

ABSTRACT
Malaria is caused by parasites of the genus Plasmodium, which are transmitted to humans by the bites of Anopheles mosquitoes. After the elimination of Plasmodium falciparum, it is predicted that Plasmodium vivax will remain an important cause of morbidity and mortality outside Africa, stressing the importance of developing a vaccine against P. vivax malaria. In this study, we assessed the immunogenicity and protective efficacy of two P. vivax antigens, apical membrane antigen 1 (AMA1) and the 42-kDa C-terminal fragment of merozoite surface protein 1 (MSP142) in a plasmid recombinant DNA prime/adenoviral (Ad) vector boost regimen in Aotus monkeys. Groups of 4 to 5 monkeys were immunized with plasmid DNA alone, Ad alone, prime/boost regimens with each antigen, prime/boost regimens with both antigens, and empty vector controls and then subjected to blood-stage challenge. The heterologous immunization regimen with the antigen pair was more protective than either antigen alone or both antigens delivered with a single vaccine platform, on the basis of their ability to induce the longest prepatent period and the longest time to the peak level of parasitemia, the lowest peak and mean levels of parasitemia, the smallest area under the parasitemia curve, and the highest self-cure rate. Overall, prechallenge MSP142 antibody titers strongly correlated with a decreased parasite burden. Nevertheless, a significant proportion of immunized animals developed anemia. In conclusion, the P. vivax plasmid DNA/Ad serotype 5 vaccine encoding blood-stage parasite antigens AMA1 and MSP142 in a heterologous prime/boost immunization regimen provided significant protection against blood-stage challenge in Aotus monkeys, indicating the suitability of these antigens and this regimen for further development.
KEYWORDS: Plasmodium vivax, malaria vaccine, plasmid DNA vaccines, recombinant adenovirus vaccines, prime/boost immunization, Aotus monkeys, animal models, immunology, adenovirus vectors, antimalarial vaccines, malaria, plasmid DNA
INTRODUCTION
Recent reports indicate a decrease in malaria cases worldwide; nevertheless, approximately 0.4 million deaths from malaria still occurred in 2015, with these deaths largely being caused by Plasmodium falciparum and mainly occurring in sub-Saharan Africa (1, 2). While Plasmodium vivax is responsible for fewer deaths, it is more widespread across the globe than P. falciparum and also exacts a heavy toll. Even if elimination of P. falciparum were to be successful, it is predicted that P. vivax will remain an important cause of morbidity and mortality (3), especially in Asia and Central and South America, in part due to the relapses occurring months to years after infection, which are characteristic of this species of Plasmodium and which make the infection harder to eliminate from the human reservoir (4).
The development of vaccines against malaria is considered a priority by international health experts (5). A phase 3 trial in 6- to 12-week-old infants and 5- to 17-month-old children with the most developed vaccine candidate (RTS,S/AS01) showed that it conferred partial protection against clinical disease (vaccine efficacy in the two age groups, 25.9% [95% confidence interval {CI}], 19.9 to 31.5] and 36.3% [95% CI, 31.8 to 40.5], respectively) but little protection against infection (6–9), and a 6-year follow-up of individuals who participated in an earlier RTS,S trial showed an increase in rebound malaria cases in the fifth and sixth years in highly exposed children, reducing the 7-year efficacy to 4.4%. A positive scientific opinion on the use of RTS,S/AS01 was provided by the European Medicines Agency's Committee for Medicinal Products for Human Use on the basis of its review of the results of the phase 3 study; however, the World Health Organization has recommended that additional studies be performed before the vaccine is licensed for use, in particular, to address concerning safety signals arising in the phase 3 trial. The slow progress in the development of malaria vaccines reflects the challenges facing vaccine developers, which include the identification of protective antigens, the selection of optimal vaccine delivery systems, and formulation of the vaccine with appropriate adjuvants to consistently elicit protective immune responses. One approach to improve protection is to combine multiple antigens, and a second is to use heterologous prime/boost immunization strategies (10).
The blood-stage antigens apical membrane antigen 1 (AMA1) and the 42-kDa C-terminal fragment of merozoite surface protein 1 (MSP142) have individually been shown to induce partially protective immune responses in monkeys of the genus Aotus, which are New World primates that support the blood-stage development of these parasites (11–14); immunization with the P. falciparum AMA1 protein conferred delayed or undetected patency and lower peak levels of parasitemia (5), while immunization with P. falciparum MSP142 provided various degrees of protection in this model (13). AMA1 has also induced protection in humans: when it was delivered as a recombinant protein, strain-specific protection was documented in an area of endemicity (15), and when it was delivered as a plasmid DNA prime/adenovirus (Ad) vector boost regimen in combination with Plasmodium circumsporozoite protein (CSP), 27% of individuals were sterilely protected against controlled human malaria parasite infection (16, 17). Protection in this clinical trial appeared to be associated primarily with CD8+ T cell responses to AMA1, which is also expressed in the sporozoite and liver stages (16, 17). MSP142, delivered as a recombinant protein, has induced antibody (Ab) responses in humans that are inhibitory to blood-stage parasites in vitro (18), and when it was delivered in a gene-based adenovirus vector prime/modified vaccinia virus Ankara boost regimen in combination with AMA1, it induced partial protection in humans (19). Based on these data, we elected to combine AMA1 and MSP142 and deliver them in a heterologous gene-based prime/boost regimen in order to improve protection in the Aotus model of challenge with blood-stage P. vivax parasites, anticipating that the antibody responses to these blood-stage antigens would be protective.
The ability of replication-deficient recombinant adenovirus (rAd) vectors to elicit strong cellular (CD8+ T cell), humoral, and innate immune responses (1) makes these vectors an ideal vaccine platform. Adenovirus serotype 5 (Ad5) vectors have been used for immunization against malaria with relative success (1). The immunogenicity of Ad5 encoding CSP or liver-stage antigen 1 (LSA1) in a heterologous prime/boost vaccine schedule with Ad35 elicited enhanced T cell responses in rhesus macaques (20). Other experiments using different heterologous prime/boost platforms that included priming with plasmids (DNA) or alphavirus replicons (VRP) and boosting with attenuated Ad5 or attenuated poxvirus (COPAK) encoding preerythrocytic antigens CSP and SSP2/TRAP and erythrocytic antigens AMA1 and MSP142 achieved 60% sterile but short-lived protection in Plasmodium knowlesi sporozoite-challenged rhesus monkeys (21). Also, as mentioned above, protection was achieved in humans when a regimen consisting of a plasmid DNA prime and a boost with Ad5 encoding the antigens CSP and AMA1 was used (16). Priming with DNA can markedly increase the antibody responses to adenovirus vectors in humans, although it may not be better than priming with an initial dose of an adenovirus vector (22).
Previous trials of malaria vaccine candidates have shown the Panamanian Aotus monkey/P. falciparum model to be useful for supporting malaria vaccine development, as it allows the assessment of the humoral immunogenicity of vaccine antigens, different routes of administration, and various vaccine platforms, including gene-based vaccines. For instance, immunogenicity studies of a plasmid DNA vaccine encoding the Plasmodium yoelii (rodent malaria) CSP (PyCSP) in Panamanian Aotus monkeys demonstrated that the intradermal (i.d.) route of inoculation induced higher levels of antibodies than the intramuscular (i.m.) route. Antibody levels were comparable to those generated with a multiple-antigen synthetic peptide (MAP) vaccine delivered with an adjuvant and were increased 4-fold when a booster was given 46 weeks after the primary immunization (23). We have used the plasmid DNA platform to test the immunogenicity of single or multigene plasmid vaccines in this model (24). Subsequently, different vaccine formulations and different routes and methods of administration with a comparable hepatitis B virus plasmid DNA vaccine were explored in Panamanian Aotus lemurinus lemurinus monkeys in order to elucidate the best route and methods of immunization for a plasmid DNA vaccine (25). Further studies with multiple plasmids encoding EBA175, MSP142, and AMA1 did not show antigenic competition when these vaccines were delivered as a mixture to Panamanian Aotus monkeys (24). Also tested in Panamanian Aotus monkeys was the induction of antibodies by recombinant protein-based vaccines, such as the ligand EBA175 region II (RII), capable of blocking the binding of RII to erythrocytes and inhibiting parasite invasion of erythrocytes (26). When EBA175 was tested in Panamanian Aotus monkeys using a plasmid DNA prime/recombinant protein boost approach, no protection against challenge with the P. falciparum FVO strain was observed. However, in the same experiment, animals immunized with a recombinant MSP142 protein/peptide vaccine formulation were partially protected (unpublished data). These results contrasted with those obtained previously in Aotus nancymae monkeys from Peru, where partial protection was achieved with EBA175 RII using the same formulation and approach (27, 28). Finally, in the Aotus monkey model, high-grade (sterile) protection can be achieved by repeated blood-stage challenges with P. falciparum (29), indicating the suitability of the model for assessing whole-parasite vaccines. These sterilely protected animals were partially protected against challenge with a heterologous strain.
In the study described here, based on these rationales for the choice of antigens, viral vector, and animal model, we assessed the immunogenicity and protective efficacy of the P. vivax AMA1 (PvAMA1) and P. vivax MSP142 (PvMSP142) antigens administered in a plasmid DNA prime/Ad5 boost regimen to Aotus l. lemurinus monkeys. Our hypothesis was that the heterologous prime/boost, gene-based approach using two antigens would induce an immune response stronger and broader than that induced by other vaccines and better engage humoral immunity than other vaccines do, improving protection, as shown by reduced levels of parasitemia in Aotus monkeys challenged with blood-stage P. vivax parasites.
RESULTS
Parasitemia.
All animals became parasitemic following blood-stage challenge (no sterile protection). To evaluate partial protection, we compared the active immunization regimens (groups B to F) to the control regimens (groups A and G) for the day of parasitemia onset, the mean and peak levels of parasitemias, and the rate of self-cure (in which no rescue treatment was required). To evaluate the benefit of antigen combinations, we compared the partial protection obtained following prime/boost immunization in the group receiving the AMA1-MSP142 antigen pair (group D) to the protection obtained in the groups receiving a single antigen (groups B and C). To evaluate the benefit of a heterologous regimen, we compared the results for animals receiving both platforms in sequence (group D) to those for animals receiving only the plasmid DNA prime (group E) or the Ad boost (group F).
(i) Prepatent period.
Figure 1 shows the mean parasitemia counts for each group, and Fig. 2 shows the parasitemia counts for each monkey, the results for which are color coded and separated into separate panels by group. All six control animals (groups A and G; Fig. 1 and 2a and g) became positive and remained actively parasitemic until day 35 postinoculation (p.i.), indicating a successful challenge and an inability to self-cure. Five of the six groups that received active immunization regimens (groups B to F), however, showed various patterns of self-curing infections, with differences in the proportion of self-cures without rescue treatment and in the peak and mean densities of parasitemia being seen and with group D showing a highly significant difference in the proportion of self-cured monkeys compared with that for control group A (P < 0.001, chi-square test) (Table 1; Fig. 1 and 2d). Immunized monkeys became parasitemic at between days 5 and 9 according to the findings on Giemsa-stained thick smears, with one animal in the AMA1 group being an outlier; that animal turned positive on day 15. Once this animal became positive, however, it followed the same course of infection as the others in group B. The majority of these animals became positive at between days 5 and 7, but three animals (in addition to the outlier in group B) showed delays: in group A (control), Aotus monkey MN13165 was positive on day 9 p.i., in group B (in which the animals were immunized with AMA1 alone), monkey MN13141 was positive on day 9 p.i., and in group E (in which the animals were immunized with a plasmid DNA prime only), monkey MN13090 was positive on day 9 p.i. In summary, there did not appear to be any significant delays in the prepatent period.
FIG 1.
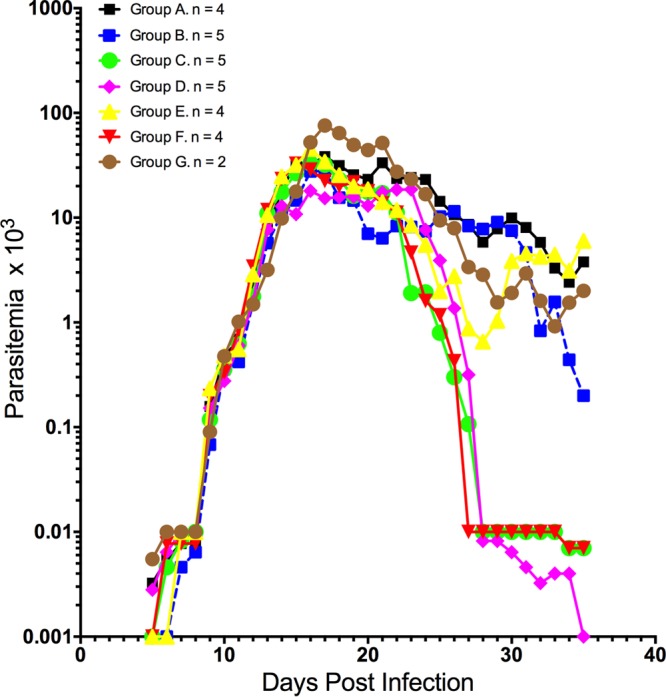
Mean levels of parasitemia in Aotus monkeys immunized in a prime/boost regimen with plasmid DNA/Ad5 encoding P. vivax blood-stage antigens AMA1 and MSP142 and challenged with P. vivax SAL-1. Group A, plasmid/virus control (n = 4); group B, AMA1/AMA1 (n = 5); group C, MSP142/MSP142 (n = 5); group D, AMA1-MSP142/AMA1-MSP142 (n = 5); group E, control/AMA1-MSP142 (n = 4); group F, AMA1-MSP142/control (n = 4); group G, infection control (n = 2). A level of parasitemia of 0.001 × 103 parasites/μl was considered negative or undetectable by microscopy.
FIG 2.

Levels of parasitemia in individual Aotus monkeys immunized in a prime/boost regimen with plasmid DNA/Ad5 encoding P. vivax blood-stage antigens AMA1 and MSP142 and challenged with P. vivax SAL-1. (a) Group A, plasmid/virus control (n = 4); (b) group B, AMA1/AMA1 (n = 5); (c) group C, MSP142/MSP142 (n = 5); (d) group D, AMA1-MSP142/AMA1-MSP142 (n = 5); (e) group E, control/AMA1-MSP142 (n = 4); (f) group F, AMA1-MSP142/control (n = 4); (g) group G, infection control (n = 2). (h) Kaplan-Meier survival plot of animals not treated with MQ, color coded by group, to day 35 PoC. Red arrows, MQ rescue treatment for anemia (HCT less than 50% of that at the baseline); a, the animal was treated for anemia; T, the animal was treated to the end of the experiment on day 35 PoC; d, the animal died of causes unrelated to malaria. A level of parasitemia of 0.001 × 103 parasites/μl was considered negative or undetectable by microscopy.
TABLE 1.
Parasitemia outcome in Aotus monkeys immunized with plasmid DNA/Ad5 encoding AMA1 and MSP142 in a prime/boost regimen against a Plasmodium vivax SAL-1 challengea
| Group and monkey no. | Sexb | Wt (g) | Mean (SD) prepatent period (days) | Parasitemia |
Day of MQ treatment and no. of monkeys treated/total no. of monkeys in groupc: |
Day of SC and no. of monkeys with SC/total no. of monkeys in group | Day of deathd | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day and mean (SD) day of peak | Peak and mean (SD) peak no. of parasites (103)/μl | Group mean (SD) no. of parasites (103)/μl | Individual AUC and total group AUC | Before day 28 | Days 29–34 | Day 35 | ||||||
| A | ||||||||||||
| 13096 | 1 | 769 | 5 | 21 | 50.4 | 273 | 35 | |||||
| 13107 | 2 | 830 | 6 | 15 | 39.1 | 203 | 35 | |||||
| 12955 | 1 | 964 | 5 | 16 | 95.0 | 825 | 35 | |||||
| 13165 | 1 | 840 | 8 | 21 | 35.3 | 290 | 35 | |||||
| Summary values | 7 (2) | 18 (3) | 54.9 (27.4) | 13 (12) | 398 | 0/4 | 0/4 | 4/4 | 0/4 | |||
| B | ||||||||||||
| 13144 | 1 | 774 | 6 | 17 | 12.8 | 47 | 21e | |||||
| 13130 | 1 | 910 | 8 | 26 | 34.5 | 216 | 35 | |||||
| 13131 | 2 | 845 | 7 | 16 | 30.0 | 132 | 21e | |||||
| 13143 | 1 | 803 | 6 | 17 | 75.8 | 344 | 28e | |||||
| 13141 | 1 | 967 | 8 | 17 | 31.5 | 151 | 35 | |||||
| Summary values | 9 (3) | 19 (4) | 36.9 (23.3) | 7.0 (7.4)f | 218 | 3/5 | 0/5 | 2/5 | 0/5 | |||
| C | ||||||||||||
| 13135 | 2 | 876 | 5 | 15 | 59.2 | 272 | 35 | |||||
| 13168 | 1 | 819 | 5 | 17 | 56.5 | 267 | 35 | |||||
| 13166 | 1 | 924 | 7 | 20 | 45.6 | 217 | 23 | |||||
| 13145 | 1 | 866 | 6 | 16 | 49.5 | 150 | 35 | 34 | ||||
| 13146 | 2 | 830 | 6 | 16 | 25 | 108 | 21e | |||||
| Summary values | 7 (1) | 17 (2) | 47.16 (13.5) | 6.8 (10)f | 209 | 1/5 | 0/5 | 3/5 | 1/5 | |||
| D | ||||||||||||
| 13173 | 1 | 862 | 5 | 23 | 39.5 | 199 | 34e | 32 | 35 | |||
| 13156 | 2 | 805 | 6 | 19 | 33.5 | 230 | 31 | 30 | ||||
| 13149 | 1 | 849 | 6 | 16 | 36.8 | 226 | 35 | |||||
| 13157 | 1 | 796 | 5 | 22 | 27.0 | 177 | 34 | 31 | ||||
| 13158 | 2 | 760 | 5 | 17 | 10.7 | 67 | 31 | 27 | ||||
| Summary values | 6 (1) | 19 (3) | 29.4 (11.5) | 5.8 (7.3)g | 180 | 0/5 | 4/5 | 1/5 | 4/5f | |||
| E | ||||||||||||
| 13090 | 1 | 758 | 8 | 19 | 16.8 | 119 | 35 | 34 | ||||
| 13091 | 2 | 781 | 5 | 21 | 24.8 | 146 | 35 | |||||
| 13178 | 2 | 890 | 5 | 16 | 72.4 | 297 | 35 | |||||
| 13179 | 1 | 848 | 5 | 14 | 74.0 | 90 | 15 | |||||
| Summary values | 6 (1) | 17 (3) | 47.0 (30.4) | 7.0 (10)h | 216 | 0/4 | 0/4 | 3/4 | 1/4 | |||
| F | ||||||||||||
| 13134 | 1 | 698 | 6 | 16 | 48.3 | 315 | 35 | |||||
| 13076 | 1 | 936 | 6 | 16 | 50.2 | 267 | 28f | |||||
| 13013 | 2 | 916 | 6 | 16 | 22.5 | 94 | 31 | 28 | ||||
| 13177 | 1 | 800 | 6 | 16 | 60.5 | 431 | 35 | |||||
| Summary values | 6 | 16 | 45.38 (16.2) | 9.3 (12)i | 287 | 1/4 | 1/4 | 2/4 | 1/4 | |||
| G | ||||||||||||
| 13190 | 2 | 855 | 4 | 18 | 49.9 | 337 | 35 | |||||
| 13174 | 1 | 764 | 5 | 17 | 111.0 | 612 | 35 | |||||
| Summary values | 4 (1) | 17 (1) | 80.5 (43.20) | 15 (22)i | 474 | 0/2 | 0/2 | 2/2 | 0/2 | |||
AUC, area under the parasitemia curve; MQ, mefloquine at 20 mg/kg orally once; SC, self-cured. Boldface indicates statistically significant different data.
1, male; 2, female.
Rescue treated.
The animals died of causes unrelated to malaria.
The animal had anemia.
P < 0.01.
P < 0.0001.
P < 0.001.
ns, not significant.
(ii) Parasitemia levels and self-cure.
Parasitemia data are presented in Table 1. As with the prepatent period, there were no statistically significant differences among the groups in the number of days to peak parasitemia, with the longest interval being shown by groups B and D (day 19). However, the parasite densities varied between the groups, and in some cases they showed statistically significant differences. The control animals showing the highest peak parasite levels (54.9 × 103 and 80.5 × 103 parasites/μl for groups A and G, respectively) and the total areas under the parasitemia curve (AUCs) (398 and 474 for groups A and G, respectively), while group D showed the lowest peak level of parasitemia (29.4 × 103 parasites/μl), mean level of parasitemia (5.8 × 103 parasites/μl) (P < 0.0001, 2-way analysis of variance [ANOVA] with the Bonferroni multiple-comparison test), and total AUC (Table 1). Moreover, individual monkeys in groups C, D, E, and F controlled their parasitemias during follow-up, including 4/5 monkeys in group D (P < 0.01, chi-square test), while no monkey in group A, B, or G was able to do so (Table 1). One animal from group C (monkey MN13145) exhibited self-cure and was negative by day 34 postchallenge (PoC); in the same way, four monkeys in group D self-cured and were negative between days 27 and 32, while one animal each from groups E (monkey MN13090) and F (monkey MN13013) self-cured on days 34 and 28, respectively. All the others monkeys in groups A to F remained positive at levels of ≥10 × 103 parasites/μl until rescue treatment on day 35 p.i. All monkeys but one from group C (MSP142 prime/MSP142 boost) and one from group E (DNA prime only) showed suppression of parasitemia to low levels but remained positive until day 35. During the experiment, three animals died of Klebsiella pneumoniae peritonitis (30) (Table 1).
When the peak, mean, and cumulative levels of parasitemia at day 35 PoC were analyzed for each group, statistically significant differences emerged among some of the immunized groups. For instance, animals in group D had the lowest peak level of parasitemia (29.4 × 103 parasites/μl) (P = 0.0992, t test), the lowest mean level of parasitemia (5.8 × 103 parasites/μl) (P < 0.0001, 2-way ANOVA with the Bonferroni multiple-comparison test), and the smallest total AUC by day 35 PoC (Table 1). In the same way, the mean levels of parasitemia between control group A and groups B and C were significantly different at the level of a P value of <0.01 and group E at the level of a P value of <0.001 (Table 1).
During the 35-day follow-up period PoC, 6 of the 23 immunized monkeys required rescue treatment for anemia (Table 1; Fig. 2h). Notably, 3/5 animals from group B (AMA1 prime/AMA1 boost), 1/5 from group C (MSP142 prime/MSP142 boost), and 1/4 from group F (DNA control/AMA-MSP142 boost) needed treatment before day 28 PoC and were considered to have early cases of anemia. Nevertheless, the proportion of anemic animals from group B (3/5, 0.60) was borderline significantly different from that of anemic animals from control group A (P = 0.0578, chi-square test) (Table 1). On the other hand, 1/5 animals from group D (AMA1-MSP142 prime/AMA1-MSP142 boost) developed anemia between days 29 and 34 PoC and were considered to have late cases of anemia. No case of anemia was detected among the animals in group E. Remarkably, none of the 6 control animals from groups A and G (malaria naive) developed anemia until the end of the experiment on day 35 PoC, when they were treated for persistent parasitemia (Table 1; Fig. 1 and 2).
In summary, animals in group D showed the lowest peak and group mean levels of parasitemia, the smallest total AUC, and the highest self-cure rate among the immunized groups. Group D animals also had a relatively long prepatent period and a relatively long period to peak parasitemia (19 ± 3 days). Some monkeys in the group immunized with the single MSP142 antigen (group C) and the group immunized only with the plasmid DNA antigen pair (group E) had a pattern of self-limited infection similar to that of monkeys in group D. The group immunized with Ad5 only (group F) appeared to be less protected than the other immunized groups, with half of the monkeys still being parasitemic at the end of the challenge at day 35 p.i. Table 1 shows that the results for this group were not significantly different from those for control group A (P > 0.05, t test).
Serology.
Figures 3Aa to e show the antibody responses to AMA1 in group A (control), group B (AMA prime/AMA boost), group D (AMA1-MSP142 prime/AMA1-MSP142 boost), group E (AMA1-MSP142 prime/control), and group F (control/AMA1-MSP142 boost). Statistically significant antibody responses were found in the animals in group B (AMA1 prime/AMA1 boost) after two plasmid DNA immunizations (P = 0.0284, t test) and in their prechallenge (PC) serum samples after the Ad5 boost (P = 0.0038, t test) (Fig. 3Ab). In the same way, the PC serum samples from the animals in group D (Fig. 3Ac) (AMA1-MSP142 prime/AMA1-MSP142 boost) developed statistically significant antibody responses after the Ad5 boost (P = 0.0061, t test). No significant antibody responses were detected in the animals in groups A, E, and F (Fig. 3Aa, d, and e). A 6-fold lower PC antibody response to AMA1 was observed in the group immunized with the antigen pair (group D) than in the group immunized with AMA1 alone (group B) (Fig. 3Ab and c), indicating potential interference by the inclusion of MSP142 in group D.
FIG 3.

ELISA antibody titers (relative Ab units) for Aotus monkeys immunized in a prime/boost regimen with plasmid DNA/Ad5 encoding P. vivax blood-stage antigens AMA1 and MSP142. (A) Levels of Ab against AMA1; (B) levels of Ab against MSP142.
Figures 3Ba to e show the antibody responses to MSP142 in group A (control), group C (MSP142 prime/MSP142 boost), group D (AMA1-MSP142 prime/AMA1-MSP142 boost), group E (AMA1-MSP142 prime/control), and group F (control/AMA1-MSP142 boost), respectively. Again, there were no detectable antibody responses following three plasmid DNA immunizations in groups A, C, E, and F (Fig. 3Ba, b, d, and e), antibody responses were detected in group D only after the second plasmid DNA immunization (P < 0.05, t test) (Fig. 3Bc), and Ad5 provided a strong boost, as shown in Fig. 3Bb and c for groups C and D, although the increase was statistically significant only for group D. However, unlike with AMA1, there was no apparent inhibition of the response to MSP142 in the group receiving both antigens (group D).
When the levels of Ab against AMA1 in the prime/boost regimens were compared for groups A, D, E, and F (Fig. 3Aa, c, d, and e), the single-platform regimen (three plasmid DNA immunizations or one Ad5 immunization) also did not generate a significant antibody response against AMA1, indicating that the heterologous prime/boost greatly enhanced the antibody responses. Figures 3Ba, c, d, and e show the same data for MSP142, comparing groups A, D, E, and F. As with AMA1, neither single-platform regimen (three plasmid DNA immunizations or one Ad5 immunization) generated a significant antibody response against MSP142, while the heterologous regimen greatly enhanced the antibody responses.
Postchallenge antibody responses.
Exposure to parasites markedly boosted the antibody titers in the vaccinated groups. Figure 3A shows the effect of the immunization regimens on the levels of antibodies against AMA1 postchallenge, revealing the following hierarchy of response: AMA1-MSP142 plasmid DNA/Ad encoding AMA1-MSP142 (group D) > plasmid DNA of a single antigen/Ad encoding a single antigen (group B or C) > AMA1-MSP1423 plasmid DNA (group E) or Ad encoding AMA-MSP142 (group F) > control (group A). The antibody titers (group means) in the vaccinated groups increased up to 1,800-fold compared with those detected prechallenge. Antibody titers (group means) increased up to 980-fold compared with those in the controls postchallenge. Thus, exposure to whole parasites strongly boosted the antibody responses in all of the immunized groups. Even though there was no measurable antibody response prechallenge, the titers in both monkeys immunized with plasmid DNA alone and monkeys immunized with Ad5 alone were much higher than those in the controls PoC (P < 0.01, t test), indicating that both platforms can prime for boosting on exposure to parasites.
Figure 4 shows the correlation between PC enzyme-linked immunosorbent assay (ELISA) antibody titers and the levels of parasitemia to day 35 PoC by immunization group. As shown in Fig. 4a, there was a weak negative correlation (r = −0.36; reference r values for a weak correlation, 0.20 to 0.39) between the antibody titers and the mean levels of parasitemia to day 35 PoC for AMA1 but a strong negative correlation (r = −0.68; reference r values for a strong negative correlation, 0.60 to 0.79) for MSP142. Similarly, as shown in Fig. 4c, a weak negative correlation between Ab titers and the cumulative levels of parasitemia to day 35 PoC was detected for AMA1 (r = −0.35) and a strong negative correlation was detected for MSP142 (r = −0.67). As seen in Fig. 4b, a moderate negative correlation (reference r values for a moderate negative correlation, 0.40 to 0.59) between Ab titers and peak levels of parasitemia was detected for both AMA1 (r = −0.45) and MSP142 (r = 0.59).
FIG 4.

Correlation between prechallenge antibody titers and levels of parasitemia and those at day 35 postchallenge. (a) Mean level of parasitemia; (b) peak level of parasitemia; (c) cumulative level of parasitemia. The level of parasitemia is given as the number (103) of parasites per microliter. r, Pearson's correlation coefficient; PC, prechallenge.
DISCUSSION
Adenovirus vaccine delivery systems, when used alone or in heterologous prime/boost immunization regimens, have shown favorable safety and tolerability profiles and have induced strong antimalarial immunity targeting both preerythrocytic and blood-stage malaria parasite antigens in animals (31) and humans (19, 32, 33). It has been thought for some time that both humoral and cellular immunity likely contribute to protection, and this concept has recently been tested in clinical trials with some success, although the protection against preerythrocytic stages observed with adenovirus vectors appears to be primarily mediated by CD8+ T cell responses and not by antibodies (19, 34), and the antibody responses in humans following the use of heterologous regimens are relatively weak compared to the findings obtained in animal models (35). Efforts are now focused on inducing stronger antibody responses in combination with an already robust T cell response (36). This is particularly important for inducing protective responses against blood-stage antigens, where antibodies are known to play an important role (37).
In this study, we tested a plasmid DNA prime/Ad5 boost regimen in Aotus monkeys, using two P. vivax antigens, AMA1, which is both a preerythrocytic and blood-stage antigen, and MSP142, which is a blood-stage antigen. We focused on the induction of antibody responses, comparing the results obtained with the combined antigen prime/boost regimen to those obtained with priming alone, boosting alone, or prime/boost regimens with each individual antigen. Based on the findings of prior studies in Aotus monkeys showing the more robust induction of antibodies using intradermal injection, we selected this route for vaccine administration. The monkeys were assessed, via blood-stage challenge, for sterile immunity or for partial protection, with the latter being determined from the delays in the onset of parasitemia, the time to the peak level of parasitemia, the density of parasites at the time of the peak level of parasitemia, the group mean level of parasitemia, the overall level of parasitemia determined by the area under the parasitemia curve, and the ability of the monkeys to self-cure without the need for rescue treatment. Because the study focused on antibody responses, blood-stage challenge seemed the most appropriate due to the importance of humoral immunity for limiting parasitemia in nature (37). While no animals were sterilely protected, the full two-antigen heterologous prime/boost regimen (group D) was superior to all other regimens by measures of partial protection, showing the longest delay to the peak level of parasitemia, the lowest parasite densities (peak and group mean levels of parasitemia [P < 0.0001, t test]), and the highest frequency of self-cures (P < 0.01, chi-square test). We also found a strong negative correlation between MSP142 antibody titers and the level of parasitemia and a weak to moderate negative correlation between AMA1 antibody titers and the level of parasitemia, suggesting that prechallenge antibody titers correlated with a decreased parasite burden. This indicated that both heterologous immunization regimens and the antigen combination were beneficial. Overall, the results support the multiantigen, prime/boost approach to vaccine design.
However, the study also indicated the limitations of this approach. For example, there appeared to be significant interference in the antibody response to AMA1 when AMA1 was combined with MSP142, mirroring the results seen more recently in humans (38). This interference was not detected in an earlier study (24), but this may have been due to the fact that the earlier study examined antibody responses only after plasmid DNA immunization, whereas in our study the interference was manifest after the Ad boost. Antibody responses to MSP142, in contrast, were not inhibited by combination with AMA1, indicating a hierarchy of antigen interference and the importance of identifying compatible antigen mixtures for vaccine development (17, 39). Because the results obtained with animal models may not translate to humans, clinical trials of subunit combinations should compare groups immunized with single antigens and antigens in combination to ascertain whether interference is an issue and downselect compatible antigens accordingly.
We used 10 intradermal injections for each vaccine administration, a regimen that would be impractical for use in humans. While this seemed appropriate in our proof-of-concept study, clinical application would require a more practical approach, and clinical studies of other malaria vaccines have shown that more efficient methods of intradermal injection, such as the use of jet injection devices, may be able to circumvent this concern (40).
An important limitation relates to the known extensive polymorphisms of P. vivax AMA1 (41) and the relative lack of cross-strain immunity induced by immunization with P. falciparum AMA1 in field protection studies (15), a finding which would presumably apply to P. vivax AMA1 as well. Overcoming the limitations of strain-specific immunity is an active area of investigation in the field, with several promising avenues being pursued (42).
Another potential issue is that the prior exposure of all animals to a single blood-stage infection with P. falciparum may have affected the immune responses, despite evidence in humans that cross-species protection is minimal after a single exposure (43). However, as the intent of the study was to compare regimens, we assumed that any such effect would have been equalized among the groups. In addition, prior to immunization, all animals were negative when they were tested for antibody responses to P. vivax antigens.
A striking finding of this study was that none of the controls animals in groups A and G developed anemia during infection, but those immunized with AMA1 (group B) or MSP142 (group C) alone developed early cases of anemia (before day 28 PoC), while those immunized with the dual-antigen vaccines (AMA1 and MSP142) (group D) developed anemia only at the end of infection (days 29 to 34 PoC) (Fig. 2h). This finding suggests that early anemia appears to be an indication of incomplete protection, manifested in this study by a low level of parasitemia in the presence of antibodies against a single antigen, as has been observed in other single-immunogen P. falciparum vaccine efficacy trials using AMA1 (44), RH5 (31), and EBA175 (45), which partially protected Aotus monkeys. However, during a recent P. falciparum blood-stage vaccine trial, partial control of parasitemia was not considered a likely explanation for the anemia observed in AMA1-immunized individuals (46), raising questions about the safety of the intervention. On the other hand, a correlation between low-level anti-AMA1 antibodies and protection against severe anemia in Kenyan children less than 2 years old has been reported (47). Other likely explanations for the early cases of anemia observed in this study may involve autoimmune or other immune-mediated events that led to the lysis of noninfected erythrocytes (45). An understanding of the vaccine-induced anemia in this nonhuman primate model could yield important information on the contribution of specific immunogens to the development of anemia in human malaria vaccine trials.
Significantly, we found that immunization primed for boosting by exposure to parasites, with the full prime/boost regimen that was the most effective at enhancing the immune response induced by challenge being a plasmid DNA prime/Ad5 boost, which was more effective than Ad5 alone or plasmid DNA alone, partially overcoming the antigen interference noted prior to challenge. This characteristic could be advantageous if it was applied to humans living in areas where malaria is endemic. The appearance of marked differences between regimens in antibody titers (up to 128-fold) following challenge indicates the need for caution when interpreting the readouts of immunological assays, such as those of ELISAs, in vaccine trials when challenge is not performed (which presents a risk of not identifying the most immunogenic regimens).
In summary, the combination of PvAMA1 and PvMSP142 in a plasmid DNA prime/Ad5 boost regimen was partially efficacious in an Aotus monkey model of blood-stage challenge, significantly suppressing parasitemia compared to that in the controls, and appeared to be more efficacious than either antigen or either platform alone, despite a degree of antigen interference. Our results support the use of this antigen combination and suggest that it will be a useful approach for increasing the protection provided by candidate vaccines. Further development of PvAMA1 and PvMSP1 should be considered, especially if additional blood-stage antigens that add to protection can be identified. The study also demonstrated the value of heterologous prime/boost regimens for increasing vaccine potency.
MATERIALS AND METHODS
Ethics statement.
The experimental protocol entitled Efficacy and immunogenicity of a plasmid DNA vaccine against Plasmodium vivax infection in Aotus monkeys was approved and registered at the ICGES Institutional Animal Care and Use Committee (CIUCAL) under accession number 2006/02 and was approved by the WRAIR/NMRC Institutional Animal Care and Use Committee (protocol number NP42). It was conducted in accordance with the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council (48), and the laws and regulations of the Republic of Panama.
Animals.
The study used 30 adult male and female spleen-intact Aotus l. lemurinus Panamanian owl monkeys of karyotypes VIII and IX (49). The weight range was from 698 to 967 g. All monkeys had previously been experimentally exposed to P. falciparum once between the years 1999 and 2003 and had been cured of their infections. Previous experiments indicated that exposure of Aotus monkeys to P. falciparum blood-stage challenge does not cross-protect against challenge with P. vivax (unpublished data). Similarly, no cross protection against P. falciparum, as measured by the frequency of fever or by the parasite density in blood, was observed in human patients treated for neurosyphilis with P. vivax sporozoites (43). Even though we could not ensure that there was residual protection from the previous challenge with P. falciparum, the fact that the preimmunization serum was negative for the P. vivax blood-stage antigens tested indicated to us that these animals were suited for use in vaccine studies. In addition, since each animal had received a single prior infection with P. falciparum, any effect or preexisting immunity would be equalized across groups. The animals were assigned by weight and sex into 6 experimental groups of 4 or 5 monkeys each. Briefly, the animals were first listed by weight in ascending order. Male and female animals were then assigned to each experimental group, alternating between low- and high-weight animals. Two animals were assigned to the infection control group. One animal was used as a donor of blood for use in the challenge inoculation. The animals were cared for and maintained as previously described (50).
DNA plasmid vaccines.
Full-length genes for the P. vivax SAL-1 strain apical membrane antigen 1 (AMA1; GenBank accession number AF063138) and the 42-kDa C-terminal fragment of merozoite surface protein 1 (MSP142; GenBank accession number AF435593) were cloned into the VR1020 mammalian expression vector (Vical Inc., San Diego, CA). This vector contains a human cytomegalovirus (CMV) promoter and a human tissue plasminogen activator (TPA) signal sequence. Each gene was cloned into a separate plasmid. Both plasmids were previously shown to be immunogenic after immunization in mice (51). The plasmids were diluted in phosphate-buffered saline (PBS; pH 7.2) prior to injection.
Viral vectors.
The same sequences of the P. vivax genes were cloned into the shuttle plasmid pShuttle-CMV (52) downstream from the CMV early promoter in the correct orientation. Homologous recombination between the pShuttle-CMV-P. vivax antigen plasmids and the adenoviral backbone plasmid pAdEasy-1 was allowed to occur in Escherichia coli BJ5183 cells, followed by generation of the replication-incompetent E1/E3-defective Ad5 vectors encoding the P. vivax antigens in HEK293 cells as described previously (52, 53). A separate Ad5 vector was generated for each P. vivax antigen and subsequently purified by ultracentrifugation over a cesium chloride gradient.
Immunization regimens.
Previous studies performed to optimize the antibody responses in Aotus monkeys after immunization with plasmid DNA vaccines demonstrated that the intradermal (i.d.) route of immunization in multiple sites in the lower back was better than the intramuscular route (23); moreover, the antibody responses obtained by the intradermal route were augmented 6-fold by the addition of E. coli DNA and were more potent than the antibody responses obtained by the i.m. route with a plasmid expressing DNA encoding a hepatitis B virus antigen (25). For these reasons, the intradermal route was selected.
Each group of monkeys was immunized as shown in Table 2. Group A (n = 4 monkeys) was the negative-control group, and the monkeys in this group received three immunizations with the empty plasmid and one boost with the empty adenovirus 5 vector with no P. vivax sequence insertion. Monkeys in group B (n = 5) received an AMA1 plasmid DNA prime and a boost with adenovirus encoding AMA1. Monkeys in group C (n = 5) received an MSP142 plasmid DNA prime and a boost with adenovirus encoding MSP142. Monkeys in group D (n = 5) received an AMA1-MSP142 plasmid DNA prime and a boost with adenovirus encoding AMA1-MSP142. Monkeys in group E (n = 4) received an AMA1-MSP142 plasmid DNA prime and a boosted with the empty adenovirus control. Monkeys in group F (n = 4) received priming immunizations with the empty DNA control and a boost with adenovirus encoding AMA1-MSP142. Monkeys in group G (n = 2) received no immunizations (infectivity controls). Each regimen consisted of three priming plasmid DNA immunizations administered 4 weeks apart, as described previously (23, 25), followed by a recombinant Ad5 boost 12 weeks after the last plasmid DNA immunization (Fig. 5).
TABLE 2.
Immunization regimen
| Group | Monkey no. | DNA primea | Ad5 boost |
|---|---|---|---|
| A | 13096, 13107, 12955, 13165 | Control plasmid | Control Ad5 vector |
| B | 13144, 13130, 13131, 13143, 13141 | AMA1 | AMA1 |
| C | 13135, 13168, 13166, 13145, 13146 | MSP142 | MSP142 |
| D | 13173, 13156, 13149, 13157, 13158 | AMA1-MSP142 | AMA1-MSP142 |
| E | 13090, 13091, 13178, 13179 | AMA1-MSP142 | Control Ad5 vector |
| F | 13134, 13076, 13013, 13177 | Control plasmid | AMA1-MSP142 |
| G | 13190, 13174 | Infection control | Infection control |
Three DNA prime immunizations were used.
FIG 5.
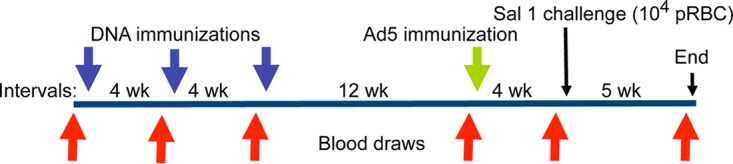
Immunization and bleeding schedule for Aotus monkeys immunized in a prime/boost regimen with plasmid DNA/Ad5 encoding P. vivax blood-stage antigens AMA1 and MSP142. Animals that were parasitemic received mefloquine treatment on day 35 (End). pRBC, parasite-infected red blood cells.
The doses administered were as follows: for plasmid DNA, a total of 500 μg (250 μg AMA1 and 250 μg MSP142) per immunization delivered by i.d. injection with a 29-gauge insulin needle in the skin of the lower back in an amount not to exceed 0.1 ml at each of 10 different sites for a total of 1 ml as described previously (27), and for Ad5, a total of 1 × 1010 viral particles (vp) per immunization (0.5 × 1010 vp AMA1 and 0.5 × 1010 vp MSP142) delivered i.d. in the same way described above. For those groups receiving a single antigen, an amount of the second antigen equivalent to that in the vector control was administered: 250 μg vector DNA or 0.5 × 1010 vp expressing no antigen. Prior to immunization, the animals were sedated with ketamine (100 mg/ml) at a dose of 10 to 20 mg/kg of body weight i.m.
Serology.
Serum samples were collected before each immunization and 30 days after challenge, as shown in Fig. 5 (a few samples were not obtained from the monkeys in groups B, C, and F). Serum samples were collected before rescue treatment with mefloquine (MQ), which was administered once at 10 mg/kg on day 35 postchallenge or earlier in the case of anemia (Table 1). Antibody titers were measured using a standard enzyme-linked immunosorbent assay (ELISA) as previously described (54) with slight modifications. Briefly, ELISA plates were coated with 100 units of recombinant AMA1 expressed in E. coli (equivalent to 0.4 μg/ml) and 100 units of MSP142 (equivalent to 0.2 μg/ml) in PBS and incubated for 6 h at room temperature. The antigen wells were then blocked with a blocking buffer (PBS, pH 7.0, containing 0.5% Tween 20 and 3% nonfat dried skim milk) at 4°C overnight. A standard consisting of a pool of serum samples known to be positive, test sera, the internal control, and blank samples, all of which were diluted in PBS, pH 7.0, were added, and the plates were incubated for 2 h at room temperature. The wells were washed three times with PBS-Tween 20 before the plates were incubated for 1 h with a 1:1,000 dilution of conjugated goat anti-human immunoglobulin G (IgG) for 2 h at room temperature. The wells were then washed three times and incubated with the respective substrate solution for 20 min. The color reaction was measured in a micro-ELISA reader by determination of the optical density (OD) at 405 nm. The assays were performed in quadruplicate. The absorbance of individual test samples was converted into ELISA units using a standard curve, generated by serially diluting the standard in the same plate, and the results are shown as the OD of a 1:100 dilution of the serum sample on a relative scale based on the results obtained with a pool of serum samples known to be positive.
Cellular immunity.
Human and chimpanzee adenovirus vectors have been used to deliver malaria vaccine candidates. These vectors have been tested in mice (55), rhesus macaques (56), and humans showing consistent antimalarial T cell immune responses (57). However, the small size of the Aotus monkeys (weight, 700 to 1,000 g), the small amount of blood that could be safely obtained from the Aotus monkeys (less than 3 ml of blood every 10 to 15 days), and the lack of species-specific reagents at the time that the study was conducted precluded the use of this metric to correlate the level of parasitemia and cellular immune responses to the candidate malaria vaccines in this animal model. Therefore, T cell immune responses were not measured in this experiment.
Malaria parasite challenge.
Previous studies in Aotus monkeys have demonstrated that the best time for challenge after immunization with repeated P. falciparum blood-stage infections is ∼4 to 5 weeks after the last immunization, when all animals challenged with an heterologous strain are partially protected (29). A frozen stabilate of erythrocytes infected with P. vivax SAL-1 (originally obtained from William C. Collins at CDC in 1997) was thawed, washed three times with RPMI medium, and resuspended in 1 ml of RPMI medium for intravenous (i.v.) inoculation into the saphenous vein of donor animal MN13127 using a 25-gauge butterfly needle catheter attached to a 3-ml syringe. When the level of parasitemia reached a peak of 39,800 parasites/μl, a dilution of blood was made in RPMI to get a total inoculum of 10,000 parasites/ml. All animals received 1 ml of the inoculum through the saphenous vein as described above.
Animal monitoring.
Following infection, all monkeys were monitored daily for parasitemia beginning at day 5 postchallenge (PoC). A complete physical exam, determination of weight, complete blood count, and chemistry profile were performed for each animal on days −7, 6, 13, 17, 21, and 28. Anemia was defined as a hematocrit (HTC) of less than 50% of that at the baseline. This HTC level triggered rescue treatment with MQ at 20 mg/kg once orally. The proportion of anemic animals before day 28 (early anemia) and between days 29 and 34 PoC (late anemia) was recorded for each group to determine the contribution of AMA1 or MSP142 alone or AMA1 and MSP142 in combination to the development of anemia in the immunized group compared to the control group. Correspondingly, a Kaplan-Meier survival curve was plotted to determine the percentage of animals remaining without treatment for anemia at the end of the experiment on day 35 PoC.
Parasitemia determination and follow-up.
Daily thick Giemsa-stained blood smears were prepared from blood obtained from a prick in the marginal ear vein for determination of the level of parasitemia by the method of Earle and Perez (58). Parasitemia outcomes were defined as cleared if the blood was negative for two consecutive days (parasitemia, <10 parasites/μl or undetectable by microscopy), cleared and self-cured if the blood was negative for more than 2 days without recrudescence, or recrudescent if the blood was cleared and was then positive 2 days after it became negative. If the level of parasitemia reached >150,000 parasites/μl, the hematocrit dropped to 50% below the baseline level, or the platelet count reached <50,000/μl prior to day 28 p.i., the animals were rescue treated with a single oral dose of 20 mg/kg of MQ. If an animal was treated for anemia on the second day of clearance, the parasitemia was considered cleared. Otherwise, follow-up analysis for parasitemia continued until day 35 p.i. when any monkeys not previously treated were treated. At between days 28 (the last day that the CBC was performed) and 35 p.i., if an animal presented with persistent parasitemia (10 parasites/μl), severe thrombocytopenia (<50,000 platelets), or anemia (an HCT less than 50% of that at the baseline) (59), a decision was left to the veterinarian to treat the animal with MQ before the end of the experiment on day 35 PoC. The reason for extending the experiment to day 35 rather than ending it on day 28, as in prior studies, was to provide a longer period over which to obtain self-cure or parasite clearance data before the end of the experiment.
Protection was classified as sterile (no parasites observed) or partial. Partial protection was measured as a statistically significant difference between the immunized groups and the controls for the number of days to patency (prepatent period), the number of days to the peak level of parasitemia, the group mean level of parasitemia (the group mean level of parasitemia was calculated from the mean daily level of parasitemia for the group during follow-up), or the total area under the parasitemia curve (AUC) or differences in the rate of self-cure from that for the controls (need for rescue treatment due to a level of parasitemia of >150,000 parasites/μl, a drop in the HTC of >50% of that at the baseline, or the persistence of parasitemia on day 35 PoC) (Table 1).
Statistical analysis.
To analyze the data obtained during this experiment, the Prism (version 5) plotting and statistical software package (GraphPad Software Inc., La Jolla, CA) was used to compare means using unpaired t tests and a 2-way ANOVA with the Bonferroni correction for multiple comparisons. Similarly, the chi-square test was used to determine the statistical significance of the difference between proportions. Prism (version 5) software was also used to measure the total AUC for each individual animal and the mean level of parasitemia for each treatment group, as well as to generate Kaplan-Meier survival curves for treatment outcomes and determine Pearson's correlation coefficients for prechallenge ELISA antibody levels at the time of the mean, peak, and cumulative levels of parasitemia at day 35 PoC for each immunized group.
ACKNOWLEDGMENTS
We thank the following individuals at the Center for the Evaluation of Antimalarial Drugs and Vaccines, Tropical Medicine Research/Gorgas Memorial Institute of Health Studies, in Panama City, Panama: Camilo Marin and the animal caretakers for their assistance with animal handling and care, Maritza Brewer for secretarial assistance, and Gladys Calviño and Bertha de Gordon for administrative assistance. We also thank Gines Sanchez and Ceferino Sanchez at TMR; Jorge Motta, secretary of the Secretaria Nacional de Ciencia, Tecnología e Innovación (SENACYT) of Panama; Scot Robert for his review of the manuscript; Stephen Hoffman at Sanaria, Rockville, MD; and Nestor Sosa and Rafael Samudio at the Gorgas Memorial Institute of Health Studies in Panama for logistical support.
This work was supported by the Naval Medical Research Center and the U.S. Army Medical Research and Materiel Command (USAMRMC; contract number DAMD-17-01-C0039) and in part by a distinguished investigator award to N.O. from the Sistema Nacional de Investigación de Panamá (SNI), SENACYT of Panama, Republic of Panama, and the Department of Immunology and Infectious Diseases, Harvard University T. H. Chan School of Public Health, Boston, MA.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the U.S. Department of the Navy, the U.S. Department of Defense, or the U.S. government.
Michael G. Stockelman and Thomas L. Richie were military service members and Yupin Charoenvit and Denise L. Doolan were employees of the U.S. government at the time that this research was conducted. This work was prepared as part of their official duties.
N.O. and M.G.S. planned the experiments. N.O., M.G.S., W.O., and J.A.C. performed the experiments. N.O., M.G.S., and T.L.R. analyzed the data. N.O., M.G.S., and T.L.R. wrote the paper.
REFERENCES
- 1.Schuldt NJ, Amalfitano A. 2012. Malaria vaccines: focus on adenovirus based vectors. Vaccine 30:5191–5198. doi: 10.1016/j.vaccine.2012.05.048. [DOI] [PubMed] [Google Scholar]
- 2.WHO. 2015. World malaria report 2015. WHO, Geneva, Switzerland. [Google Scholar]
- 3.Baird JK. 2009. Severe and fatal vivax malaria challenges ‘benign tertian malaria’ dogma. Ann Trop Paediatr 29:251–252. doi: 10.1179/027249309X12547917868808. [DOI] [PubMed] [Google Scholar]
- 4.Baird JK. 2009. Resistance to therapies for infection by Plasmodium vivax. Clin Microbiol Rev 22:508–534. doi: 10.1128/CMR.00008-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz L, Brown GV, Genton B, Moorthy VS. 2012. A review of malaria vaccine clinical projects based on the WHO rainbow table. Malar J 11:11. doi: 10.1186/1475-2875-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein SL, Shann F, Moss WJ, Benn CS, Aaby P. 2016. RTS,S malaria vaccine and increased mortality in girls. mBio 7:e00514-16. doi: 10.1128/mBio.00514-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olotu A, Fegan G, Wambua J, Nyangweso G, Leach A, Lievens M, Kaslow DC, Njuguna P, Marsh K, Bejon P. 2016. Seven-year efficacy of RTS,S/AS01 malaria vaccine among young African children. N Engl J Med 374:2519–2529. doi: 10.1056/NEJMoa1515257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.RTS,S Clinical Trials Partnership. 2015. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet 386:31–45. doi: 10.1016/S0140-6736(15)60721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.WHO. 2015. RTS,S/AS01 malaria vaccine. Wkly Epidemiol Rec 50:693–696. [Google Scholar]
- 10.Hill AV, Reyes-Sandoval A, O'Hara G, Ewer K, Lawrie A, Goodman A, Nicosia A, Folgori A, Colloca S, Cortese R, Gilbert SC, Draper SJ. 2010. Prime-boost vectored malaria vaccines: progress and prospects. Hum Vaccin 6:78–83. doi: 10.4161/hv.6.1.10116. [DOI] [PubMed] [Google Scholar]
- 11.Lyon JA, Angov E, Fay MP, Sullivan JS, Girourd AS, Robinson SJ, Bergmann-Leitner ES, Duncan EH, Darko CA, Collins WE, Long CA, Barnwell JW. 2008. Protection induced by Plasmodium falciparum MSP1(42) is strain-specific, antigen and adjuvant dependent, and correlates with antibody responses. PLoS One 3:e2830. doi: 10.1371/journal.pone.0002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gozalo AS, Lucas CM, Qin J, Hall BT, Magill AJ. 2007. Anemia and antibodies to the 19-kDa fragment of MSP1 during Plasmodium falciparum infection in Aotus monkeys. Comp Med 57:396–401. [PubMed] [Google Scholar]
- 13.Singh S, Miura K, Zhou H, Muratova O, Keegan B, Miles A, Martin LB, Saul AJ, Miller LH, Long CA. 2006. Immunity to recombinant Plasmodium falciparum merozoite surface protein 1 (MSP1): protection in Aotus nancymai monkeys strongly correlates with anti-MSP1 antibody titer and in vitro parasite-inhibitory activity. Infect Immun 74:4573–4580. doi: 10.1128/IAI.01679-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stowers AW, Kennedy MC, Keegan BP, Saul A, Long CA, Miller LH. 2002. Vaccination of monkeys with recombinant Plasmodium falciparum apical membrane antigen 1 confers protection against blood-stage malaria. Infect Immun 70:6961–6967. doi: 10.1128/IAI.70.12.6961-6967.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thera MA, Doumbo OK, Coulibaly D, Laurens MB, Ouattara A, Kone AK, Guindo AB, Traore K, Traore I, Kouriba B, Diallo DA, Diarra I, Daou M, Dolo A, Tolo Y, Sissoko MS, Niangaly A, Sissoko M, Takala-Harrison S, Lyke KE, Wu Y, Blackwelder WC, Godeaux O, Vekemans J, Dubois MC, Ballou WR, Cohen J, Thompson D, Dube T, Soisson L, Diggs CL, House B, Lanar DE, Dutta S, Heppner DG Jr, Plowe CV. 2011. A field trial to assess a blood-stage malaria vaccine. N Engl J Med 365:1004–1013. doi: 10.1056/NEJMoa1008115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chuang I, Sedegah M, Cicatelli S, Spring M, Polhemus M, Tamminga C, Patterson N, Guerrero M, Bennett JW, McGrath S, Ganeshan H, Belmonte M, Farooq F, Abot E, Banania JG, Huang J, Newcomer R, Rein L, Litilit D, Richie NO, Wood C, Murphy J, Sauerwein R, Hermsen CC, McCoy AJ, Kamau E, Cummings J, Komisar J, Sutamihardja A, Shi M, Epstein JE, Maiolatesi S, Tosh D, Limbach K, Angov E, Bergmann-Leitner E, Bruder JT, Doolan DL, King CR, Carucci D, Dutta S, Soisson L, Diggs C, Hollingdale MR, Ockenhouse CF, Richie TL. 2013. DNA prime/adenovirus boost malaria vaccine encoding P. falciparum CSP and AMA1 induces sterile protection associated with cell-mediated immunity. PLoS One 8:e55571. doi: 10.1371/journal.pone.0055571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sedegah M, Hollingdale MR, Farooq F, Ganeshan H, Belmonte M, Kim Y, Peters B, Sette A, Huang J, McGrath S, Abot E, Limbach K, Shi M, Soisson L, Diggs C, Chuang I, Tamminga C, Epstein JE, Villasante E, Richie TL. 2014. Sterile immunity to malaria after DNA prime/adenovirus boost immunization is associated with effector memory CD8+ T cells targeting AMA1 class I epitopes. PLoS One 9:e106241. doi: 10.1371/journal.pone.0106241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ockenhouse CF, Angov E, Kester KE, Diggs C, Soisson L, Cummings JF, Stewart AV, Palmer DR, Mahajan B, Krzych U, Tornieporth N, Delchambre M, Vanhandenhove M, Ofori-Anyinam O, Cohen J, Lyon JA, Heppner DG, MSP-1 Working Group. 2006. Phase I safety and immunogenicity trial of FMP1/AS02A, a Plasmodium falciparum MSP-1 asexual blood-stage vaccine. Vaccine 24:3009–3017. doi: 10.1016/j.vaccine.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 19.Sheehy SH, Duncan CJ, Elias SC, Choudhary P, Biswas S, Halstead FD, Collins KA, Edwards NJ, Douglas AD, Anagnostou NA, Ewer KJ, Havelock T, Mahungu T, Bliss CM, Miura K, Poulton ID, Lillie PJ, Antrobus RD, Berrie E, Moyle S, Gantlett K, Colloca S, Cortese R, Long CA, Sinden RE, Gilbert SC, Lawrie AM, Doherty T, Faust SN, Nicosia A, Hill AV, Draper SJ. 2012. ChAd63-MVA-vectored blood-stage malaria vaccines targeting MSP1 and AMA1: assessment of efficacy against mosquito bite challenge in humans. Mol Ther 20:2355–2368. doi: 10.1038/mt.2012.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez A, Mintardjo R, Tax D, Gillissen G, Custers J, Pau MG, Klap J, Santra S, Balachandran H, Letvin NL, Goudsmit J, Radosevic K. 2009. Evaluation of a prime-boost vaccine schedule with distinct adenovirus vectors against malaria in rhesus monkeys. Vaccine 27:6226–6233. doi: 10.1016/j.vaccine.2009.07.106. [DOI] [PubMed] [Google Scholar]
- 21.Jiang G, Shi M, Conteh S, Richie N, Banania G, Geneshan H, Valencia A, Singh P, Aguiar J, Limbach K, Kamrud KI, Rayner J, Smith J, Bruder JT, King CR, Tsuboi T, Takeo S, Endo Y, Doolan DL, Richie TL, Weiss WR. 2009. Sterile protection against Plasmodium knowlesi in rhesus monkeys from a malaria vaccine: comparison of heterologous prime boost strategies. PLoS One 4:e6559. doi: 10.1371/journal.pone.0006559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Enama ME, Ledgerwood JE, Novik L, Nason MC, Gordon IJ, Holman L, Bailer RT, Roederer M, Koup RA, Mascola JR, Nabel GJ, Graham BS, VRC 011 Study Team. 2014. Phase I randomized clinical trial of VRC DNA and rAd5 HIV-1 vaccine delivery by intramuscular (i.m.), subcutaneous (s.c.) and intradermal (i.d.) administration (VRC 011). PLoS One 9:e91366. doi: 10.1371/journal.pone.0091366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gramzinski RA, Maris DC, Doolan D, Charoenvit Y, Obaldia N, Rossan R, Sedegah M, Wang R, Hobart P, Margalith M, Hoffman S. 1997. Malaria DNA vaccines in Aotus monkeys. Vaccine 15:913–915. doi: 10.1016/S0264-410X(96)00270-8. [DOI] [PubMed] [Google Scholar]
- 24.Jones TR, Gramzinski RA, Aguiar JC, Sim BK, Narum DL, Fuhrmann SR, Kumar S, Obaldia N, Hoffman SL. 2002. Absence of antigenic competition in Aotus monkeys immunized with Plasmodium falciparum DNA vaccines delivered as a mixture. Vaccine 20:1675–1680. doi: 10.1016/S0264-410X(01)00513-8. [DOI] [PubMed] [Google Scholar]
- 25.Gramzinski RA, Millan CL, Obaldia N, Hoffman SL, Davis HL. 1998. Immune response to a hepatitis B DNA vaccine in Aotus monkeys: a comparison of vaccine formulation, route, and method of administration. Mol Med 4:109–118. [PMC free article] [PubMed] [Google Scholar]
- 26.Sim BK, Narum DL, Liang H, Fuhrmann SR, Obaldia N III, Gramzinski R, Aguiar J, Haynes JD, Moch JK, Hoffman SL. 2001. Induction of biologically active antibodies in mice, rabbits, and monkeys by Plasmodium falciparum EBA-175 region II DNA vaccine. Mol Med 7:247–254. [PMC free article] [PubMed] [Google Scholar]
- 27.Jones TR, Narum DL, Gozalo AS, Aguiar J, Fuhrmann SR, Liang H, Haynes JD, Moch JK, Lucas C, Luu T, Magill AJ, Hoffman SL, Sim BK. 2001. Protection of Aotus monkeys by Plasmodium falciparum EBA-175 region II DNA prime-protein boost immunization regimen. J Infect Dis 183:303–312. doi: 10.1086/317933. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman SL, Doolan DL. 2000. Can malaria DNA vaccines on their own be as immunogenic and protective as prime-boost approaches to immunization? Dev Biol 104:121–132. [PubMed] [Google Scholar]
- 29.Jones TR, Obaldia N III, Gramzinski RA, Hoffman SL. 2000. Repeated infection of Aotus monkeys with Plasmodium falciparum induces protection against subsequent challenge with homologous and heterologous strains of parasite. Am J Trop Med Hyg 62:675–680. [DOI] [PubMed] [Google Scholar]
- 30.Obaldia N., III 1991. Detection of Klebsiella pneumoniae antibodies in Aotus l. lemurinus (Panamanian owl monkey) using an enzyme linked immunosorbent assay (ELISA) test. Lab Anim 25:133–141. doi: 10.1258/002367791781082603. [DOI] [PubMed] [Google Scholar]
- 31.Douglas AD, Baldeviano GC, Lucas CM, Lugo-Roman LA, Crosnier C, Bartholdson SJ, Diouf A, Miura K, Lambert LE, Ventocilla JA, Leiva KP, Milne KH, Illingworth JJ, Spencer AJ, Hjerrild KA, Alanine DG, Turner AV, Moorhead JT, Edgel KA, Wu Y, Long CA, Wright GJ, Lescano AG, Draper SJ. 2015. A PfRH5-based vaccine is efficacious against heterologous strain blood-stage Plasmodium falciparum infection in Aotus monkeys. Cell Host Microbe 17:130–139. doi: 10.1016/j.chom.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sedegah M, Tamminga C, McGrath S, House B, Ganeshan H, Lejano J, Abot E, Banania GJ, Sayo R, Farooq F, Belmonte M, Manohar N, Richie NO, Wood C, Long CA, Regis D, Williams FT, Shi M, Chuang I, Spring M, Epstein JE, Mendoza-Silveiras J, Limbach K, Patterson NB, Bruder JT, Doolan DL, King CR, Soisson L, Diggs C, Carucci D, Dutta S, Hollingdale MR, Ockenhouse CF, Richie TL. 2011. Adenovirus 5-vectored P. falciparum vaccine expressing CSP and AMA1. Part A. Safety and immunogenicity in seronegative adults. PLoS One 6:e24586. doi: 10.1371/journal.pone.0024586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamminga C, Sedegah M, Regis D, Chuang I, Epstein JE, Spring M, Mendoza-Silveiras J, McGrath S, Maiolatesi S, Reyes S, Steinbeiss V, Fedders C, Smith K, House B, Ganeshan H, Lejano J, Abot E, Banania GJ, Sayo R, Farooq F, Belmonte M, Murphy J, Komisar J, Williams J, Shi M, Brambilla D, Manohar N, Richie NO, Wood C, Limbach K, Patterson NB, Bruder JT, Doolan DL, King CR, Diggs C, Soisson L, Carucci D, Levine G, Dutta S, Hollingdale MR, Ockenhouse CF, Richie TL. 2011. Adenovirus-5-vectored P. falciparum vaccine expressing CSP and AMA1. Part B. Safety, immunogenicity and protective efficacy of the CSP component. PLoS One 6:e25868. doi: 10.1371/journal.pone.0025868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheehy SH, Duncan CJ, Elias SC, Biswas S, Collins KA, O'Hara GA, Halstead FD, Ewer KJ, Mahungu T, Spencer AJ, Miura K, Poulton ID, Dicks MD, Edwards NJ, Berrie E, Moyle S, Colloca S, Cortese R, Gantlett K, Long CA, Lawrie AM, Gilbert SC, Doherty T, Nicosia A, Hill AV, Draper SJ. 2012. Phase Ia clinical evaluation of the safety and immunogenicity of the Plasmodium falciparum blood-stage antigen AMA1 in ChAd63 and MVA vaccine vectors. PLoS One 7:e31208. doi: 10.1371/journal.pone.0031208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruder JT, Stefaniak ME, Patterson NB, Chen P, Konovalova S, Limbach K, Campo JJ, Ettyreddy D, Li S, Dubovsky F, Richie TL, King CR, Long CA, Doolan DL. 2010. Adenovectors induce functional antibodies capable of potent inhibition of blood-stage malaria parasite growth. Vaccine 28:3201–3210. doi: 10.1016/j.vaccine.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 36.Hodgson SH, Choudhary P, Elias SC, Milne KH, Rampling TW, Biswas S, Poulton ID, Miura K, Douglas AD, Alanine DG, Illingworth JJ, de Cassan SC, Zhu D, Nicosia A, Long CA, Moyle S, Berrie E, Lawrie AM, Wu Y, Ellis RD, Hill AV, Draper SJ. 2014. Combining viral vectored and protein-in-adjuvant vaccines against the blood-stage malaria antigen AMA1: report on a phase 1a clinical trial. Mol Ther 22:2142–2154. doi: 10.1038/mt.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boyle MJ, Reiling L, Osier FH, Fowkes FJ. 2017. Recent insights into humoral immunity targeting Plasmodium falciparum and Plasmodium vivax malaria. Int J Parasitol 47:99–104. doi: 10.1016/j.ijpara.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheehy SH, Douglas AD, Draper SJ. 2013. Challenges of assessing the clinical efficacy of asexual blood-stage Plasmodium falciparum malaria vaccines. Hum Vaccin Immunother 9:1831–1840. doi: 10.4161/hv.25383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sedegah M, Charoenvit Y, Minh L, Belmonte M, Majam VF, Abot S, Ganeshan H, Kumar S, Bacon DJ, Stowers A, Narum DL, Carucci DJ, Rogers WO. 2004. Reduced immunogenicity of DNA vaccine plasmids in mixtures. Gene Ther 11:448–456. doi: 10.1038/sj.gt.3302139. [DOI] [PubMed] [Google Scholar]
- 40.Epstein JE, Gorak EJ, Charoenvit Y, Wang R, Freydberg N, Osinowo O, Richie TL, Stoltz EL, Trespalacios F, Nerges J, Ng J, Fallarme-Majam V, Abot E, Goh L, Parker S, Kumar S, Hedstrom RC, Norman J, Stout R, Hoffman SL. 2002. Safety, tolerability, and lack of antibody responses after administration of a PfCSP DNA malaria vaccine via needle or needle-free jet injection, and comparison of intramuscular and combination intramuscular/intradermal routes. Hum Gene Ther 13:1551–1560. doi: 10.1089/10430340260201644. [DOI] [PubMed] [Google Scholar]
- 41.Ord RL, Tami A, Sutherland CJ. 2008. AMA1 genes of sympatric Plasmodium vivax and P. falciparum from Venezuela differ significantly in genetic diversity and recombination frequency. PLoS One 3:e3366. doi: 10.1371/journal.pone.0003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ouattara A, Barry AE, Dutta S, Remarque EJ, Beeson JG, Plowe CV. 2015. Designing malaria vaccines to circumvent antigen variability. Vaccine 33:7506–7512. doi: 10.1016/j.vaccine.2015.09.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collins WE, Jeffery GM. 1999. A retrospective examination of sporozoite- and trophozoite-induced infections with Plasmodium falciparum in patients previously infected with heterologous species of Plasmodium: effect on development of parasitologic and clinical immunity. Am J Trop Med Hyg 61:36–43. doi: 10.4269/tropmed.1999.61-036. [DOI] [PubMed] [Google Scholar]
- 44.Egan AF, Blackman MJ, Kaslow DC. 2000. Vaccine efficacy of recombinant Plasmodium falciparum merozoite surface protein 1 in malaria-naive, -exposed, and/or -rechallenged Aotus vociferans monkeys. Infect Immun 68:1418–1427. doi: 10.1128/IAI.68.3.1418-1427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones TR, Stroncek DF, Gozalo AS, Obaldia N III, Andersen EM, Lucas C, Narum DL, Magill AJ, Sim BK, Hoffman SL. 2002. Anemia in parasite- and recombinant protein-immunized Aotus monkeys infected with Plasmodium falciparum. Am J Trop Med Hyg 66:672–679. [DOI] [PubMed] [Google Scholar]
- 46.Ellis RD, Fay MP, Sagara I, Dicko A, Miura K, Guindo MA, Guindo A, Sissoko MS, Doumbo OK, Diallo D. 2011. Anaemia in a phase 2 study of a blood-stage falciparum malaria vaccine. Malar J 10:13. doi: 10.1186/1475-2875-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murungi LM, Sonden K, Llewellyn D, Rono J, Guleid F, Williams AR, Ogada E, Thairu A, Farnert A, Marsh K, Draper SJ, Osier FH. 2016. Targets and mechanisms associated with protection from severe Plasmodium falciparum malaria in Kenyan children. Infect Immun 84:950–963. doi: 10.1128/IAI.01120-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.National Research Council. 1996. Guide for the care and use of laboratory animals, 7th ed National Academies Press, Washington, DC. [Google Scholar]
- 49.Ma NS, Rossan RN, Kelley ST, Harper JS, Bedard MT, Jones TC. 1978. Banding patterns of the chromosomes of two new karyotypes of the owl monkey, Aotus, captured in Panama. J Med Primatol 7:146–155. [DOI] [PubMed] [Google Scholar]
- 50.Obaldia N III, Otero W, Marin C, Aparicio J, Cisneros G. 2011. Long-term effect of a simple nest-box on the reproductive efficiency and other life traits of an Aotus lemurinus lemurinus monkey colony: an animal model for malaria research. J Med Primatol 40:383–391. doi: 10.1111/j.1600-0684.2011.00489.x. [DOI] [PubMed] [Google Scholar]
- 51.Rogers WO, Gowda K, Hoffman SL. 1999. Construction and immunogenicity of DNA vaccine plasmids encoding four Plasmodium vivax candidate vaccine antigens. Vaccine 17:3136–3144. doi: 10.1016/S0264-410X(99)00146-2. [DOI] [PubMed] [Google Scholar]
- 52.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. 1998. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A 95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeng M, Smith SK, Siegel F, Shi Z, Van Kampen KR, Elmets CA, Tang DC. 2001. AdEasy system made easier by selecting the viral backbone plasmid preceding homologous recombination. Biotechniques 31:260–262. [DOI] [PubMed] [Google Scholar]
- 54.Rogers WO, Baird JK, Kumar A, Tine JA, Weiss W, Aguiar JC, Gowda K, Gwadz R, Kumar S, Gold M, Hoffman SL. 2001. Multistage multiantigen heterologous prime boost vaccine for Plasmodium knowlesi malaria provides partial protection in rhesus macaques. Infect Immun 69:5565–5572. doi: 10.1128/IAI.69.9.5565-5572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reyes-Sandoval A, Sridhar S, Berthoud T, Moore AC, Harty JT, Gilbert SC, Gao G, Ertl HC, Wilson JC, Hill AV. 2008. Single-dose immunogenicity and protective efficacy of simian adenoviral vectors against Plasmodium berghei. Eur J Immunol 38:732–741. doi: 10.1002/eji.200737672. [DOI] [PubMed] [Google Scholar]
- 56.Capone S, Reyes-Sandoval A, Naddeo M, Siani L, Ammendola V, Rollier CS, Nicosia A, Colloca S, Cortese R, Folgori A, Hill AV. 2010. Immune responses against a liver-stage malaria antigen induced by simian adenoviral vector AdCh63 and MVA prime-boost immunisation in non-human primates. Vaccine 29:256–265. doi: 10.1016/j.vaccine.2010.10.041. [DOI] [PubMed] [Google Scholar]
- 57.Ogwang C, Kimani D, Edwards NJ, Roberts R, Mwacharo J, Bowyer G, Bliss C, Hodgson SH, Njuguna P, Viebig NK, Nicosia A, Gitau E, Douglas S, Illingworth J, Marsh K, Lawrie A, Imoukhuede EB, Ewer K, Urban BC, Hill AVS, Bejon P, MVVC Group. 2015. Prime-boost vaccination with chimpanzee adenovirus and modified vaccinia Ankara encoding TRAP provides partial protection against Plasmodium falciparum infection in Kenyan adults. Sci Transl Med 7:286re285. doi: 10.1126/scitranslmed.aaa2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Earle W, Perez M. 1932. Enumeration of parasites in the blood of malarial patients. J Lab Clin Med 17:1124–1130. [Google Scholar]
- 59.Obaldia N., III 2007. Clinico-pathological observations on the pathogenesis of severe thrombocytopenia and anemia induced by Plasmodium vivax infections during antimalarial drug efficacy trials in Aotus monkeys. Am J Trop Med Hyg 77:3–13. [PubMed] [Google Scholar]