equipment and software are from Axon Instruments/Molecular Devices. Averaged mEPSC amplitude and frequency were calculated from at least 100 events for each cell and cumulative distributions of the amplitude and inter-event interval are made from the first 30 mEPSC events from each cell. Access resistance (Ra) was monitored through out the recording and only cells with Ra < 30 mOhm with < 15% changes were included for quantification. WT data were collected from 19 neurons/ 7 animals and KO data were from 18 neurons/ 6 animals from 6 litters of GRASP1 animals. For mEPSC amplitude, WT= 13.6 ± 0.5 and KO= 13.0 ± 0.2pA; p= 0.28, Mann-Whitney test; p= 0.42, Kolmogorov-Smirnov test. For frequency WT= 0.32 ± 0.03 and KO= 0.23 ± 0.02 Hz; p= 0.03, Mann-Whitney test; p< 0.001, Kolmogorov-Smirnov test). All data were presented as mean ± SEM.
Extracellular field recordings
Pairs of Graps1 WT and KO male mice (3–4 weeks old) were recorded at one animal per day in in an interleaved manner with randomized sequence. Mice were anesthetized with the inhalation anesthetic isoflurane prior to decapitation. Brains were rapidly dissected out and placed in ice-cold, oxygenated (95% O2 and 5% CO2) low-Ca2+/high-Mg2+ dissection buffer (2.6 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 211 mM sucrose, 11 mM glucose, 0.5 mM CaCl2 and 7 mM MgCl2). 350 μm transverse slices from dorsal hippocampus were prepared using a vibratome (Leica; VT1200s) in dissection buffer and a cut between CA3 and CA1 was made to minimize recurrent activity during recording. Slices were then transferred to a static submersion chamber filled with oxygenated ACSF2 (125 mM NaCl, 5 mM KCl, 1.25 mM NaH2PO4, 2 mM CaCl2, 1 mM MgCl2, 25 mM NaHCO3 and 11 mM glucose) at 30°C for recovery for at lea st 1 hour before LTP or 2 hours before LTD recording. Prior to recording, slices were transferred to a recording chamber where they were perfused continuously with oxygenated ACSF2 at a flow rate of ~3 ml/min at 30°C. Hippocampal CA1 fEPSP was evoked at 0.033 Hz with a 125 μm platinum/ iridium concentric bipolar electrode (FHC, Bowdoinham, ME) placed in the middle of stratum radiatum of CA1. Synaptic responses were recorded with ACSF2-filled microelectrodes (1–2 MQ), positioned 200 μm away (orthodromic) from the stimulating electrode, and were quantified as the initial slopes of fEPSPs. Input/output relationships were obtained for each slice with various stimulus intensity and responses were set to 45% max for LTP experiments and □55% max for LTD experiments. LTP was induced by TBS consists of 4 trains of 10 bursts at 5 Hz, with each burst consisting of four stimuli given at 100 Hz and 10-second inter-train interval. LTD were induced by LFS consists of 900 single pulses at 1 Hz. Slices with unstable baseline were discarded without further induction of LTP or LTD. All plasticity experiments are presented as responses normalized to the average of the 20-minute baseline. 5-minute averages taken at the indicated time were used to calculate the magnitude of plasticity and for statistical tests. For LTP, WT data were collected from 24 slices/ 8 animals and KO data were from 24 slices/ 7 animals from 5 litters of GRASP1 animals. For the magnitudes of LTP at the initial phase (5–10 min), WT= 225.3 ± 13.9 and KO= 178.3 ± 7.9%, p= 0.008, unpaired t-test. For the magnitude of LTP at the maintenance phase (55–60 min), WT= 167.7 ± 7.9 and KO= 139.4 ± 5.0%, p= 0.005, unpaired t-test). For LTD: WT data were collected from 14 slices/ 5 animals and KO data were from 11 slices/ 5 animals from 5 litters of GRASP1 animals. For the magnitudes of LTD (55–60 min), WT= 80.1 ± 1.9 and KO= 76.8 ± 5.9%, p=0.60, unpaired t-test). Prior to LTP and LTD recording, paired-pulse responses (PPR) were recorded with inter-stimulus intervals of 25–250 ms. PPR data were presented as a ratio of the second response slope relative to the first. 34 slices from 6 WT and 32 slices from 6 KO littermates were collected for quantifications (25ms: WT= 2.0 ± 0.04 and KO= 2.0 ± 0.05; 50ms: WT= 1.9 ± 0.03 and KO= 2.0 ± 0.04; 100ms: WT= 1.8 ± 0.03 and KO=1.8 ± 0.03; 150ms: WT= 1.6 ± 0.02 and KO = 1.6 ± 0.03; 250ms: WT= 1.4 ± 0.01 and KO= 1.4 ± 0.02; p> 0.05, two-way ANOVA).
Morris water maze (MWM)
The MWM was performed as previously described with minor modification (Volk et al., 2013). Briefly, Graps1 WT and KO male littermates were group housed at 3–5 animals (containing both genotypes) per cage after wean and an independent examiner, blind to animal genotypes, performed the MWM experiment when the animals were at their adult age of 2–5 month old. Mice after handling (3 minutes each day for 4 consecutive days) were trained to find a submerged platform in a water maze using four visual cues surrounding the pool to test their spatial learning followed by a probe trial to test their memory retention. Prior to the first training trial, mice were given a single habituation trial without the platform to assess any spatial bias and their basal swim speed. For training (day1–4), mice were randomly introduced to different start locations of the pool for each trial with the hidden platform maintained in the same quadrant (target quadrant). Swim path and latency to locate the platform was tracked and determined by a computerized video tracking system (Any-maze). Mice were trained 4 trials per sessions, 2 sessions per day over 4 days and the averaged performance per session were plotted to show the learning curve and used for statistical analysis. For probe test (day5), the platform was removed and the swimming in each quadrant and specifically the preference for the target quadrant was measured to evaluate spatial memory. Visual and sensorimotor skills were assessed with a visible platform placed at various locations after the probe test. Two independent experiments from two cohorts of animals, each containing 10 pairs of Grasp1 WT/KO littermates, were used for quantification. All values and statics were listed in Table S1& S2.
Inhibitory avoidance (IA)
The step-through IA was performed as previously described (Volk et al., 2010). Briefly, Grasp1 WT and KO male littermates were group housed at 2–5 animals per cage containing both genotypes after wean and the examiner performing IA experiments was blind to animal genotype until the end of IA testing. Adult or juvenile mice were handled for 3 minutes each day for 4 consecutive days before beginning experiments. The IA apparatus (Coulbourn Instruments) consisted of a metal grid floor as well as a light chamber and a dark chamber connected by a guillotine-style door was used to test associative learning and memory in Grasp1 mice. For habituation (day 1), a mouse was placed in the light chamber for explore freely until it crossed to the dark side, which triggered the door to be closed. The latency to crossover was automatically recorded and the mouse was returned promptly to the home cage. For training (day 2), the mouse was reintroduced to the light chamber. Similarly, the door was closed and the latency of crossover side was recorded. Additionally, the mouse received a 2-second, scrambled, mild 0.4-mA foot shock following the entry of the dark chamber. The mouse remained in the dark chamber for 15 seconds after shock before returning to the home cage. For testing (day 3), 24 hours after training the mouse was reintroduced to the light chamber. The latency of crossover to the dark chamber was recorded as a measure of associative learning and memory performance. The maximum latency was set at 5-minute. For adult Grasp1 mice (~3 month), data were collected from 13 WT and 10 KO mice from 5 litters and 3 independent experiments. The latency to crossover to the dark chamber before shock were WT= 21.5 ± 6.2 and KO= 16.3 ± 3.3 sec; and after shock were WT= 237.8 ± 24.2 and KO= 104.5 ± 29.4 sec (p< 0.001, two-way ANOVA). For juvenile mice (p21–22), data were collected from 16 WT and 16 KO from 9 litters and 5 independent experiments. The latency to crossover to the dark chamber before shock were WT= 27.4 ± 6.2 and KO= 20.2 ± 6.1 sec; and after shock were WT= 175.9 ± 27.0 and KO= 97.4 ± 21.4 sec (p< 0.001, two-way ANOVA).
Glycine-induced LTP
For cLTP experiments, cultured hippocampal or cortical neurons were first incubated for 15–20 min at 37°C in culture ACSF (143 mM NaCl, 5mM KCl, 2 mM CaCl2, 10 mM HEPES, 10 mM Glucose, pH7.2, Osm 305) supplemented with 1 mM MgCl2, 500 nM TTX, 20 μM Bicuculine and 1 μM Strychnine, followed by a 5-minute cLTP induction with 200 μM glycine in culture ACSF with same supplements but not MgCl2, and then returned to the culture ACSF for 20–25 minutes prior to lysis or homogenate for co-immunoprecipitation (cortical or hippocampal neurons) or PSD preparation (cortical neurons).
Culture neuron Imaging and Image analysis
The AMPA-induced AMPAR recycling assay was performed as previously described with minor modifications (Hoogenraad et al., 2010). Briefly, rat hippocampal neurons at DIV16–18 were transfected with GFP and a control vector (vector ctrl), shRNA for GRASP1 protein knockdown (KD), or KD plus either shRNA-resistant GRASP1 WT, S73N, or R822Q as previous described. Three days after transfection, neurons were treated with either growth medium alone or medium containing 100 μM AMPA plus 100 μM D,L-APV for 2 minutes at 37°C. Neurons were wash once and returned to original growth medium for another 58 minutes at 37C to allow internalized AMPAR to recycle before a light fix of 4 minutes with parafix (4% paraformaldehyde/ 4% sucrose in PBS) at room temperature. After washing with PBS and blocking with 3% BSA, surface AMPARs were labeled with 15F1 (mouse antibodies against GluA2 extracellular N-terminal domain) in PBS. Cells were then permeabilized with 0.1% Triton X-100 for 15min, blocked again, and incubated with rabbit anti-GluA2/3 antibodies against the intracellular C-terminal domain to probe total GluA2/3 (tGluA2/3) and chicken anti-GFP to amplify the cell-fill in PBS at 4°C for overnight. Finally, neurons were rinsed, incubated with fluorescent-labeled secondary antibodies and mounted onto glass slides. Z-series images at resolution of 1024× 1024 pixels with an optical interval of 1 μm covering the entire cells were obtained and the maximum intensity were projected for image analysis using MetaMorph software (Universal Imaging). Four dendritic segments (excluding primary dendrites) per neuron were outlined and thresholded for each channel with same parameters within an experiment to isolate transfected neurons and exclude diffuse background staining. Surface levels of endogenous GluA2 (sGluA2) were used as an indicator to evaluate the effect of GRASP1 ID mutations on AMPAR recycling. Integrated signals from sGluA2 were normalized to tGluA2/3 to reduce variability in GluA2 expression between cells. Data were collected from 3 independent experiments and presented as % mock-treated vector ctrl. For basal state, vector ctrl = 99.9 ± 4.3% quantified from 96 dendritic segments/ 24 neurons; KD= 102.8 ± 4.5% from 96 dendritic segments/ 24 neurons; KD+WT= 100.1 ± 4.8% from 96 dendritic segments/ 24 neurons; KD+S73N= 98.2 ± 5.4% from 96 dendritic segments/ 24 neurons; KD+R822Q= 110.0 ± 4.6% from 96 dendritic segments/ 24 neurons; p>0.05; one-way ANOVA). For recycling state, vector ctrl= 86.4 ± 4.2% from 92 dendritic segments/ 23 neurons and KD= 74.0 ± 3.3% from 100 dendritic segments/ 25 neurons, p= 0.04; KD+WT= 92.1 ± 4.8% from 96 dendritic segments/ 24 neurons and KD, p=0.002; KD+WT and KD+S73N= 77.4 ± 4.2% from 88 dendritic segments/ 22 neurons, p=0.015; KD+WT and KD+R822Q= 70.3 ± 4.1% from 92 dendritic segments/ 23 neurons, p=0.0002; one-way ANOVA).
To analyze endosomal distribution of internalized AMPAR, internalized GluA2 (iGluA2) were labeled and their colocalization with early, recycling and late endosomes systematically examined with well-known markers, early endosome antigen 1 (EEA1), Stx13 and lysosomal-associated membrane protein 1 (LAMP1), respectively in neurons transfected with GFP, KD, and shRNA-resistant GRASP1 WT, S73N or R822Q. Endogenous sGluA2 were first labeled with 15F1 in growth medium for 10 minutes, rinsed off non-bound antibodies, stimulated with AMPA to induce AMPAR internalization, rinsed again and returned to original growth medium for 43 minutes allowing internalized AMPAR to recycle back to the surface (all steps performed at 37°C). AMPAR trafficking was then stopped by rin sing neurons with pre-cooled 3% BSA-containing ACSF once and the sGluA2 was saturated with non-conjugated mouse secondary antibodies in 3% BSA containing ACSF at 10°C for 30 minutes. Neurons were then fixed for 15 minutes, permeablized, blocked and incubated with rabbit antibodies against various endosomal markers along with chicken-anti-GFP for overnight. iGluA2-containing AMPAR, endosomal markers and GFP were visualized with corresponding secondary antibodies conjugated with Alexa Flour 647, 546 and 488, respectively. Colocalization of internalized AMAPR with each marker was measured in the “colocalization module” in MetaMorph as described (Lee et al., 2004) which calculated % integrated values of internalized GluA2 overlapping with endosomal markers. Somatic area was used for colocolizaiton measurement due to the limitation of week fluorescent signals from endosomal markers in dendrites. To minimize false positive colocalization from the Z planes, a single optical section from the most iGluA2 signals was used to determine the degree of colocllization in each cell. For early endosome colocalization, data were collected from 3 independent experiments and presented as % iGluA2 colocolization with EEA1 (KD+WT= 8.4 ± 1.0% quantified from 24 neurons; KD+S73N= 9.3 ± 1.4% from 24 neurons; KD+R822Q= 12.0 ± 1.5% from n= 23 neurons, p> 0.05, one-way ANOVA). For recycling endosome colocalization, data were collected from 3 independent experiments and presented as % iGluA2 colocolization with stx13 (KD+WT= 21.4 ± 3.2% quantified from 19 neurons and KD+S73N= 36.9 ± 1.8% from 17 neurons, p= 0.002; KD+WT and KD+R822Q= 32.9 ± 3.9% from 17 neurons, p= 0.021, one-way ANOVA). For late endosome colocalization, data were collected from 3 independent experiments and presented as % iGluA2 colocolization with LAMP1 (KD+WT= 22.8 ± 2.5% and KD+S73N= 23.6 ± 3.1%; KD+WT and KD+R822Q=19.8 ± 2.1, quantified from 17 neurons each group, p> 0.05, one-way ANOVA).
Glutamate uncaging-induced LTP
For uncaging experiments, rat hippocampal neurons at DIV16 were co-transfected with SEP-GluA1/2 (1:1 ratio) and DsRed2 to monitor dynamic changes of AMPAR content and spine size during LTP as well as either vector ctrl, KD, KD+WT, KD+S73N, KD+ R822Q to study their effects on activity-dependent synaptic and structural plasticity. 3–4 days after transfection, neurons were perfused with 2.5 mM MNI-caged-L-glutamate in Mg++ free culture ACSF supplemented with 1mM TTX, 50μM picrotoxin, 0.5 μM glycine and 1 μM strychnine at 37°C. Z-series of secondary and tertiary dendrites were acquired with a 20X water immersion objective lens (20X/1.0 NA, Zeiss) in a custom-build two photon microscope controlled by ScanImage written in MATLAB (Pologruto et al Biomed. Eng. Online, 2003). SEP-GluA1/2 and dsRed2 signals were excited at 910 nm with a tunable Ti:sapphire laser (Coherent) and images were acquired at 1,024 × 1,024 pixels with a voxel size of 0.09μm in x and y, and a z-step of 1 μm. To uncage glutamate, the laser was set to a wavelength of 730 nm and a power of 20 mW at the back aperture of the objective. Custom software was used to position the uncaging laser using the imaging galvo mirrors and to correct for spatial offset between imaging and stimulation due to chromatic aberration. To induce plasticity, 30 6ms laser pulses were delivered at 0.5 Hz to photolyse the caged glutamate at the spine head. Laser was tuned back to 910 nm for imaging at indicated times. For quantification, Z-projections of sum intensities were made and fixed regions of interest (ROIs) for uncaged spines and adjacent dendrites were determined by the dsRed2 signal and applied to images of all time points. Spine sizes were measured as the integrated spine intensities normalized to their own adjacent dendrite region to reduce the variability in intensity between time points. Only spines showing a greater than 10% increase over baseline in spine size right after uncaging were included for further analysis. To measure AMPAR content, the same spine/dendrite ROIs determined in the dsRed channel were applied to the SEP channel, and signals from spines were normalized to their own dendritic signals to avoid image artifacts. Data were collected from 11 spines from 8 neurons for vector ctrl, 11 spines from 8 neurons for KD, 10 spines from 7 neurons for KD+WT, 10 spines from 7 neurons for KD+SN and 11 spines from 7 neurons from 5 independent hippocampal cultures. Statistical analysis of AMPAR content at 40-minute after uncaging, normalized to baseline, are vector ctrl = 138.5 ± 6.5% and KD = 107.4 ± 8.5%, p< 0.001; KD+WT= 137.7 ± 16.0% and KD, p= 0.003; KD+WT and KD+S73N = 99.3 ± 7.0%, p= 0.03; KD+WT and KD+R822Q = 103.5 ± 5.9%, p< 0.001; two-way ANOVA. Statistical analysis of spine size at 40-minute are vector ctrl = 145.7 ± 7.5% and KD = 106.8 ± 9.4%, p= 0.010; KD+WT= 153.7 ± 13.9% and KO, p= 0.006; KD+WT and KD+S73N = 112.1 ± 7.7%, p< 0.001; KD+WT and KD+R822Q = 110.5 ± 7.3%, p= 0.001; two-way ANOVA). Representative images shown in figures were median filtered, up-scaled, and contrast enhanced.
Mutation Analysis and Genotyping
Genomic DNA was extracted from lymphoblast cell lines. For each sample, 25 exons of GRASP1 (GRIP1-associated protein 1, a.k.a. GRIPAP1; NM020137; (GRCh38/hg38; December 2013) with 100 bp flanking introns were PCR-amplified using HotMaster TaqDNA polymerase (Eppendorf) following the manufacturer’s instructions. PCR amplicons and primer sequences are provided in Table S3. PCR products were purified by Exo1/SAP following a standard protocol (Applied Biosystems). Sanger sequencing was performed using ABI PRISM BigDye Terminator Cycle Sequencing kit on an ABI3100 automatic DNA sequencer (Applied Biosystems). Sequence alignment and variant calls were completed using MacVector (http://www.macvector.com).
X Chromosome Exome Sequencing
Affected Sib-Pairs with XL-ID were further subjected for X chromosome Exome sequencing as previously described (Niranjan et al., 2015). Sequencing libraries were prepared using a TruSeq™ DNA Sample Preparation kit following a standard protocol from the manufacturer (Illumina). X chromosome exome was enriched using a SureSelect Human X Chromosome Exome Kit (Agilent) and sequenced using 75 bp pair-end sequence module in HiSeq2000 Sequencer (Illumina). Sequence data analysis and variant calls were completed following standard algorithms.
DNA Constructs
cDNAs encoding full-length mouse GRASP1 were sub-cloned into a vector downstream of CMV promoter in a pEGFP backbone (Clonetech). N-terminal HA-tagged GRASP1 was generated using a standard overlap extension PCR protocol. GRASP1 shRNA with an upstream H1 RNA polymerase III promoter were inserted into the pEGFP backbone or FuGW backbone (Takamiya et al., 2008) for bicistronic expression of shRNA and GFP simultaneously. The shRNA targeting sequence is designed against position 673–691 of the rat/mouse GRASP1 open reading frame (5′-GCTAAGCTCTCTGAGAAAT-3′). To gnerate a shRNA resistant wild-type GRASP1, we created 2 silent mutations by replacing T to C and C to T at positions 675 and 681, respectively. ID point mutations were introduced to the sh673 resistant GRASP1 gene by replacing a G to A at either 251 or 2453 positions to substitute a serine at amino acid sequence 73 with an asparagine or arginine at position 822 with a glutamine residue. All mutations were generated by the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene) and all mutated sequences were confirmed with DNA sequencing. GFP-tagged GRIP1 were constructed as previously described (Thomas et al., 2012).
Co-Immunoprecipitation
Cultured neurons or transfected HEK cells were lysed in lysis buffer (1% NP-40, 200 mM NaCl, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 20 mM HEPES (pH 7.4) and protease inhibitors. Cell lysates were collected, passed through a 26-gauge needle for 12 times and centrifuged at maximum speed in the cold room for 15 minutes. The supernatant was precleared with protein A or G beads for 1 hour before protein quantification with BCA Protein Assay Kit (Pierce). 300–500 ug protein was first incubated with 2–5 μl purified antibody or serum against GFP, Stx13 or GluA1 for 1 hour followed by adding 10 μl Protein A or G beads at 4°C for 2 hours to overnight. Beads were washed four times with lysis buffer without protease inhibitors and immune complexes were eluted in a 2x SDS (sodium dodecyl sulfate)-sample buffer with heating at 65°C for 10 min. Eluates were resolved by 8 or 10% SDS-polyacrylamide gel eletrophoresis (PAGE), transferred to PVDF membrane and immunoblotted to examine protein of interest using specific primary antibody. The corresponding secondary antibodies used were HRP-coupled whole antibodies (GE Healthcare Life Sciences) or light-chain specific antibodies (Jackson Immuno Research). Proteins were visualized by Luminata Forte Western HPR substrate (Millipore) and the intensity of each band on the developed films was background subtracted and measured in ImageJ software.
For experiments testing activity-induced changes in GRASP1/GRIP1 and AMPAR interactions, we immunoprecipitated AMPARs by GluA1 antibodies (clone 4.9D) from cultured cortical neurons before or after a 5-minute glycine treatment at various times. IP eluents were used to analyze GRASP1, GRIP1 and GluA2 protein contents in GluA1-containing AMPAR protein complexes using respective antibodies. For GRASP1, data were collected from 7 independent experiments, presented as % NT and statistically compared to NT (NT=100.0 ± 7.3; Gly+0= 130.6 ± 9.8, p=0.09; Gly+5= 145.0 ± 8.6, p= 0.005; Gly+10= 148.4 ± 4.3, p= 0.002; Gly+25= 123.1 ± 12.6, n=0.31; one-way ANOVA). For GRIP1, data were collected from 7 independent experiments, presented as % NT and statistically compared to NT (NT: 99.9 ± 10.1; Gly+0= 115.9 ± 8.1, p>0.99; Gly+5= 144.0 ± 9.9, p= 0.019; Gly+10= 148.0 ± 6.2, p= 0.009; Gly+25= 145.4 ± 14.8, p= 0.015; one-way ANOVA). For GluA2, data were collected from 5 independent experiments, presented as % NT and statistically compared to NT (NT= 100.2 ± 6.8; Gly+0= 110.4 ± 6.23; Gly+5= 108.8 ± 7.6; Gly+10= 94.4 ± 3.4; Gly+25= 98.0 ± 10.3; p> 0.99 between all groups, one-way ANOVA).
To analyze interactions of GRASP1 ID mutants with GRIP1, HEK cells were first co-transfected with GFP tagged GRIP1 with either GRASP1 WT, S73N, R822Q or GBD deletion (dGBD) mutants. We then immunoprecipitated GRIP1-containing protein complexes with GFP antibodies (Santa Cruz) and performed western blot analysis to quantify the amount of GRASP1 in the GRIP1 protein complexes. Data were collected from 5 independent experiments, presented as % WT and statistically compared to WT. (WT = 100.0 ± 4.5; S73N= 206.0 ± 24.1, p< 0.001; R822Q= 43.7 ± 6.6, p= 0.011; dGBD = 42.6 ± 3.6, p= 0.007, one-way ANOVA). Similarly, HEK cells were co-transfected with GFP tagged Rab4 with GRASP1 WT mutants and GFP antibodies were used to immunoprecipitate GFP-Rab4 to analyze its interactions with different GRASPIs. Data were collected from 5 independent experiments, presented as % WT and statistically compared to WT (WT= 100.0 ± 12.0; S73N= 98.2 ± 12.2, p=0.99; R822Q= 105.0 ± 9.2, p=0.55; dGBD= 34.8 ± 10.2; p< 0.001; one-way ANOVA). For interactions with Stx13, rabbit sera against endogenous Stx13 (Synaptic Systems) were used to immunoprecipitate and analyze Stx13 interactions with WT or mutant GRASP1s. Data were collected from 7 independent experiments, presented as % WT and statistically compared to WT (WT= 100.0 ± 7.4; S73N= 552.6 ± 137.3, p< 0.001; R822Q= 83.2 ± 7.1, p=0.86; dGBD= 62.7 ± 8.9, p=0.66, one-way ANOVA).
PSD Preparation
Cultured cortical neurons or dorsal hippocampus tissue was collected by scraping and homogenized by passage through a 26g needle, 12 times, in homogenization buffer (320mM sucrose, 5mM sodium pyrophosphate, 1 mM EDTA, 10 mM HEPES (pH 7.4), 200 nM okadaic acid and protease inhibitors). The homogenate was centrifuged at 800xg for 10 minutes at 4°C to yield post-nuclear pelleted fraction 1 (P1) and supernatant fraction 1 (S1). S1 was further centrifuged at 15,000xg for 20 minutes at 4°C to yi eld P2 and S2. P2 was resuspended in milliQ water, adjusted to 4 mM HEPES (pH 7.4) from a 1 M HEPES stock solution, and incubated with agitation at 4°C for 30 minutes. The suspended P2 was centrifuged at 25,000×g for 20 minutes at 4°C to yield LP1 and LS2. LP1 was resuspended in 50 mM HEPES (pH 7.4), mixed with an equal volume of 1% triton X-100, and incubated with agitation at 4°C for 15 minutes. The PSD was generated by centrifugation at 32,000×g for 20 minutes at 4°C. The final PSD pellet was resuspended in 50 mM HEPES followed by protein quantification and Western blot. For IA induced synaptic AMPAR analysis, IA habituation and training were performed as previously described except that half of the mice were not given any food shock when they entering the dark chamber. Dorsal hippocampi from lA-trained (+ shock) or work-though (− shock) littermates were isolated, snap froze in liquid nitrogen at 30 minutes following IA training and stored at −80°C before PSD preparation. Data were collected from 6 animals each group (WT+ shock, WT− shock, KO+ shock and KO− shock) from 6 independent experiments, and presented as % non-shocked controls for each protein analyzed with western bolts (GluAI: WT= 130.3 ± 10.5 and KO= 97.0 ± 4.4%, p=0.03; GluA2: WT= 125.6 ± 8.1 and KO= 98.8 ± 4.8%, p=0.03; GluA3: WT= 120.8 ± 2.8 and KO= 98.0 ± 3.4%, p< 0.001; GluN1: WT= 107.3 ± 7.3 and KO= 90.4 ± 5.4%, p=0.12; mGluR5: WT= 94.1 ± 7.1 and KO= 99.2 ± 5.2%, p=0.59; PSD95: WT= 96.8 ± 4.2 and KO mice= 114.9 ± 10.5%, p=0.10; unpaired t-test). For cLTP induced PSD enrichment experiments, PSD were isolated from cortical neurons at DIV13 either not treated (NT) or 20 minutes after a 5-minute glycine treatment. PSD enrichments were quantified from 4 independent experiments and presented as % NT for each protein analyzed (GRASP1= 213.8 ± 32.7, p= 0.04; GRIP1= 195.5 ± 21.0%, p= 0.02; GluA1= 131.9 ± 6.0, p= 0.013; GluA2= 123.6 ± 1.3, p< 0.001; GluA3= 128.3 ± 8.6, p= 0.046; Stx13= 109.9 ± 12.1; PICK1= 116.0 ± 10.0; NR1 = 109.5 ± 3.9; NR2B= 110.0 ± 9.6; PSD95= 121.0 ± 6.4, p= 0.045; one-way ANOVA).
In Utero Electroporation
In utero electroporation were performed at E14.5–16.5 mouse pups. Timed pregnant Grasp1 females were anesthetized with Avertin via intraperitoneal injection, and surgical procedures were performed to expose the uterus and embryos. A glass microcapillary pipette was used to deliver ~1 μl of fast-green/DNA mixtures containing either dsRed alone or with various GRASP1 constructs in PBS into the lateral ventricles of the embryos. Five electrical pulses at 36 V, each with a 50 ms duration, were delivered at 1-second intervals with a tweezers electrode to both hemispheres of the injected embryos. The uterus with the electroporated embryos were then replaced in the abdominal cavity and the surgical opening was sutured.
Spine Density Measurement
To quantify spine density in Grasp1 animals, pairs of WT and KO male mice at p21 were delivered to the examiner blinded to the genotype. Mice were perfused with PBS followed by 4% paraformaldehyde in PBS at room temperature. Brains were fixed for overnight and cut into 200 pm-thickness coronal slices with a vibratome. Two slices from dorsal hippocampus were mount in PermaFluor mounting medium (Thermo Scientific) and Z-series images of CA1 dendrites from Thy1-GFP mice were obtained using a Zeiss 510 laser scanning confocal microscope with a digital zoom 2 at 1,024 × 1,024 in x and y, and a z-step of 0.5 pm under a 63X objective lens. First few splits of apical secondary dendrites (around 50–150 μm away from the soma) that are parallel to the imagining plane with total dendrite length greater than 200 μm per neurons were quantified. All apparent protrusions from dendrites regardless of the shape were blindly counted as spines in the z-series images and normalized to the length of dendrite in image J. Data were quantified from 12 neurons each genotype from four pairs of Thy1-GFPxGrasp1 WT/KO littermates from 4 litters (WT= 1.50 ± 0.03 and KO= 1.28 ± 0.04 spines/μm; p < 0.001, unpaired t-test). For slices obtained from in utero-electroporated mice, dsRed antibodies were used to amplify the dsRed2 signal for better spine visualization. Nucleus staining with DAPI was sometimes performed to highlight hippocampal structure and evaluate electroporation efficiency. 10 neurons each group were quantified from 4–5 animals derived from 2–3 litters each conditions (WT/dsR alone= 1.53 ± 0.04 and KO/dsR alone= 1.33 ± 0.02, p< 0.001; KO/WT= 1.48 ± 0.02 and KO/dsR alone, p= 0.005; KO/WT and KO/S73N= 1.36 ± 0.03, p= 0.048; KO/WT and KO/R822Q= 1.28 ± 0.04; p<0.001, one-way ANOVA). Tail samples for all animals used were saved and their genotypes were confirmed after the completion of the experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were presented as mean ± SEM (standard error of the mean). All statistical details and statistical significance, calculated using Mann-Whitney test, Kolmogorov-Smirnov test, un-paired t-test, one-way or two-way ANOVA, were indicated in the figure legends. Fisher’s LSD and Boferonni post-hoc tests were used following one-way and two-way ANOVA, respectively. *, p<0.05; **, p<0.01, ***, p< 0.001.
DATA AND SOFTWARE AVAILABILITY
N/A
ADDITIONAL RESOURCES
N/A
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-GluAI (clone 4.9D) | Dong et al., 1997 | N/A |
| Mouse anti-GluA2 (clone 15F1) | Eric Gouaux, Vollum Institute | N/A |
| Rabbit anti-GluA2/3 (JH4854) | Xia et al., 1999 | N/A |
| Rabbit anti-GluA3 (JH4300) | Mao et al., 2010 | N/A |
| Rabbit anti-GluN1 (JH2590) | Mao et al., 2010 | N/A |
| Rabbit anti-GRASPI (JH2730) | Ye et al., 2000 | N/A |
| Rabbit anti-GRIPI (JH2260) | Dong et al., 1997 | N/A |
| Rabbit anti-PICKI (JH2906) | Xia et al., 1999 | N/A |
| Rabbit anti-Syntaxin13 | Synaptic Systems | Cat# 110 113, RRID: AB_10639254 |
| Rabbit anti-EEA1 | Cell Signaling Technology | Cat# 3288S, RRID: AB_2096811 |
| Rabbit anti-LAMP1 | Abcam | Cat# ab62562, RRID: AB_2134489 |
| Mouse anti-GFP | Santa Cruz | Cat# sc-69779, RRID: AB_1123603 |
| Chicken anti-GFP | Aves Labs | Cat# GFP-1020, RRID: AB_10000240 |
| Rabbit anti-dsRed | Clontech | Cat# 632496, RRID: AB_10013483 |
| Mouse anti-HA (clone 16B12) | Covance | Cat# MMS-101P, RRID: AB_2314672 |
| Mouse anti-GRIP1 | BD Biosciences | Cat# 611319, RRID: AB_398845 |
| Mouse anti-Rab4 | BD Biosciences | Cat# 610889, RRID: AB_398206 |
| Mouse anti-PSD95 | UC Davis/NIH NeuroMab | Cat# 75–028, RRID: AB_2307331 |
| Mouse anti-mGluR5 | Abcam | Cat# ab76316, RRID: AB_1523944 |
| Mouse monoclonal anti-αTubulin (Clone B-5-1-2) | Sigma-Aldrich | Cat# T6074; RRID: AB_47758 |
| Biological Samples | ||
| Human cell line BH0092 (GRASP1 _S73N) | Wu et al., 2007 | European Collection of Authenticated Cell Culture (ECACC) |
| Human cell line 5897 (GRASP1 _R822Q) | Wu et al., 2007 | Greenwood Genetic Center |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tetrodotoxin citrate | Tocris | Cat# 1069; CAS: 18660-81-6 |
| Bicuculline methochloride | Abcam | Cat# ab120110; CAS: 53552-05-9 |
| Glycine | Tocris | Cat# 0219; CAS: 5640–6 |
| Strychnine hydrochloride | Abcam | Cat# ab120416; CAS: 1421-86-9 |
| Picrotoxin | Abcam | Cat# ab120315; CAS: 124-87-8 |
| AMPA | Tocris | Cat# 0254; CAS: 83643-88-3 |
| DL-APV | Tocris | Cat# 0105; CAS: 76326-31-3 |
| MNI-caged-L-glutamate | Tocris | Cat# 1490; CAS: 295325-62-1 |
| Critical Commercial Assays | ||
| N/A | N/A | N/A |
| Deposited Data | ||
| N/A | N/A | N/A |
| Experimental Models: Cell Lines | ||
| Human: Human Embryonic Kidney (HEK) 293T cells | ATCC | Cat# CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Grasp1 KO in C57BL/6 background | This paper | N/A |
| Mouse: Thy1-GFP (line-M) in C57BL/6 background | Feng et al., 2000 | N/A |
| Recombinant DNA | ||
| pEGFP-N1 | Clontech | Cat# 632162 |
| shGRASP1 (shRNA against GRASP1) on pEGFP backbone | This paper | N/A |
| HA:GRASP1 WT (from mouse) on pEGFP backbone | This paper | N/A |
| HA:GRASP1 S73N on pEGFP backbone | This paper | N/A |
| HA:GRASP1 R822Q on pEGFP backbone | This paper | N/A |
| pFUGW | Lien et al., 2008 | Addgene plasmid# 37632 |
| shGRASP1 on pFUGW backbone | This paper | |
| HA:GRASP1 WT on pFUGW backbone | This paper | N/A |
| HA:GRASP1 S73N on pFUGW backbone | This paper | N/A |
| HA:GRASP1 R822Q on pFUGW backbone | This paper | N/A |
| GFP:Rab4 | Gu et al., 2016 | N/A |
| GRIP1 :myc | Thomas et al., 2012 | N/A |
| SEP:GluA1 | Lin et al., 2009 | N/A |
| SEP:GluA2 | Thomas et al., 2012 | N/A |
| pDsRed2-N1 | Clontech | Cat# 632406 |
| Sequence-Based Reagents | ||
| shRNA targeting sequence: shGRASPI: 5′-GCTAAGCTCTCTGAGAAAT-3′ | This paper | N/A |
| Primers for human GRASP1 mutation analysis, see Table S3 | This paper | N/A |
| Software and Algorithms | ||
| MetaMorph | Molecular Devices | https://www.moleculardevices.com/systems/metamorph-research-imaging |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| ScanImage | HHMI | https://openwiki.janelia.org/wiki/display/ephus/ScanImage |
| Other | ||
| N/A | N/A | N/A |
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institute of Health (NS36715, HD052680 and NS073854 to R.L.H., T.W and C.E.S), the Howard Hughes Medical Institute (to R.L.H.) and the Brain and Behavior Research Foundation (19607 to S.L.C.). S.-L.C. and G.H.D were supported by the NARSAD Young Investigator Grant and the Canadian Institute for Health Research postdoctoral fellowship award, respectively. We thank Dr. Eric Gouaux for the GluA2 antibody, all members of the Huganir lab for helpful discussion and critical reading of the manuscript, and Drs. Lenora Volk, Se-Young Choi, Ingie Hong, Richard Roth and Rebecca Rose for experimental supports. R.L.H. is a paid consultant to Millipore Corporation. Under a licensing agreement between Millipore Corporation and the Johns Hopkins University, R.L.H. is entitled to a share of royalties received by the University on sales of products described in this article.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
S. -L.C. and R.H. designed research. S.-L.C. and G.H.D. performed experiments and analyzed data. K.T and B.Y. generated Grasp1 KO mice. C.-M.C. generated molecular tools. T.W., Y.J., T.N. and C.E.S identified GRASP1 ID mutations. S.-L.C. and R.H. wrote the paper.
The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict-of-interest policies.
Supplemental information
Supplemental Information includes four figures and three tables.
References
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Current opinion in neurobiology. 2012 doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Kandel ER. Structural changes accompanying memory storage. Annu Rev Physiol. 1993;55:397–426. doi: 10.1146/annurev.ph.55.030193.002145. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Brown TC, Correia SS, Petrok CN, Esteban JA. Functional compartmentalization of endosomal trafficking for the synaptic delivery of AMPA receptors during long-term potentiation. J Neurosci. 2007;27:13311–13315. doi: 10.1523/JNEUROSCI.4258-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarota M, Bernabeu R, Levi De Stein M, Izquierdo I, Medina JH. Learning-specific, time-dependent increases in hippocampal Ca2+/calmodulin-dependent protein kinase II activity and AMPA GluR1 subunit immunoreactivity. Eur J Neurosci. 1998;10:2669–2676. doi: 10.1046/j.1460-9568.1998.00254.x. [DOI] [PubMed] [Google Scholar]
- Chung BH, Drmic I, Marshall CR, Grafodatskaya D, Carter M, Fernandez BA, Weksberg R, Roberts W, Scherer SW. Phenotypic spectrum associated with duplication of Xp11.22-p11.23 includes Autism Spectrum Disorder. Eur J Med Genet. 2011;54:e516–520. doi: 10.1016/j.ejmg.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Dev KK, Henley JM. The schizophrenic faces of PICK1. Trends Pharmacol Sci. 2006;27:574–579. doi: 10.1016/j.tips.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, O’Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386:279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhang P, Song I, Petralia RS, Liao D, Huganir RL. Characterization of the glutamate receptor-interacting proteins GRIP1 and GRIP2. J Neurosci. 1999;19:6930–6941. doi: 10.1523/JNEUROSCI.19-16-06930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edens AC, Lyons MJ, Duron RM, Dupont BR, Holden KR. Autism in two females with duplications involving Xp11.22-p11.23. Dev Med Child Neurol. 2011;53:463–466. doi: 10.1111/j.1469-8749.2010.03909.x. [DOI] [PubMed] [Google Scholar]
- Esteban JA. Intracellular machinery for the transport of AMPA receptors. Br J Pharmacol. 2008;153(Suppl 1):S35–43. doi: 10.1038/sj.bjp.0707525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteves da Silva M, Adrian M, Schatzle P, Lipka J, Watanabe T, Cho S, Futai K, Wierenga CJ, Kapitein LC, Hoogenraad CC. Positioning of AMPA Receptor-Containing Endosomes Regulates Synapse Architecture. Cell reports. 2015;13:933–943. doi: 10.1016/j.celrep.2015.09.062. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Giorda R, Bonaglia MC, Beri S, Fichera M, Novara F, Magini P, Urquhart J, Sharkey FH, Zucca C, Grasso R, et al. Complex segmental duplications mediate a recurrent dup(X)(p11.22–p11.23) associated with mental retardation, speech delay, and EEG anomalies in males and females. Am J Hum Genet. 2009;85:394–400. doi: 10.1016/j.ajhg.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger AJ, Shi Y, Lu W, Cerpas M, Nicoll RA. LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature. 2013;493:495–500. doi: 10.1038/nature11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Chiu SL, Liu B, Wu PH, Delannoy M, Lin DT, Wirtz D, Huganir RL. Differential vesicular sorting of AMPA and GABAA receptors. Proc Natl Acad Sci U S A. 2016;113:E922–931. doi: 10.1073/pnas.1525726113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirling H, Steiner P, Chaperon C, Marsault R, Regazzi R, Catsicas S. Syntaxin 13 is a developmentally regulated SNARE involved in neurite outgrowth and endosomal trafficking. Eur J Neurosci. 2000;12:1913–1923. doi: 10.1046/j.1460-9568.2000.00076.x. [DOI] [PubMed] [Google Scholar]
- Hoogenraad CC, Popa I, Futai K, Sanchez-Martinez E, Wulf PS, van Vlijmen T, Dortland BR, Oorschot V, Govers R, Monti M, et al. Neuron specific Rab4 effector GRASP-1 coordinates membrane specialization and maturation of recycling endosomes. PLoS biology. 2010;8:e1000283. doi: 10.1371/journal.pbio.1000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Goswami D, Zhang Y, Molina AJM, Sudhof TC, Malenka RC. LTP requires a unique postsynaptic SNARE fusion machinery. Neuron. 2013;77:542–558. doi: 10.1016/j.neuron.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien WH, Gelfand VI, Vasioukhin V. Alpha-E-catenin binds to dynamitin and regulates dynactin-mediated intracellular traffic. J Cell Biol. 2008;183:989–997. doi: 10.1083/jcb.200805041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Makino Y, Sharma K, Hayashi T, Neve R, Takamiya K, Huganir RL. Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat Neurosci. 2009;12:879–887. doi: 10.1038/nn.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron. 2011;72:1001–1011. doi: 10.1016/j.neuron.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Takamiya K, Thomas G, Lin DT, Huganir RL. GRIP1 and 2 regulate activity-dependent AMPA receptor recycling via exocyst complex interactions. Proc Natl Acad Sci U S A. 2010;107:19038–19043. doi: 10.1073/pnas.1013494107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of longterm potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- Mejias R, Adamczyk A, Anggono V, Niranjan T, Thomas GM, Sharma K, Skinner C, Schwartz CE, Stevenson RE, Fallin MD, et al. Gain-of-function glutamate receptor interacting protein 1 variants alter GluA2 recycling and surface distribution in patients with autism. Proc Natl Acad Sci U S A. 2011;108:4920–4925. doi: 10.1073/pnas.1102233108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsushima D, Ishihara K, Sano A, Kessels HW, Takahashi T. Contextual learning requires synaptic AMPA receptor delivery in the hippocampus. Proc Natl Acad Sci U S A. 2011;108:12503–12508. doi: 10.1073/pnas.1104558108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Niranjan T, Skinner C, May M, Turner T, Rose R, Stevenson R, Schwartz C, Wang T. Affected Kindred Analysis of Human X Chromosome Exomes to Identify Novel X-Linked Intellectual Disability Genes. PLoS One. 2015;10:e0116454. doi: 10.1371/journal.pone.0116454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson MA, Szatmari EM, Yasuda R. AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a Ras-ERK-dependent manner during long-term potentiation. Proc Natl Acad Sci U S A. 2010;107:15951–15956. doi: 10.1073/pnas.0913875107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. PICK1 targets activated protein kinase Calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci. 2001;21:5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, Choquet D. Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron. 2009;63:92–105. doi: 10.1016/j.neuron.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prekeris R, Klumperman J, Chen YA, Scheller RH. Syntaxin 13 mediates cycling of plasma membrane proteins via tubulovesicular recycling endosomes. J Cell Biol. 1998;143:957–971. doi: 10.1083/jcb.143.4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert KO, Focking M, Prehn JH, Cotter DR. Hypothesis review: are clathrin-mediated endocytosis and clathrin-dependent membrane and protein trafficking core pathophysiological processes in schizophrenia and bipolar disorder? Mol Psychiatry. 2012;17:669–681. doi: 10.1038/mp.2011.123. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Sonnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol. 2000;149:901–914. doi: 10.1083/jcb.149.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner P, Alberi S, Kulangara K, Yersin A, Sarria JCF, Regulier E, Kasas S, Dietler G, Muller D, Catsicas S, et al. Interactions between NEEP21, GRIP1 and GluR2 regulate sorting and recycling of the glutamate receptor subunit GluR2. EMBO J. 2005;24:2873–2884. doi: 10.1038/sj.emboj.7600755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamiya K, Mao L, Huganir RL, Linden DJ. The glutamate receptorinteracting protein family of GluR2-binding proteins is required for long-term synaptic depression expression in cerebellar Purkinje cells. J Neurosci. 2008;28:5752–5755. doi: 10.1523/JNEUROSCI.0654-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Hayashi T, Chiu SL, Chen CM, Huganir RL. Palmitoylation by DHHC5/8 targets GRIP1 to dendritic endosomes to regulate AMPA-R trafficking. Neuron. 2012;73:482–496. doi: 10.1016/j.neuron.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluijs P, Hoogenraad CC. New insights in endosomal dynamics and AMPA receptor trafficking. Semin Cell Dev Biol. 2011;22:499–505. doi: 10.1016/j.semcdb.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Volk L, Chiu SL, Sharma K, Huganir RL. Glutamate Synapses in Human Cognitive Disorders. Annu Rev Neurosci. 2015 doi: 10.1146/annurev-neuro-071714-033821. [DOI] [PubMed] [Google Scholar]
- Volk L, Kim CH, Takamiya K, Yu Y, Huganir RL. Developmental regulation of protein interacting with C kinase 1 (PICK1) function in hippocampal synaptic plasticity and learning. Proc Natl Acad Sci U S A. 2010;107:21784–21789. doi: 10.1073/pnas.1016103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. PKM-zeta is not required for hippocampal synaptic plasticity, learning and memory. Nature. 2013;493:420–423. doi: 10.1038/nature11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao Y, Zhang X, Badie H, Zhou Y, Mu Y, Loo LS, Cai L, Thompson RC, Yang B, et al. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down’s syndrome. Nat Med. 2013;19:473–480. doi: 10.1038/nm.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Edwards JG, Riley N, Provance DW, Jr, Karcher R, Li XD, Davison IG, Ikebe M, Mercer JA, Kauer JA, et al. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson DJ, Ostroff L, Cao G, Parker PH, Smith H, Harris KM. LTP enhances synaptogenesis in the developing hippocampus. Hippocampus. 2015 doi: 10.1002/hipo.22536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Wu Y, Arai A, Rumbaugh G, Srivastava A, Turner G, Hayashi T, E S, Jiang Y, Zhang L, Rodriguez J, et al. Mutations in Ionotropic AMPA receptor 3 (iGluR3) alter channel properties and are associated with moderate cognitive impairment in humans. Proc Natl Acad Sci USA. 2007;104:18163–18168. doi: 10.1073/pnas.0708699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- Ye B, Liao D, Zhang X, Zhang P, Dong H, Huganir RL. GRASP-1: a neuronal RasGEF associated with the AMPA receptor/GRIP complex. Neuron. 2000;26:603–617. doi: 10.1016/s0896-6273(00)81198-8. [DOI] [PubMed] [Google Scholar]
- Zhang L, Jie C, Obie C, Abidi F, Schwartz C, Stevenson R, Valle D, Wang T. X-chromosome cDNA microarray screening identifies a functional PLP2 promoter polymorphism enriched in patients with X-linked mental retardation (XLMR) Genome Res. 2007;17:641–648. doi: 10.1101/gr.5336307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Jeyifous O, Munro C, Montgomery JM, Green WN. Synaptic activity regulates AMPA receptor trafficking through different recycling pathways. Elife. 2015;4 doi: 10.7554/eLife.06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.