

Abstract
Sef (similar expression to fgf), also know as IL17RD, is a transmembrane protein shown to inhibit fibroblast growth factor signaling in developmental and cancer contexts, however its role as a tumor suppressor remains to be fully elucidated. Here we show that Sef regulates epithelial-mesenchymal transition (EMT) in breast cancer cell lines. Sef expression was highest in the normal breast epithelial cell line MCF10A, intermediate expression in MCF-7 cells and lowest in MDA-MB-231 cells. Knockdown of Sef increased the expression of genes associated with EMT, and promoted cell migration, invasion, and a fibroblastic morphology of MCF-7 cells. Overexpression of Sef inhibited the expression of EMT marker genes and inhibited cell migration and invasion in MCF-7 cells. Induction of EMT in MCF10A cells by TGF-β and TNF-α resulted in downregulation of Sef expression concomitant with upregulation of EMT gene expression and loss of epithelial morphology. Overexpression of Sef in MCF10A cells partially blocked cytokine induced EMT. Sef was shown to block β-catenin mediated luciferase reporter activity and to cause a decrease in the nuclear localization of active β-catenin. Furthermore, Sef was shown to co-immunoprecipitate with β-catenin. In a mouse orthotopic xenograft model, Sef overexpression in MDA-MB-231 cells slowed tumor growth and reduced expression of EMT marker genes. Together, these data indicate that Sef plays a role in the negative regulation of EMT in a β-catenin dependent manner and that reduced expression of Sef in breast tumor cells may be permissive for EMT and the acquisition of a more metastatic phenotype.
Keywords: Breast cancer, Epithelial mesenchymal transition, Sef, Migration, Snail, Slug
Receptor tyrosine kinase signaling (RTK) is essential to cell proliferation, differentiation, migration and survival [Friesel and Maciag, 1995; Korc and Friesel, 2009; Lemmon and Schlessinger, 2010]. Under normal physiological situations, RTKs are tightly regulated, however when RTK expression or activity becomes deregulated through genetic mutations or other means, developmental abnormalities or disease states such as cancer are often the result [Korc and Friesel, 2009]. In addition to deregulation of RTKs, tumorigenesis may also result from suppression of the negative regulators of RTK pathways [Cabrita and Christofori, 2008; Dikic and Giordano, 2003; Thisse and Thisse, 2005].
Sef (similar expression to FGF genes), also referred to as IL17RD, was originally identified as an inhibitor of FGF signaling in zebrafish development [Furthauer et al., 2002; Tsang et al., 2002]. In humans, the hSef gene gives rise to at least two isoforms, hSef-a, which is a type I transmembrane protein and hSef-b which encodes a cytosolic isoform [Preger et al., 2004]. The mouse and human transmembrane isoforms of Sef inhibit RTK-mediated ERK and Akt signaling pathways [Kovalenko et al., 2003; Ziv et al., 2006]. In the case of FGFR signaling, evidence suggests that this occurs in part through binding of Sef to the FGFR and inhibiting its activation [Kovalenko et al., 2003]. The cytosolic isoform of hSef has been reported to cause aberrant cellular localization of Ras and MEK1, thus disrupting normal ERK signaling [Torii et al., 2004]. Given these properties, Sef may be considered a tumor suppressor gene. In support of this notion, several recent reports indicate that hSef expression is down regulated in human carcinomas [Zisman-Rozen et al., 2007], including prostate [Darby et al., 2009; Darby et al., 2006] and breast carcinomas [Yang et al., 2003; Zisman-Rozen et al., 2007]. Indeed, the most aggressive and metastatic forms of carcinomas have the lowest levels of expression of hSef [Darby et al., 2006; Zisman-Rozen et al., 2007]. It has also been reported that downregulation of hSef enhances FGF signaling in prostate cancer cell lines [Korc and Friesel, 2009; Tsang et al., 2002]. Together, these data suggest that loss of Sef function may contribute to the acquisition of the metastatic phenotype in carcinomas. However, because there remains doubt about the mechanisms of action of Sef we sought to characterize its functions in breast carcinoma cell lines.
Epithelial to mesenchymal transition (EMT) is the loss of the epithelial phenotype due to the down regulation of E-cadherin, loss of cell-cell junctions, increased migration and acquisition of a fibroblastic morphology [Kalluri and Weinberg, 2009]. E-cadherin is down regulated by several transcriptional repressors such as Snail, Slug, and Zeb1, which are induced by activation of the ERK and Akt pathways. Because the most aggressive carcinomas are thought to undergo EMT to acquire their metastatic potential [Kalluri and Weinberg, 2009], and because Sef is significantly down regulated in many carcinomas [Zisman-Rozen et al., 2007], we reasoned that Sef might play a role in regulating EMT. In this study, we show that overexpression of Sef in breast carcinomas with low or moderate levels of Sef expression have reduced EMT marker gene expression and that knockdown of Sef in these cells results in the induction of EMT markers. Furthermore we show that Sef regulates EMT in part through a β-catenin dependent mechanism.
Materials and Methods
Cell lines and cell culture
MCF-10A cells (ATCC) were cultured in DMEM/F12 medium (Invitrogen) with 5% horse serum (Atlanta Biologicals, Inc.), 1% penicillin/streptomycin (Invitrogen), and 20ng/ml EGF, 0.5mg/ml hydrocortisone, 100ng/ml cholera toxin, 10 μg/ml insulin (all were from Sigma). MCF-7 cells (ATCC) were cultured in Eagle’s MEM (Invitrogen), 10% fetal bovine serum (FBS, Atlanta Biologicals, Inc.), 10μg/ml insulin, and 1% penicillin/streptomycin. MDA-MB-231 cells (ATCC) were cultured in alpha MEM with 10% FBS and 1% penicillin/streptomycin.
Expression vectors and stable cell lines
Plasmids encoding SefFL, SefICpTM (SefIC) (amino acids 321–738 with added PDGFR transmembrane domain) and SefEC (SefEC) (amino acids 1–325) were cloned into pcDNA3.1 / V5-His TOPO vector were described previously [Kovalenko et al., 2006]. The preparation of SefFL, SefIC and SefEC adenoviruses (AdSef) was also described previously [Kovalenko et al., 2006].
SefFL, SefIC and SefEC were cloned into the retroviral vector pWZL, and VSV-G pseudotyped retroviruses produced by the amphotropic packaging cell line 293GPG. These retroviruses were used for MCF-7 and MDA-MB-231 cell transduction to generate stable cell lines. Hygromycin (Invitrogen) selection (100 μg/mL for MCF-7 and 500 μg/ml for MDA-MB-231) was started 2 days after transfection, and maintained throughout the culture period.
Small interfering RNA transfection, shRNA transduction and adenoviral infections
MCF-7 cells (5 × 104) were seeded in 6-well plates for 24h, and then transfected with Sef siRNA or NT siRNA (Sigma) using Lipofectamine (Invitrogen). Silencing was examined 48h after transfection by real-time RT-PCR. For shRNA knockdown studies, MCF-7 cells were transduced with Sef shRNA lentiviruses and selected with 1μg/ml puromycin for 48h. MCF-10A cells were transduced with adeonviruses AdLacZ, AdSefFL, AdSefIC or AdSefEC (104 viral particles/cell) complexed with poly-lysine. Six hours after transduction, the cells were treated with TGFβ/TNFα (5ng/ml or 10ng/ml) for another 24h.
Reverse transcription, PCR, and real-time PCR assays
Total RNA was isolated using an RNeasy Plus Mini kit (QIAGEN). 1μg of total RNA was used to synthesize cDNA using the cloned AMV First-stand synthesis kit (New England Biolabs). Real-time PCR was performed using SYBR Green PCR Master Mix (SABioscience) in triplicate. Relative expression levels of the genes of interest were normalized to β-actin.
Immunoblot analysis and immunoprecipitation assay
Cells were rinsed in PBS and lysed with HNTG lysis buffer (20mm HEPES, pH 7.4, 150mM NaCl, 10% glycerol, 1% Triton X-100, 1.5mM MgCl, 1.0mM EGTA, 1.0mM NaVO4, proteinase inhibitor cocktail (Roche)). Cell lysates were either directly subjected to immunoblot analysis, or incubated with the indicated antibodies overnight for immunoprecipitation. Immune complexes were captured on protein A/G-agarose (Santa Cruz), eluted with 2 × SDS sample buffer and separated by 10% SDS-PAGE. For some experiments subcellular fractionation was performed using the Qproteome kit (Qiagen) according to the supplier’s protocol. Antibodies used in these experiment were V5 (Invitrogen), E-cadherin, active β-catenin (BD-Transduction Laboratories), N-cadherin, β-catenin (Santa Cruz), Snail, Slug (Cell Signaling), hSef (R&D Systems), and tubulin (Sigma).
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde for 15 min at room temperature, washed with PBS, and then permeabilized with 0.2% Triton X-100 and blocked with 1% bovine serum albumin for 1h. Cells were stained with primary antibodies overnight at 4°C. After extensive washing with PBS, the cells were incubated with Cy3 or FITC conjugated secondary antibodies for 1h at room temperature, washed extensively and mounted for immunofluorescence using VECTASHIELD anti-fade mounting medium. Images were captured with a Leica SP1 confocal microscope.
In vitro scratch wound healing assay
MCF-7 cells were grown to confluence in 6-well plates. After being treated with mitomycin C (10μg/ml, Sigma) for 20 min, cells were washed and scratched with a pipet tip and cultured in complete medium at 37°C. Photographs were taken 72h after scratching using an Axiovert 40C microscope (Zeiss). MCF-7 cell migration was quantified by measuring the width between the edges of the injured monolayer of cells.
Invasion assay
Cell invasion was measured using Matrigel-coated multi-well inserts (Falcon BD). 5 × 104 MCF-7 cells (0.5 mL) were seeded in serum-free medium in the upper chamber with serum-containing medium in the lower chamber. After 24h at 37°C, cells were scraped from the upper chamber with a cotton swab, and the undersides of the membranes were fixed in 4% PFA and stained with DAPI. MCF-7 invasion was quantified by measuring the numbers of invaded cells at 5 distinct positions using DFC 304 FX inverted fluorescent microscope.
TOPflash luciferase assay
TOPflash luciferase assays were performed to assess the effect of Sef on the β-catenin signaling pathway. Stably transduced MCF-7 cells were seeded into 24-well plates and the next day transiently transfected with 0.5μg TOPFlash TCF/β-catenin reporter plasmid and 0.05μg Renilla luciferase reporter plasmid, which is an internal control for transfection efficiency. 48h after transfection, luciferase activities were measured using the dual-luciferase reporter assay system (Promega), and normalized to Renilla luciferase relative light unit (RLU) values.
Human MDA-MB-231 breast cancer xenograft model
All animal experiments were approved by the Maine Medical Center Institutional Animal Care and Use Committee. All animals were maintained in an AAALAC accredited barrier facility in accordance with the NIH Guide for the care and use of laboratory animals. Female NOD-SCID mice (6–8 week old, Jackson Laboratory) were divided into 2 groups (4–5 mice/group), 2 ×106 MDA-MB-231 cells stably transfected with WZL control or Sef retroviruses were injected into the abdominal mammary fat pad. The primary tumor size was measured at different time points following cell injection, and the tumor volume was calculated according to the following formula: 1/2 (length × width2). The tumors were removed at day 49 after MDA-MB-231 cell injection, photographed and weighed. The tumors were then processed for real-time PCR and immunohistochemical staining.
Results
High levels of Sef expression down regulate the expression of genes associated with EMT
Low levels of Sef expression have been associated with a variety of cancers, such as breast, thyroid, prostate and ovarian carcinomas [Zisman-Rozen et al., 2007]. Sef is highly expressed in normal tissues, but undetectable or significant reduced in the tumors originating from these epithelial tissues, indicating a potential tumor suppressor role for Sef. We first compared the expression of Sef in the immortalized normal breast epithelial MCF-10A cell line, weakly metastatic MCF-7, and highly metastatic MDA-MB-231 breast tumor cell lines. We observed that both mRNA and protein expression levels of Sef were lower in MCF-7 and MDA-MB-231 cells compared with MCF-10A. MDA-MB-231 cells, which are more metastatic, showed a nearly 10-fold decrease in Sef expression compared to MCF-10A cells (Figure 1A, B).
Figure 1. Sef regulates EMT marker gene expression.
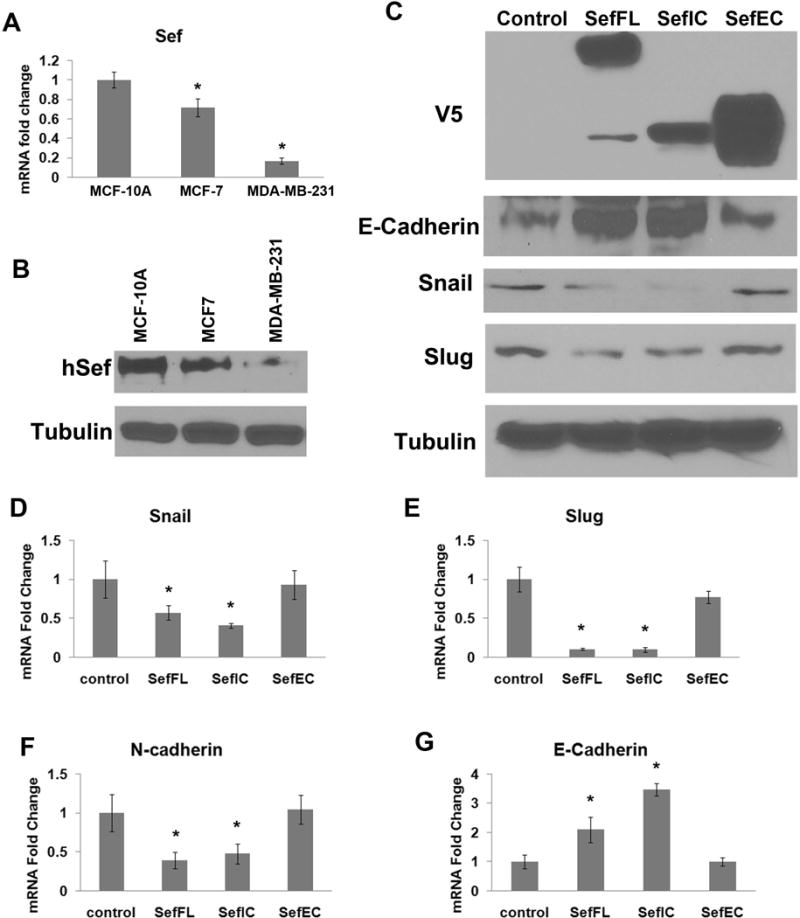
(A) Sef mRNA expression in MCF-10A, MCF-7 and MDA-MB-231 cells was determined by RT-qPCR. (B) Sef protein expression in MCF-10A, MCF-7 and MDA-MB-231 cells as determined by immunoblotting. (C) Immunoblot analysis of EMT marker expression in pWZL control, Sef, SefIC, and SefEC stably transduced MCF-7 cells. (D–G) RT-qPCR results of EMT marker expression in MCF- 7 cells overexpressing Sef, SefIC, or SefEC. Asterisks indicate significant changes at p<0.05.
Overexpression of Sef represses EMT markers
We previously reported that full-length mouse Sef (mSef), and the membrane-anchored extracellular and intracellular domains of Sef have different functions with respect to inhibiting FGF signaling [Kovalenko et al., 2006]. In order to investigate the functions of Sef domains in breast tumor cells, we first transduced MCF-7 cells with retroviruses encoding full-length wild-type mSef (Sef), its truncated mutant with intracellular and transmembrane portions (SefIC), as well as the extracellular domain mutant of Sef containing the transmembrane domain (SefEC). The empty pWZL retroviral vector served as control. Overexpression of Sef, SefIC, and SefEC in MCF-7 cells was confirmed by immunoblot analysis (Fig. 1C). Immunoblot analysis showed that overexpressed Sef or SefIC significantly decreased the expression of the transcriptional repressors Snail and Slug, whereas the expression level of E-cadherin was increased. However, overexpression of SefEC had no significant effect on Snail or Slug expression (Fig. 1C). Real-time RT-qPCR analysis of MCF-7 cells overexpression of Sef and SefIC showed a significant reduction in Snail, Slug and N-cadherin mRNA expression and increased E-cadherin mRNA, while SefEC transduced cells showed little change in the expression of these EMT markers (Fig. 1D–G).
Depletion of Sef leads to EMT-like changes in MCF-7 breast tumor cells
To characterize the effect of inactivation of Sef on epithelial-mesenchymal transition, we transfected siRNAs targeting Sef into MCF-7 cells and used a scramble non-targeting sequence (NT) as a control. Real-time RT-qPCR showed that Sef siRNA reduced Sef mRNA levels by about 70% (Fig. 2A). Knockdown of Sef significantly upregulated the expression of the EMT markers Snail, Slug, ZEB1, CyclinD1 and N-Cadherin, and repressed the expression of E-cadherin (Fig. 2A). We also examined the expression of E-cadherin by immunofluorescence staining and found that depletion of Sef using Sef siRNA reduced E-cadherin levels (Figure 2B).
Figure 2. Knockdown of Sef in MCF-7 cells promotes epithelial to mesenchymal transition.
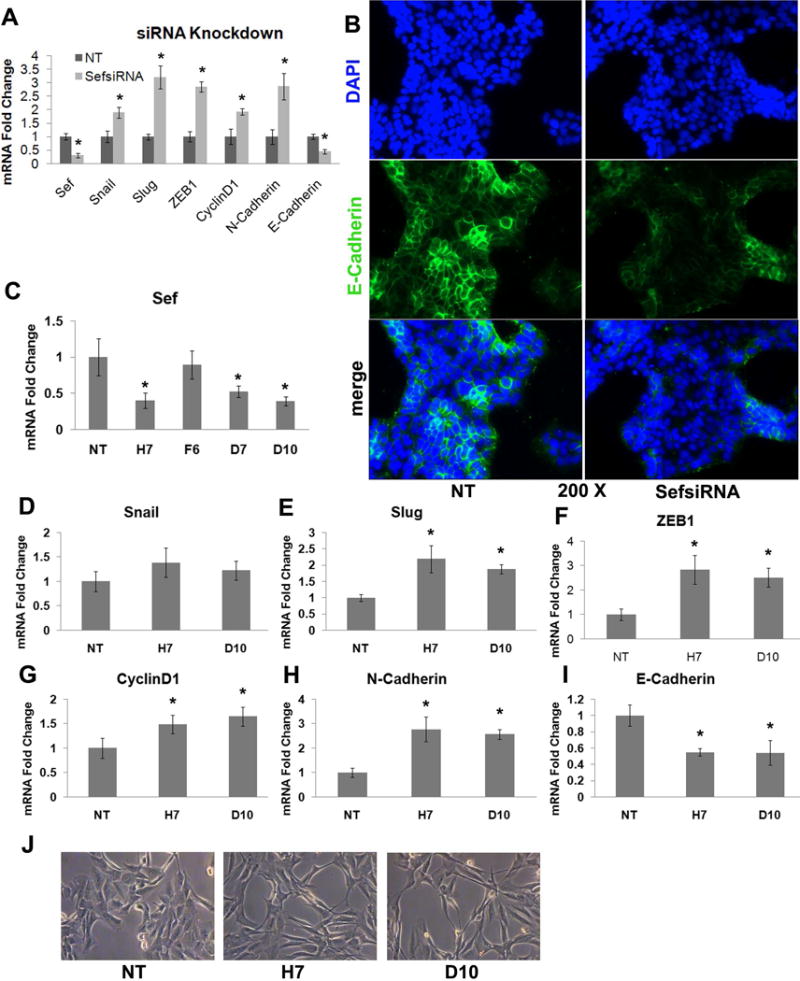
(A) MCF-7 cells were transfected with Sef siRNA or NT siRNA, and total RNA collected after 72h for RT-qPCR analysis of the indicated EMT markers (* indicated p<0.05 between Sef siRNA and NT siRNA transfected cells). (B) E-cadherin immunostaining (green) of MCF-7 cells transfected with Sef siRNA or NT siRNA for 72h. DAPI (blue) was used to visualize nuclei. (C–I) Real-time PCR of Sef shRNA knockdown MCF-7 cells. (C) RT-qPCR quantification of knockdown of Sef mRNA using individual shRNA lentiviruses (H7, F6, D7, and D10) or a non-targeting control (NT). RT-qPCR analysis of EMT marker gene expression in MCF-7 cells with stable knockdown of Sef with two independent shRNA lentiviruses (H7, D10); (D) Snail, (E) Slug, (F) ZEB1, (G) CyclinD1, (H) N-cadherin, (I) E-cadherin (* indicated p<0.05 between NT and Sef shRNA transfected samples). (J) Morphologic changes in MCF-7 cells stably transduced with Sef shRNAs for 2-weeks. (NT; non-targeting shRNA, H7; Sef shRNA, and D10; Sef shRNA).
In subsequent experiments we used lentiviruses encoding Sef targeting shRNAs to produce stable knockdown mutants in MCF-7 cells. Five different Sef shRNAs were tested, and two (H7 and D10) exhibited more than 50% reduction in Sef mRNA, and were used for subsequent experiments (Figure 2C). Consistent with the Sef siRNA results, stable knockdown of Sef in MCF-7 cells with shRNAs also resulted in increased EMT marker gene expression compared with control cells expressing scrambled shRNA sequences (Figure 2E–I). Interestingly, Snail expression remained relatively unchanged (Figure 2D), and E-cadherin expression declined. Moreover, stable knockdown of Sef induced a morphologic change in MCF-7 cells from an epithelial morphology to a more fibroblastic-spindle shape, suggesting the cells were undergoing epithelial-mesenchymal transition (Figure 2J).
Sef inhibits MCF-7 migration and invasion
To investigate whether altering Sef expression regulates cancer cell motility, a scratch assay was performed with MCF-7 cells either stably expressing Sef or Sef mutants or in which Sef was stably knocked down with shRNAs. Over a period of 48 hours, there were significant decreases in cell motility in MCF-7 cells stably expressing SefFL or SefIC compared with control cells, but no significant differences were observed between MCF-7 control and SefEC overexpressed cells (Figure 3A, B). In contrast, significantly increased migration into the denuded area was observed in MCF-7 cells where Sef was knocked down with two independent shRNAs (Figure 3C, D). These results demonstrate that Sef plays a role in regulating breast cancer cell migration.
Figure 3. Sef regulates MCF-7 cell migration and invasion.
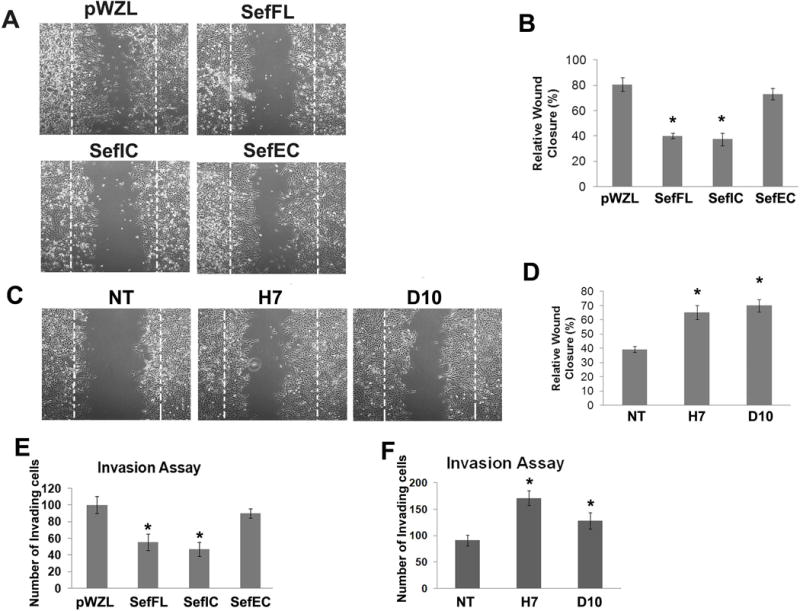
(A) Migration assay of pWZL, SefFL, SefIC and SefEC stably transduced MCF-7 cells. (B) Quantification of cell migration into denuded area (n=4). (C) Migration assay of Sef shRNA knockdown MCF-7 cells. (D) Quantification of migration activity (n=4). (E) Quantification of Boyden chamber invasion assay of pWZL, SefFL, SefIC and SefEC transfected MCF-7 cells (n=6). (F) Quantification of Boyden chamber invasion assay of Sef shRNA knockdown MCF-7 and control cells (n=6). (* indicated p<0.05).
An essential step for initiation of metastasis is invasion of cancer cells into surrounding tissues and the vasculature. We tested whether Sef affected MCF-7 cell invasion using Boyden chamber invasion assays. Overexpression of Sef and SefIC significantly reduced MCF-7 cell invasion through Matrigel-coated cell culture inserts (Figure 3E), while shRNA-mediated depletion of Sef led to an increase in cell invasiveness (Figure 3F).
Down-regulation of Sef expression during EMT in mammary epithelial cells
In the course of Sef knockdown analysis, we found that Sef expression is required for the maintenance of an epithelial morphology in MCF-7 breast cancer cells. We next investigated whether Sef expression is down regulated during the induction of EMT. MCF-10A cells, an immortalized normal mammary epithelial cell line, were treated with a combination of TGFβ1 and TNFα to induce EMT [Arima et al., 2008]. After 48h, RT-qPCR and immunoblot analysis showed TGFβ/TNFα treatment of MCF-10A cells resulted in a significant reduction in Sef expression (Figure 4A, F). TGFβ/TNFα treated MCF-10A cells also showed an increase in the expression of EMT markers such as Snail, Slug and N-cadherin, whereas E-cadherin expression was decreased (Figure 4B–F).
Figure 4. Sef inhibits TGFβ/TNFα induced EMT in MCF-10A cells.
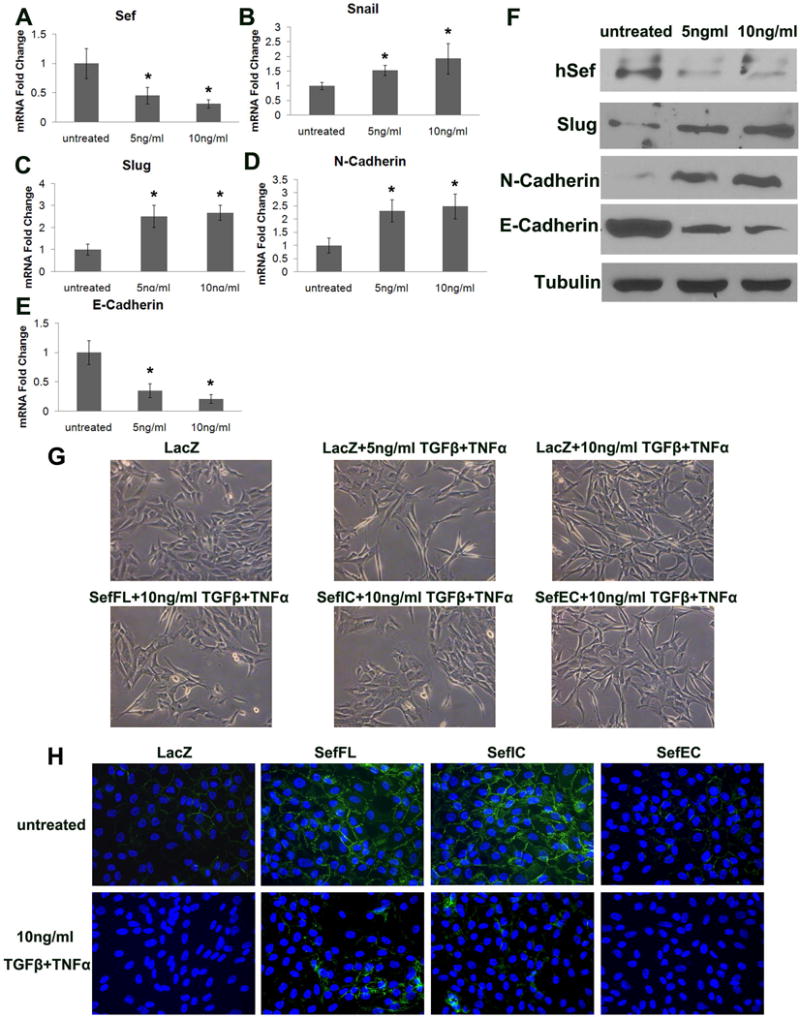
(A–E) RT-qPCR analysis of Sef and EMT marker expression in MCF-10A cells treated with 2.5ng/ml TGFβ plus 2.5 ng/ml TNFα, or 5 ng/ml TGFβ plus 5 ng/ml TNFα for 48h. (A) Sef, (B) Snail, (C) Slug, (D) N-cadherin, (E) E-cadherin (* indicates p<0.05 between NT and Sef shRNA transduced cells) (F) Immunoblot analysis of Sef and EMT markers of TGFβ/TNFα treated MCF-10A cells. (G) Morphologic changes by TGFβ/TNFα treatment and AdSefFL, AdSefIC, AdSefEC overexpression. (H) E-cadherin immunostaining (green) of TGF/TNFα treated and AdSefFL, AdSefIC, AdSefEC overexpression in MCF-10A cells. Cells were counterstained with DAPI (blue).
When MCF-10A cells were treated with TGFβ/TNFα, an epithelial-like to mesenchymal-like morphologic change was observed (Figure 4G, top row). Because Sef expression was downregulated upon induction of EMT in MCF10A cells, we tested whether Sef overexpression would inhibit cytokine induced EMT. Adenoviruses encoding Sef, SefIC or SefEC (AdSef, AdSefIC, AdSefEC) were transduced into MCF-10A cells while adenoviral LacZ transduced cells served as a control. The next day adenoviral transduced cells were treated with 5ng/ml or 10ng/ml TGFβ/TNFα for another 24h. AdSef or AdSefIC transduction repressed the epithelial to mesenchymal morphological change. AdLacZ or AdSefEC transduced MCF-10A cells exhibited a spindle-shaped morphology after TGFβ/TNFα treatment. However, AdSef and AdSefIC transduced cells showed greater cell-cell contact and displayed a more epithelial-like morphology (Figure 4G, bottom row). Immunofluorescence staining shows that E-cadherin expression in MCF10A cells is increased by adenoviral-mediated expression of SefFL and SefIC, but not SefEC (Fig. 4H, top row). Upon induction of EMT by TGFβ/TNFα treatment, E-cadherin immunofluorescence was decreased in LacZ control cells, and MCF10A cells overexpressing Sef partially maintained E-cadherin expression when treated with TGFβ/TNFα (Fig. 4H, bottom row).
Sef regulates β-catenin signaling
Because the β-catenin pathway plays a role in tumorigenesis and metastasis as well as regulating the expression of genes involved in EMT [Kalluri and Weinberg, 2009], and because β-catenin transcriptionally regulates the EMT marker Snail, we examined whether Sef inhibits β-catenin signaling. Using the TOP-Flash luciferase reporter system as an indicator of β-catenin dependent transcriptional activity we show that Sef and SefIC overexpression markedly decreased endogenous β-catenin transactivation in MCF-7 cells compared with control cells (Figure 5A). Subcellular fractionation indicated that overexpression of Sef and SefIC in MCF-7 cells resulted in increased membrane and cytoplasmic accumulation of β-catenin, and decreased nuclear localization and the active (dephosphorylated) form of β-catenin. Similar results were observed in SefIC overexpressing MCF-7 cells, with substantial cytoplasmic accumulation of β-catenin and decreased nuclear as well as active β-catenin (Figure 5B). Immunofluorescent staining of active β-catenin showed that it was expressed mainly in the nuclear compartment in control MCF-7 cells. However, the localization of active β-catenin in the nucleus was reduced when Sef or SefIC was overexpressed in MCF-7 cells (Figure 5C). In contrast, overexpression of SefEC did not change the signal intensity of active β-catenin in the nucleus while active β-catenin was also observed on the plasma membrane (Figure 5C). We next tested whether Sef and β-catenin interact. Sef and SefIC, but not SefEC, co-immunoprecipitated with β-catenin indicating that the intracellular domain of Sef is necessary for its interaction with β-catenin (Figure 5D). Taken together, our results suggest that Sef regulates β-catenin signaling in part through altering β-catenin localization to membrane and cytoplasm and blocking its activation and nuclear localization.
Figure 5. Overexpression of Sef represses the β-catenin signaling pathway.
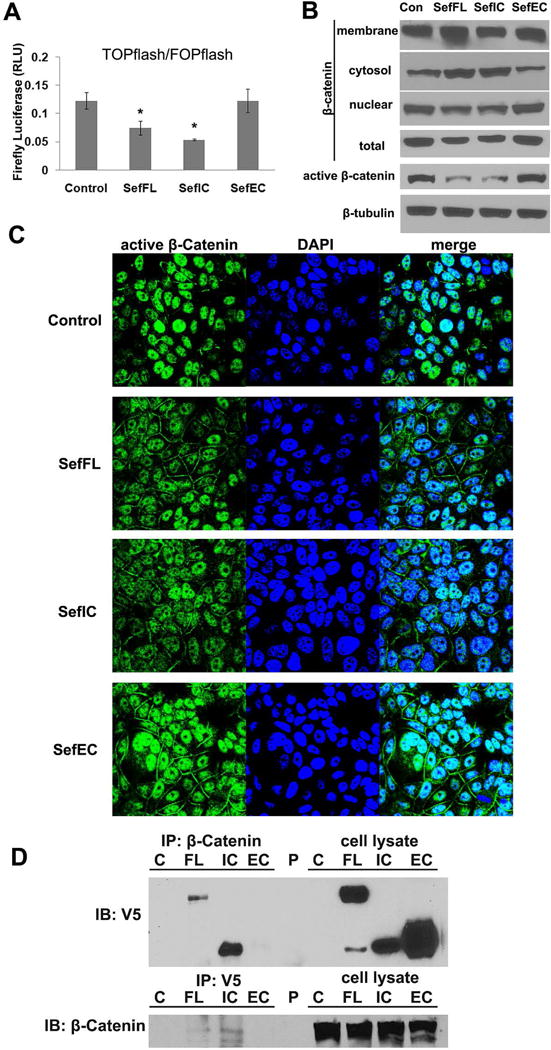
(A) TOPflash/FOPflash reporter luciferase assays were performed using SefFL, SefIC, and SefEC retrovirus transduced MCF-7 cells (* indicated p<0.05). (B) Subcellular fraction and immunoblot analysis of β-catenin localization in untransduced MCF-7 cells. Total lysates were also immunoblotted for active β-catenin. (C) Confocal images of active β-catenin immunostaining (green) in Sef and Sef mutant transduced MCF-7 cells. Nuclei were visualized with DAPI. (D) Sef interacts with β-catenin. Immunoprecipitation of cell lysates from MCF-7 cells stably transduced with pWZL (C), SefFL (FL), SefIC (IC), or SefEC (EC) followed by immunoblotting with anti-V5 antibodies (epitope-tagged Sef mutants) shows that SefFL and SefIC but not SefEC co-immunoprecipitates with β-catenin. Immunoprecipitation of the duplicate lysates with anti-V5 antibodies followed by immunoblotting with anti-β-catenin antibodies gave similar results.
Sef overexpression in MDA-MB-231 cells slows tumor growth in a murine orthotopic xenograft model of breast cancer
Because our in vitro data showed that Sef inhibits EMT, we next investigated the effects of overexpressed Sef on tumor cell growth and expression of EMT markers in vivo. 2 ×106 MDA-MB-231 cells stably transduced with pWZL-Sef or pWZL empty vector control were injected into the abdominal mammary fat pad of female NOD-SCID mice. A significant decrease in tumor size and weight was observed with Sef expressing MDA-MB-231cells compared to pWZL control cells (Figure 6A–C). At Day 49 after inoculation of tumor cells, Sef expressing tumors measured 158.43±47.26, and control tumors measured 359.29±108.54 (Figure 6A). Real-time RT-PCR analysis confirmed that overexpression of Sef inhibited EMT markers including Snail, Slug, CyclinD1 and ZEB1 in vivo (Figure 6D). Immunohistochemistry staining analysis of dissected tumors further confirmed that Sef reduced the expression of EMT markers as well as β-catenin signaling. Tumors arising from inoculation of Sef expressing MDA-MB-231 cells exhibited less expression of active β-catenin, total β-catenin, Snail and Slug by immunohistochemistry corroborating our RT-qPCR results (Figure 6E). Our data indicate that Sef inhibits tumor growth in part through regulating EMT.
Figure 6. Overexpression of Sef in MDA-MB-231 reduces tumor size in an orthotopic xenograft model of breast cancer.
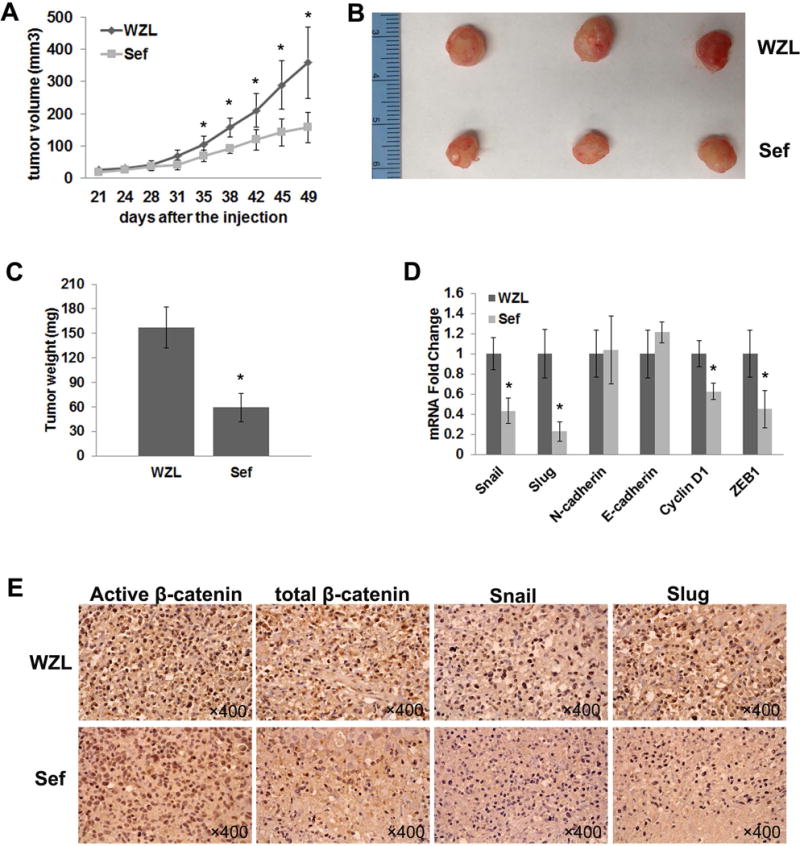
NOD/SCID mice were orthotopically engrafted with 2 ×106 MDA-MB-231 cells stably expressing either a control (pWZL) (n=4) or Sef (n=5). (A) Tumors were measured twice per week between control and Sef groups. (B) Photo of tumors extracted after 49 days of MDA-MB-231 cell implantation. (C) Tumor weight was measured after excision. (D) Real-time PCR of EMT marker gene expression in excised MDA-MB-231 control and Sef tumors. (E) Immunohistochemistry staining of active β-catenin, total β-catenin, Snail and Slug on control and Sef tumor sections.
Discussion
In this study, we show that reduced Sef expression plays a role in promoting EMT in normal and malignant epithelial cells. Knockdown of Sef in MCF-7 breast tumor cells increased the expression of EMT maker genes and increased cell migration and invasion as well as induced mesenchymal-like cell morphology. Overexpression of Sef in MCF-7 cells reduced expression of EMT marker genes and inhibited cell migration and invasion. Furthermore, induction of EMT by cytokines in the immortalized human breast epithelial cell line MCF-10A resulted in a significant decrease in Sef expression. The induction of EMT in MCF-10A cell by cytokines could be partially reversed by ectopic overexpression of Sef. In addition, overexpression of Sef in an orthotopic xenograft model slowed tumor growth and inhibited the expression of EMT markers. Taken together, these results demonstrate a crucial role for Sef in the regulation of EMT in breast epithelial cells and breast tumor cell lines.
Several recent studies have shown that hSef is highly expressed in normal epithelial tissues and at low or undetectable levels in tumors of epithelial origin and that the low Sef expression in these tumors correlated with a higher tumor grade and metastasis [Darby et al., 2006; Zisman-Rozen et al., 2007]. It was also shown that in several types of epithelial tumors, FGF receptor and ligand expression increased with tumor grade suggesting that loss of Sef and increased FGFR expression leads to unrestrained FGF signaling in these tumors [Darby et al., 2006]. While the mechanisms by which hSef is down regulated in epithelial tumors remains to be determined, our data and that from previous studies suggest that loss of hSef expression may be a prognostic marker for tumor aggressiveness and poor survival.
Previous studies showed that ectopic expression of hSef in breast [Zisman-Rozen et al., 2007], prostate [Darby et al., 2009], and endometrial carcinoma [Zhang et al., 2011] tumor cell lines inhibited FGF-induced, and in one instance EGF- induced cell proliferation. These earlier studies also showed that Sef inhibited proliferation and migration in epithelial tumor cell lines in part by inhibiting ERK and Akt signaling. In the present study we show that hSef regulates migration, invasion and EMT in part by a mechanism that involves an interaction with β-catenin. Overexpression of Sef inhibits TOPflash luciferase reporter activity and reduced nuclear accumulation of active β-catenin in vitro. Furthermore, overexpression of Sef in an MDA-MB-231 mouse xenograft model showed that Sef inhibited tumor growth, the expression of EMT markers and active β-catenin levels in vivo. Because Sef inhibits activation of Akt, and because Akt inhibits GSK3β, which phosphorylated and inactivates β-catenin [Yost et al., 1996], it is possible that reduced levels of Sef in aggressive breast tumors may result in increased Akt activation leading to decreased GSK3β activity with subsequent increases in active and nuclear β-catenin. However, we show that Sef associates with β-catenin in co-immunoprecipitation experiments, and Sef reduces nuclear levels and decreases cytosolic levels of β-catenin. It is therefore possible that Sef inhibits EMT in part by preventing nuclear localization of β-catenin. This would result in decreased formation of TCF/β-catenin transcriptional complexes that in part increase transcription of the transcriptional repressor Slug, which represses transcription of E-cadherin during EMT. Because β-catenin co-immunoprecipitated with the full length Sef and SefIC, this indicates that the interaction takes place within the cytosolic domain of Sef and this domain is necessary for Sef mediated inhibition of β-catenin signaling and transcriptional activity. Active β-catenin is a form of β-catenin that is not phosphorylated on Ser33/37/Thr41 and thus less susceptible to proteasomal degradation and mutations at these sites have been found in tumors [Morin et al., 1997]. Thus, Sef may function at multiple levels to inhibit EMT and aggressive malignant phenotypes.
In summary, we have shown for the first time that Sef plays a role regulating EMT and that loss of Sef promotes a mesenchymal phenotype in breast epithelial cells and epitheloid tumor cells. Overexpression of Sef in breast cancer cells inhibits β-catenin signaling and the expression of β-catenin target genes associated with EMT. Our findings suggest that re-expression of Sef in aggressive carcinomas may be a potential target for the treatment of breast cancer.
Acknowledgments
This work was supported by the Protein, nucleic acid analysis and cell imaging core and the Viral Vector Core for grant P30 GM103392, (R. Friesel, P.I.), and the Histopathology Core from grant numbers P30GM103392 (R. Friesel, P.I.) and P30GM103465 (D. Wojchowski, P.I.) from the National Institute of General Medical Sciences. The authors also acknowledge the generous support of Maine Medical Center.
References
- Arima Y, Inoue Y, Shibata T, Hayashi H, Nagano O, Saya H, Taya Y. Rb depletion results in deregulation of E-cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial-to-mesenchymal transition. Cancer Research. 2008;68:5104–12. doi: 10.1158/0008-5472.CAN-07-5680. [DOI] [PubMed] [Google Scholar]
- Cabrita MA, Christofori G. Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis. 2008;11:53–62. doi: 10.1007/s10456-008-9089-1. [DOI] [PubMed] [Google Scholar]
- Darby S, Murphy T, Thomas H, Robson CN, Leung HY, Mathers ME, Gnanapragasam VJ. Similar expression to FGF (Sef) inhibits fibroblast growth factor-induced tumourigenic behaviour in prostate cancer cells and is downregulated in aggressive clinical disease. British Journal of Cancer. 2009;101:1891–9. doi: 10.1038/sj.bjc.6605379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby S, Sahadevan K, Khan MM, Robson CN, Leung HY, Gnanapragasam VJ. Loss of Sef (similar expression to FGF) expression is associated with high grade and metastatic prostate cancer. Oncogene. 2006;25:4122–7. doi: 10.1038/sj.onc.1209428. [DOI] [PubMed] [Google Scholar]
- Dikic I, Giordano S. Negative receptor signalling. Curr Opin Cell Biol. 2003;15:128–35. doi: 10.1016/s0955-0674(03)00004-8. [DOI] [PubMed] [Google Scholar]
- Friesel RE, Maciag T. Molecular mechanisms of angiogenesis: fibroblast growth factor signal transduction. FASEB J. 1995;9:919–25. doi: 10.1096/fasebj.9.10.7542215. [DOI] [PubMed] [Google Scholar]
- Furthauer M, Lin W, Ang SL, Thisse B, Thisse C. Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat Cell Biol. 2002;4:170–4. doi: 10.1038/ncb750. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of Clinical Investigation. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korc M, Friesel RE. The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets. 2009;9:639–51. doi: 10.2174/156800909789057006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko D, Yang X, Chen PY, Nadeau RJ, Zubanova O, Pigeon K, Friesel R. A role for extracellular and transmembrane domains of Sef in Sef-mediated inhibition of FGF signaling. Cell Signal. 2006;18:1958–66. doi: 10.1016/j.cellsig.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Kovalenko D, Yang X, Nadeau RJ, Harkins LK, Friesel R. Sef inhibits fibroblast growth factor signaling by inhibiting FGFR1 tyrosine phosphorylation and subsequent ERK activation. J Biol Chem. 2003;278:14087–91. doi: 10.1074/jbc.C200606200. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–90. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Preger E, Ziv I, Shabtay A, Sher I, Tsang M, Dawid IB, Altuvia Y, Ron D. Alternative splicing generates an isoform of the human Sef gene with altered subcellular localization and specificity. Proc Natl Acad Sci U S A. 2004;101:1229–34. doi: 10.1073/pnas.0307952100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse B, Thisse C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev Biol. 2005;287:390–402. doi: 10.1016/j.ydbio.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Torii S, Kusakabe M, Yamamoto T, Maekawa M, Nishida E. Sef is a spatial regulator for Ras/MAP kinase signaling. Dev Cell. 2004;7:33–44. doi: 10.1016/j.devcel.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Tsang M, Friesel R, Kudoh T, Dawid IB. Identification of Sef, a novel modulator of FGF signalling. Nat Cell Biol. 2002;4:165–9. doi: 10.1038/ncb749. [DOI] [PubMed] [Google Scholar]
- Yang RB, Ng CK, Wasserman SM, Komuves LG, Gerritsen ME, Topper JN. A Novel Interleukin-17 Receptor-like Protein Identified in Human Umbilical Vein Endothelial Cells Antagonizes Basic Fibroblast Growth Factor-induced Signaling. J Biol Chem. 2003;278:33232–8. doi: 10.1074/jbc.M305022200. [DOI] [PubMed] [Google Scholar]
- Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes & Development. 1996;10:1443–54. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhao X, Yan L, Li M. Similar expression to FGF (Sef) reduces endometrial adenocarcinoma cells proliferation via inhibiting fibroblast growth factor 2-mediated MAPK/ERK signaling pathway. Gynecologic Oncology. 2011;122:669–74. doi: 10.1016/j.ygyno.2011.05.019. [DOI] [PubMed] [Google Scholar]
- Zisman-Rozen S, Fink D, Ben-Izhak O, Fuchs Y, Brodski A, Kraus MH, Bejar J, Ron D. Downregulation of Sef, an inhibitor of receptor tyrosine kinase signaling, is common to a variety of human carcinomas. Oncogene. 2007;26:6093–8. doi: 10.1038/sj.onc.1210424. [DOI] [PubMed] [Google Scholar]
- Ziv I, Fuchs Y, Preger E, Shabtay A, Harduf H, Zilpa T, Dym N, Ron D. The human Sef-a isoform utilizes different mechanisms to regulate FGFR signaling pathways and subsequent cell fate. J Biol Chem. 2006;281:39225–35. doi: 10.1074/jbc.M607327200. [DOI] [PubMed] [Google Scholar]