

Abstract
NKG2D-mediated immune surveillance is crucial for inhibiting tumor growth and metastases. Malignant tumor cells often downregulate NKG2D ligands to escape from immune surveillance. High-profile studies have shown that restoring NKG2D ligand expression via genetic engineering inhibits tumor formation and progression. However, no effective in vivo approaches are available to restore these ligands across different types of solid tumors, because the classic stress signal–dependent induction of this ligand in vitro is transient and has rarely been duplicated in solid tumors in vivo. We found that co-administration of an immune stimulatory signal (interleukin-12) and chemotherapy (doxorubicin) restored the NKG2D ligand Rae-1 in multiple tumor types, including a human tumor model. The restored expression of NKG2D ligands was associated with tumor cell death and delay of tumor progression in vivo. Induction of tumor-specific NKG2D ligands required the engagement of CD8+ T cells and was regulated by the histone acetyltransferases (HATs) GCN5 and PCAF. The tumor-specific restoration of NKG2D ligands in a variety of tumor models, including a human tumor model, resulted in NKG2D-dependent tumor regression and extended survival time. The elucidation of a CD8+ T cell–dependent mechanism suggests that activated NKG2D+CD8+ T-cell therapy alone may be able to restore NKG2D ligand in tumors.
Keywords: NKG2D ligand Rae-1, CD8+ T cells, immune surveillance, GCN5, PCAF
Introduction
Immune surveillance plays a key role in inhibiting tumorigenesis, tumor progression, and metastasis (1,2). The interaction between natural killer group 2D (NKG2D) and its ligands is a key mechanism for immune surveillance (3,4). A growing body of evidence shows that the expression of NKG2D ligands on the tumor cell surface causes NKG2D-mediated tumor cell death (3,5–7). Expression of NKG2D ligands induced by genetic engineering leads to the rejection of different types of solid tumors and extends survival time in mice (8,9). In colorectal cancer patients, large amounts of the NKG2D ligands MICA and ULBP2, in particular, were shown to correlate with a longer survival time (10). However, NKG2D ligands are shed or internalized by tumor cells during immune escape, which prevents immune cells from recognizing tumor cells through NKG2D (11). Discovery of an effective in vivo approach for restoring NKG2D ligands will be key to eliminating tumors through immune surveillance.
NKG2D ligands, which consist of Rae-1α-ε, H60, and Mult1 in mice as well as MICA/B and ULBP1–6 in humans, can be induced in vitro by heat shock, oxidative stress, DNA damage, oncogene exposure, viral infection, inflammatory context, and certain chemicals, such as chemotherapeutic agents (6,12–16). It is generally believed that NKG2D ligands are upregulated through activation of the stress-induced signaling pathway (17), whereas a number of groups reported that histone deacetylase (HDAC) inhibitors induce robust expression of NKG2D ligands on tumor cells to sensitize these cells to immune surveillance (14,15,18,19). Unfortunately, none of these in vitro observations have been duplicated in vivo in solid tumors. Therefore, identifying an effective in vivo approach to induce NKG2D ligands in tumors could be significant for enhancing immune surveillance.
To find an effective approach to induce NKG2D ligands specifically in tumors in vivo, here we describe a combination of interleukin 12 (IL12) and doxorubicin that restored tumor-specific expression of NKG2D ligands in murine and human solid tumor models in vivo, causing tumor regression via a CD8+ T cell–dependent, HAT-mediated mechanism.
Materials and Methods
Animal studies
Wild-type mice, 6–8 weeks old, were purchased from the National Cancer Institute (Bethesda, MD). Nude, Rag2−/−, NSG, and IFNγ−/−(B6.129S7-Ifngtm1Ts/J) mice of the same age range and spontaneous breast carcinoma FVB/N-Tg(MMTV-PyVT)634Mul/J model mice, 4 weeks old, were purchased from Jackson Laboratory (Bar Harbor, ME). The mouse care and handling procedures were approved by the Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center.
To create the transplant tumor mouse models, 4T-1 (breast carcinoma) or CT26 (colon cancer) cells (2 × 105 per mouse) in 30 μL of phosphate-buffered saline solution (PBS) were subcutaneously inoculated into BALB/c mice, and LLC (Lewis lung carcinoma) tumor cells (1.5 × 105 per mouse) were inoculated into C57BL/6 mice. K7M3 (osteosarcoma) tumor cells (1 × 105 per mouse) in 15 μL of PBS were inoculated into BALB/c mice via intraosseous injection. Mel 2549 (tumor-derived human melanoma) cells and autologous tumor infiltrating lymphocytes (TILs), which were expanded and maintained in Dr Bernatchez’s laboratory at MD Anderson Cancer Center (20), were inoculated subcutaneously (3 × 106 per mouse in 30 μL PBS) into NSG mice. These mice also received intraperitoneal injection of autologous TILs (5 × 106) the day after each treatment. All the cell lines were characterized by DNA fingerprinting within 6 months of initiating the experiments at MD Anderson Cancer Center’s Characterized Cell Line Core Facility.
Tumor bearing mice were subject to control DNA (10 μg/mouse), control DNA plus doxorubicin (1 mg/kg), IL12–encoding DNA (10 μg/mouse), or IL12–encoding DNA plus doxorubicin, and followed by electroporation as described previously (21). Treatment of the spontaneous breast carcinoma mice was initiated when mice were 8 weeks old, after primary tumor development was observed. Tumor volume was calculated by the formula: V = (π/8) × (ab2) where V = tumor volume in cubic centimeters, a = maximum tumor diameter, and b = diameter at 90° to a.
Plasmids and reagents
The IL12 DNA construct was purchased from Valentis, Inc. (Vilnius, Lithuania). Rae-1 constructs were PCR-amplified from mouse cDNA and cloned into pEGFP N1 vector (Clontech, Mountain View, CA). Mouse IL15 and IL4 mRNAs were purchased from Open Biosystems (Thermo Scientific, Two Rivers, WI). mIL21 pORF9 mRNA was purchased from Invivogen (San Diego, CA). mIL18 was purchased from OriGene Technologies (Rockville, MD). mIFNγ was PCR-amplified from mouse genomic DNA and mouse spleen cDNA, respectively. All constructs were confirmed by sequence analyses. DNA was prepared by using the endotoxin-free Mega preparation kit from Qiagen, Inc. (Valencia, CA) by following the manufacturer’s instructions.
Doxorubicin (Bedford Laboratories, Bedford, OH) and bleomycin (APP Pharmaceuticals, Schaumburg, IL) were purchased from the pharmacy at the Louisiana State University or The University of Texas MD Anderson Cancer Center. Cisplatin was purchased from Bristol Laboratories (Princeton, NJ). Cycloheximide, cyclophosphamide, chloroquine, methotrexate, and ifosfamide were purchased from Sigma-Aldrich (St. Louis, MO). Trichostatin A, sodium butyrate and anacardic acid were purchased from Sigma-Aldrich (St. Louis, MO). IL12 and IFNγ recombinant proteins were purchased from R&D systems (Minneapolis, MN).
Mouse GCN5 siRNA: 5′ CCAAACAAGUCUAUUUCUA 3′
Mouse PCAF siRNA: 5′ CCUAUCUGGGAUCAGGAUU 3′
Control siRNA: 5′UCCAAGUAGAUUCGACGGCGAAGTG 3′
Antibodies
Phycoerythrin (PE)-conjugated anti-mouse CD3 and its isotype control antibodies, and PE-Cy7-conjugated antibodies to mouse CD4 and mouse CD8 and their isotype control antibodies, were purchased from Biolegend (San Diego, CA); FITC-conjugated antibody to mouse NKp46, and its isotype control antibody were purchased from eBioscience (San Diego, CA). NKG2D antibody was purchased from R&D Systems. Rat antibody to mouse CD8α (clone YTS105.18) was purchased from AbD Serotec (Raleigh, NC). Rat anti-mouse CD4 (clone RM4–5) was purchased from BD Pharmingen (San Jose, CA). Rat anti-mouse NKp46 (clone 29A1.4) was purchased from Biolegend (San Diego, CA). Goat antibodies to rat Alexa fluor 488 and Alexa fluor 594 secondary antibodies were purchased from Life Technologies (Grand Island, NY). Streptavidin-conjugated Alexa fluor 594 was purchased from Life Technologies (Grand Island, NY). Mouse IL12 antibody and mouse IL12Rβ2 antibodies were purchased from R&D systems (Minneapolis, MN). GCN5 and PCAF antibodies were purchased from Cell Signaling Technology (Danvers, MA). Antibodies to β-actin and GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-mouse Rae-1 was developed by our monoclonal antibody facility (MD Anderson Cancer Center) and validated in our previous publication (Hu et al. 2014). Horseradish peroxidase–conjugated antibody to mouse IgG, rat IgG secondary, and rabbit IgG were purchased from Cell Signaling Technology. NKG2D C7 depleting antibody was generously provided by Dr. Wayne Yokoyama (Washington University School of Medicine).
Flow cytometry analysis
Cells were incubated with primary antibodies and secondary antibodies sequentially for 30 minutes each at 4°C. The stained cells were analyzed on an Attune acoustic focusing cytometer (Applied Biosystems, Carlsbad, CA). Data were analyzed with FlowJo software (BD Biosciences, San Jose, CA).
Immunoblotting assay
Frozen tissue samples were smashed before being further homogenized in 0.4 mL of ice-cold lysis buffer with five to eight silicone beads by a minibead beater (BioSpec Products, Bartlesville, OK). The protein extract was separated from tissue residues by centrifugation at the maximum speed for 20 minutes at 4°C. Sixty micrograms of total protein was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes by using the iBlot gel transfer device (Invitrogen, Grand Island, NY). The membranes were blotted with different primary and secondary antibodies to detect the proteins of interest.
Immune cell depletion in vivo
A depletion antibody against CD8+ T cells (clone 2.43), CD4+ T cells (GK1.5), or NK cells (PK136 for C57BL/6 mice, and anti-asialo GM1 for BALB/C mice), 250 μg (50 μg for anti-asialo GM1) per mouse was administered intraperitoneally into mice twice a week for 3 weeks, starting on day 7 after tumor cell inoculation. Blocking antibody to mouse NKG2D receptor (clone C7) was administered (250 μg per mouse) intraperitoneally into mice twice weekly, starting three weeks prior to tumor inoculation.
Immune cell enrichment
Mouse spleens were homogenized gently in a 40-μm nylon strainer. NK cells, CD4+ T cells and CD8+ T cells were enriched in splenocyte specimens by using the Easysep mouse NK-, CD4+ T-, or CD8+ T-cell enrichment kit (Stem Cell Technology, Vancouver, BC, Canada) by following the manufacturer’s guidelines.
Knockdown of GCN5 and PCAF in vivo
Control siRNA, or GCN5 and PCAF siRNAs (250 pmol) were injected intratumorally, followed by electroporation twice weekly. The electroporation was performed using the following parameters: two 50 ms pulses of 150 V cm−1 with a 100 ms interval between pulses.
TUNEL assay
Frozen slides were fixed in 4% formaldehyde in PBS for 25 minutes at 4°C and permeabilized in 0.2% Triton X-100 in PBS in 5 minutes. Equilibration buffer (100 μL; Promega, Madison, WI) was added to each slide at room temperature for 5–10 minutes. TdT reaction mix (50 μL; Promega) was added to each tissue section, and slides were incubated for 60 minutes at 37°C in a humidified chamber. The reaction was stopped by 2× SSC (Promega) for 15 minutes. The slides were mounted using SLOWFADE Gold antifade mountant with DAPI (Life Technologies, Grand Island, NY) and analyzed by fluorescence microscopy (Nikon, Tokyo, Japan).
Immunohistochemical/immunofluorescent analysis
Frozen tumor sections were sequentially fixed with cold acetone, acetone plus chloroform (1:1), and acetone. Tissue sections were blocked with blocking buffer (5% normal horse serum and 1% normal goat serum in PBS) and then incubated with primary antibody overnight at 4°C, secondary antibody for 1 hour at room temperature, and TUNEL staining if necessary. For immunohistochemistry staining, Nuclei were counterstained with hematoxylin (Sigma-Aldrich, St. Louis, MO), and tumor sections were mounted with Clearmount mounting solution (Life Technologies, Grand Island, NY). For immunofluorescence staining, tumor sections were mounted in SLOWFADE Gold antifade mountant with DAPI (Life Technologies, Grand Island, NY). Slides were visualized under the Nikon eclipse Ti fluorescence microscope (Nikon, Melville, NY).
Statistical analysis
The directly measured outcomes were analyzed by the two-sided Student t-test to compare two treatment groups or by one-way analysis of variance to compare more than two treatment groups. Statistical significance of each comparison was determined by GraphPad software (GraphPad Software, La Jolla, CA). We set a P value < 0.05 to indicate statistical significance. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, no statistical significance.
Results
Tumor-specific induction of Rae-1 in vivo
Many tumor cell lines express Rae-1 (Supplementary Fig. S1A). In contrast to Rae-1–positive Lewis lung carcinoma (LLC) cells in vitro, Rae-1 expression in vivo on GFP+ tumor cell surface was significantly reduced as early as 7 days after inoculation (Supplementary Fig. S1B).
Various chemotherapeutic agents have been found to induce NKG2D ligands on tumor cells in vitro (17). We confirmed these in vitro observations with both CT26 and K7M3 cell lines (Figs. S2A, S2B) in which bleomycin, cisplatin, cyclophosphamide, doxorubicin, and methotrexate elevated Rae-1 expression on the cell surface. However, these agents only induced very transient expression of Rae-1 in solid tumors in vivo (Supplementary Fig. S2C). Thus, chemotherapeutic agents alone were unable to restore long-term Rae-1 expression in solid tumors in vivo. Thus, a different approach towards restoring Rae-1 must be employed.
We treated LLC tumor-bearing mice with IL12 DNA plus doxorubicin, IL12 DNA alone, control DNA plus doxorubicin, or control DNA alone, and found that tumor volume regressed significantly and mice had significantly extended survival times, after treatment with IL12 plus doxorubicin, in an NKG2D function–dependent manner (Fig. 1A). In agreement with this result, blocking NKG2D reduced the production of the cytotoxic molecule perforin in tumors (Supplementary Fig. S3). NKG2D ligand Rae-1 expression was substantially increased, as determined by immunoblotting and flow cytometry, in tumors after two administrations of IL12 plus doxorubicin, whereas the other treatments did not have this effect (Fig. 1B). The same combination treatment did not increase Rae-1 expression in normal tissues (Supplementary Fig. S4), showing that the Rae-1 induction by IL12 plus doxorubicin was tumor specific.
Fig. 1. Treatment with IL12 and doxorubicin delayed tumor development by inducing NKG2D ligand Rae-1 in tumors in vivo.
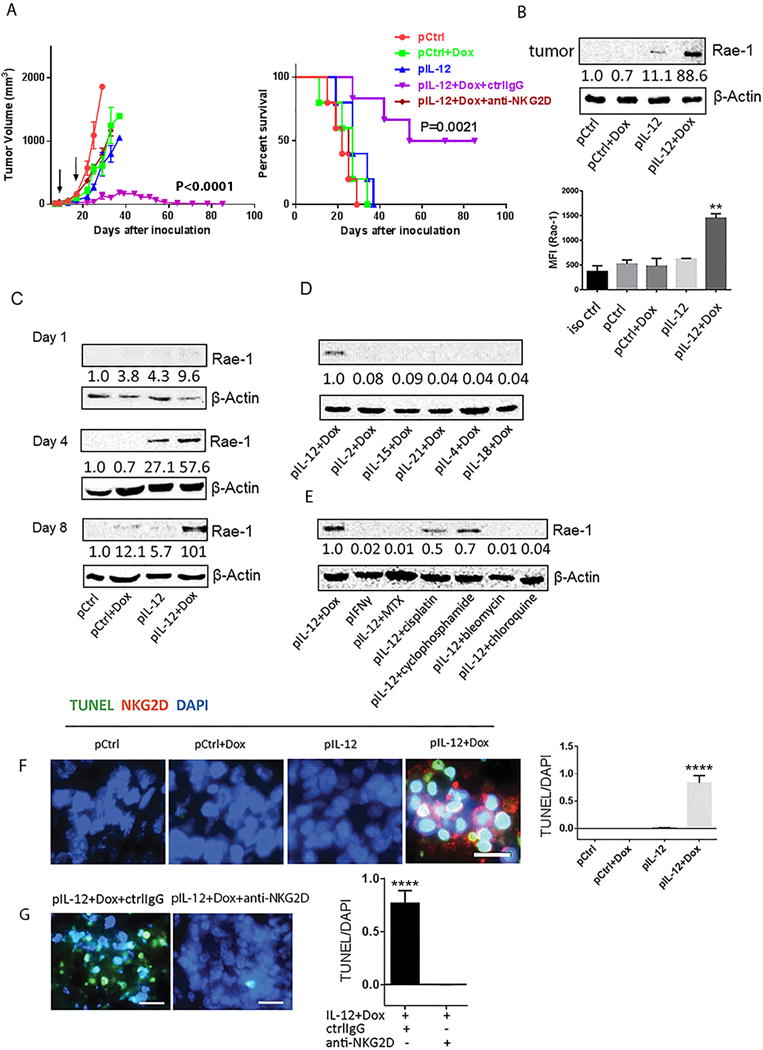
LLC tumor–bearing mice (n = 5) were subject to twice administrations (10 days apart) with the indicated cytokine DNA, the indicated chemotherapeutic agent, both, or control DNA. (A) Tumor size was measured from 5 days after inoculation twice weekly for 12 weeks. Survival time was also monitored for 12 weeks. Black arrows represent treatment dates. (B) Immunoblots (left panel) and flow cytometry (right panel) of Rae-1 in tumor samples from mice receiving one of the four indicated treatments. For (B), tumors were collected on day 4 after the second treatment. (C), tumors were collected on day 1, 4, or 8 after the second treatment. For (D) and (E), tumors were collected on day 8 after the second treatment. All results are representative of three repeated experiments. pCtrl represents control plasmid DNA; pIL12, pIL15, pIL21, pIL4 and pIL18 represent the plasmid DNAs encoding the indicated cytokines; Dox, doxorubicin; IL, interleukin; IFN, interferon. These treatment abbreviations apply to all in vivo experiments shown in the rest of the figures. Intensity quantification (intensity of Rae-1/intensity of β-Actin) shown under each blot represents the mean intensity of three repeated experiments. (F) Analysis of association between infiltration of NKG2D+ effector cells (red) and tumor cell death (green) in sections from LLC tumors treated with one of the four indicated treatments as assessed by immune staining and TUNEL assay, respectively. (G) Comparison of tumor cell death following Rae-1 induction in the presence (ctrl IgG) or absence (anti-NKG2D) of NKG2D. In both (F) and G), quantification of TUNEL signals is shown in the right panel. The scale bars equal 25 μm.
Another important question is the duration of the restoration of NKG2D ligand expression by this combination therapy. On day 1 after the second treatment, Rae-1 was weakly detectable (Fig. 1C). On day 4, Rae-1 expression was detectable in tumors treated with IL12 alone and in those treated with IL12 plus doxorubicin, although more Rae-1 was expressed in the latter. On day 8, Rae-1 remained high in tumors from mice treated with IL12 plus doxorubicin but had disappeared in tumors from mice subjected to other treatments. Thus, a combination of an immune signal (IL12) and chemotherapy (doxorubicin) could induce long-term high expression of Rae-1 on solid tumors.
Encouraged by these results, we tested whether any other combinations of immune stimulatory signals and chemotherapy could increase tumor-specific Rae-1 expression. Six different cytokines plus doxorubicin were tested in the same LLC solid tumor model. IL12 plus doxorubicin was the only one of these combinations that induced higher Rae-1 expression in tumors (Fig. 1D). Similarly, IL12 plus doxorubicin induced the highest Rae-1 tumor expression among all of the tested combinations of IL12 and a chemotherapeutic agent (cyclophosphamide [60 mg/kg], cisplatin [20 mg/kg], chloroquine [10 mg/kg], or bleomycin [5 U/kg]) (Fig. 1E). Thus, IL12 plus doxorubicin was the most reliable combination for boosting Rae-1 expression in solid tumors in this model system.
The induction of Rae-1 was associated with an accumulation of NKG2D+ immune cells and tumor cell death, which was reversed by NKG2D blocking (Fig. 1F, 1G), suggesting an induction of NKG2D immune surveillance by this combinational treatment. To identify the subtypes of the NKG2D+ cells in tumors, we costained the tumor sections with anti-NKG2D and antibodies to the immune cell markers CD8, CD4 or NKp46. We found that the majority of the NKG2D+ cells that accumulated in tumors after IL12 plus doxorubicin treatment were CD8+ T cells (Supplementary Fig. S5).
Rae-1 induction in vivo in various tumor types
To determine whether this observation was limited to the LLC tumor model, the same treatments were administered in subcutaneous colon cancer (CT26), orthotopic osteosarcoma (K7M3), and spontaneous breast carcinoma (Figs. 2A–2D). Results from immunoblotting (Figs. 2A–2D), flow cytometry (Figs. 2A–2D), immunohistochemical (Supplementary Fig. S6) assays showed that, regardless of the tumor model, IL12 plus doxorubicin substantially increased Rae-1 expression in all solid tumor models. To clarify that Rae-1 is induced only on tumor cells, but not stromal or antigen-presenting cells, Rae-1 expression was tested in LLC-GFP and CT26-GFP tumor models that received the same treatments. As expected, IL12 plus doxorubicin dramatically induced Rae-1 on the surface of GFP+ tumor cells (Supplementary Fig. S7A) but not on GFP− cells (Supplementary Fig. S7B). These results support the idea that IL12 plus doxorubicin treatment mediated the restoration of Rae-1 in tumors, and this can be applied to many types of solid tumors. To show antitumor immune surveillance, tumor sections from the same tumor models were subjected to TUNEL assay to measure tumor cell death in vivo (Supplementary Fig. S6). Indeed, Rae-1 induction by IL12 plus doxorubicin was associated with tumor cell death in vivo. Tumor volume was inversely associated with the amount of NKG2D ligand expression in tumors (Fig. 1).
Fig. 2. Rae-1 is induced by IL12 plus doxorubicin in four tumor models.
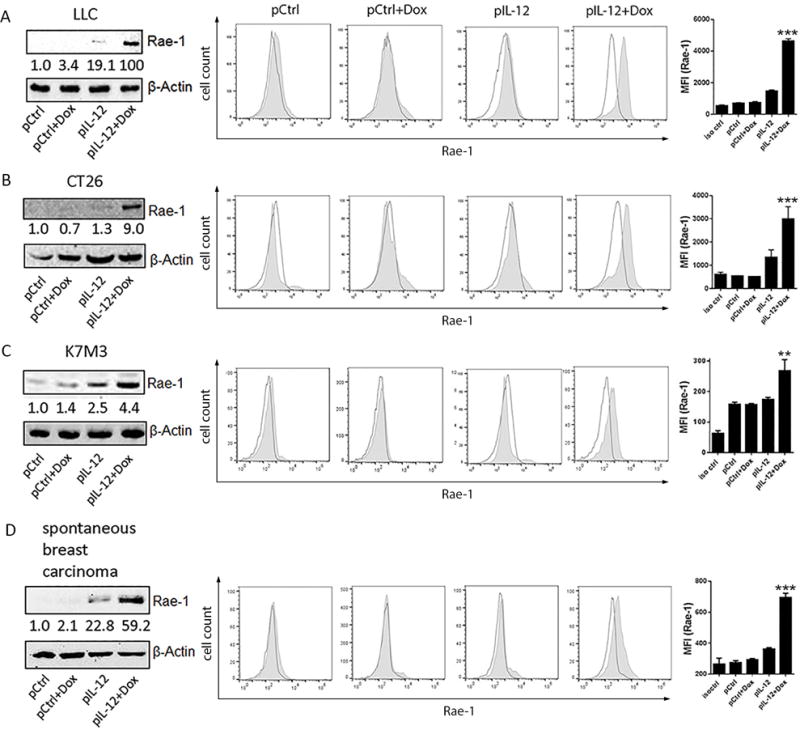
The IL12 plus doxorubicin combination was tested in four different mouse tumor models. Mice (n = 3) were treated twice (10 days apart) with one of the four indicated treatments. The tumor model is shown at the left side for each figure panel. Tumors were collected on day 4 after the second treatment. (A-D) The tumor samples were subject to immunoblotting and flow cytometry (no shade: isotype control; shade: Rae-1) of Rae-1. Intensity quantification (intensity of Rae-1/intensity of β-Actin) shown under each blot represents the mean intensity of three repeated experiments. The Bar graphs show the MFI of Rae-1 expression as mean ± SEM.
Despite the restoration of NKG2D ligands on tumor cells in vivo in several murine transplant and spontaneous tumor models (Fig. 2), we could not predict whether such treatment would also boost NKG2D immune surveillance in human cancer patients. To evaluate the potential for translating the IL12 plus doxorubicin regimen into clinical practice, we tested the IL12 plus doxorubicin regimen in a human tumor xenograft model. NSG mice were inoculated with Mel 2549, a human melanoma cell line. The expanded autologous tumor-infiltrating lymphocytes were administered to tumor-bearing mice one day after each administration of control DNA, control DNA plus doxorubicin, human IL12 DNA, or human IL12 DNA plus doxorubicin. As in the previously tested mouse tumor models, treatment with human IL12 plus doxorubicin induced MICA/B and ULBP2 in these tumors (Fig. 3A, 3B), as determined by flow cytometry (Fig. 3A) and immunoblotting (Fig. 3B). As in the murine tumor models, the restoration of NKG2D ligands in the human xenograft model was associated with a delay of tumor development (Fig. 3C). These data, together, established that treatment with IL12 plus doxorubicin not only revived NKG2D ligands in murine solid tumor models but boosted NKG2D immune surveillance in a human solid tumor model.
Fig. 3. NKG2D ligands are induced by IL12 plus doxorubicin in a human melanoma xenograft model.
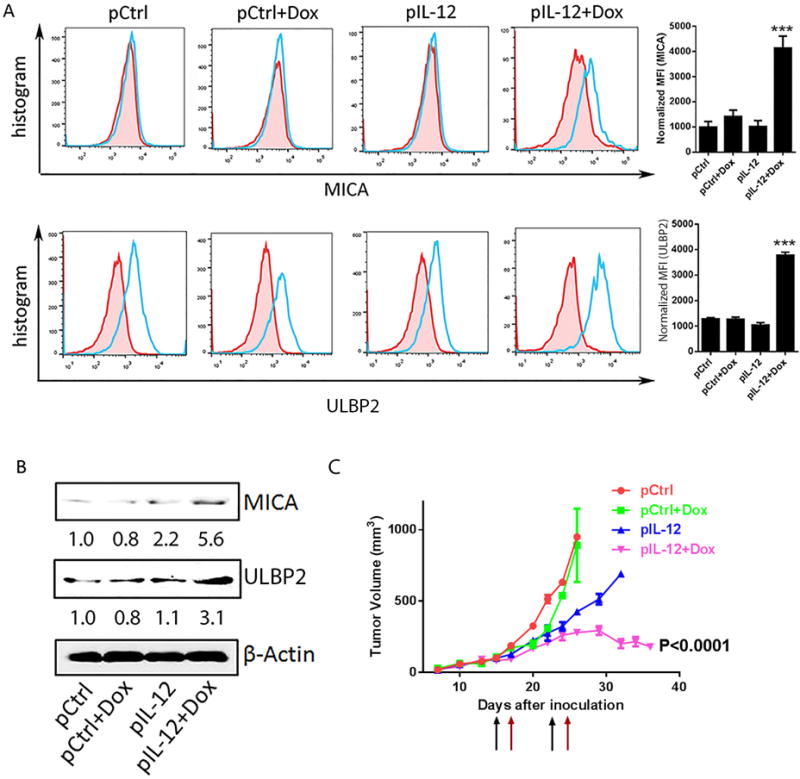
Mel 2549 tumor-bearing NSG mice (n = 3) were treated twice (10 days apart) with one of the four indicated treatments. 5 million autologous TILs were infused the days after each treatment. Tumors were collected on day 4 after the second treatment. (A) Flow cytometry analyses of NKG2D ligands MICA (top) and ULBP2 (bottom). Bar graphs show the normalized mean fluorescence index (MFI of NKG2D ligands minus MFI of isotype control). (B) Immunoblots of MICA and ULBP2 in tumor samples from mice receiving one of the four indicated treatments. Intensity quantification (intensity of Rae-1/intensity of β-Actin) shown under each blot represents the mean intensity of three repeated experiments. (C) Tumor size was measured from 7 days after inoculation twice weekly. Black arrows represent treatment dates. Red arrows represent TIL infusion dates.
CD8+ T cells facilitated the induction of NKG2D ligands on tumor cells
To decipher the mechanism underlying the induction of NKG2D ligand in tumors after treatment with IL12 plus doxorubicin, Rae-1 induction was investigated in murine tumor models. One hypothesis was that activated immune cells following IL12 plus doxorubicin treatment account for tumor specific Rae-1 restoration. To examine the role of immune cells, we administered the combination to various types of immune-deficient mice, including nude mice (which lack T cells), and Rag2−/− mice (which lack mature T and B cells), with IL12 plus doxorubicin and assayed Rae-1 in their transplant tumors. Compared with wild-type mice, nude mice (Fig. 4A) and Rag2−/− mice (Fig. 4B) had no induction of Rae-1 in tumors after treatment. To identify the T-cell subtype critical to Rae-1 restoration, we depleted different subpopulations of immune cells (CD4+ T cells, CD8+ T cells, and NK cells) by using depleting antibodies. Depletion efficiency was determined by flow cytometry analysis (Supplementary Fig. S8A–S8C). Depletion of CD8+ T cells, but not CD4+ T cells or NK cells, impaired the IL12 plus doxorubicin–mediated Rae-1 induction in tumors (Fig. 4C). Rae-1 expression on tumor cell surface was determined in flow cytometry data (Fig. 4D). In parallel with this result, depletion of NK cells neither impaired IL12 plus doxorubicin-induced tumor regression nor did it significantly decrease survival time (Fig. 4E, 4F). Thus CD8+ T cells play a critical role in Rae-1 induction in solid tumors.
Fig. 4. Tumor-specific Rae-1 elevation requires CD8+ T cells.
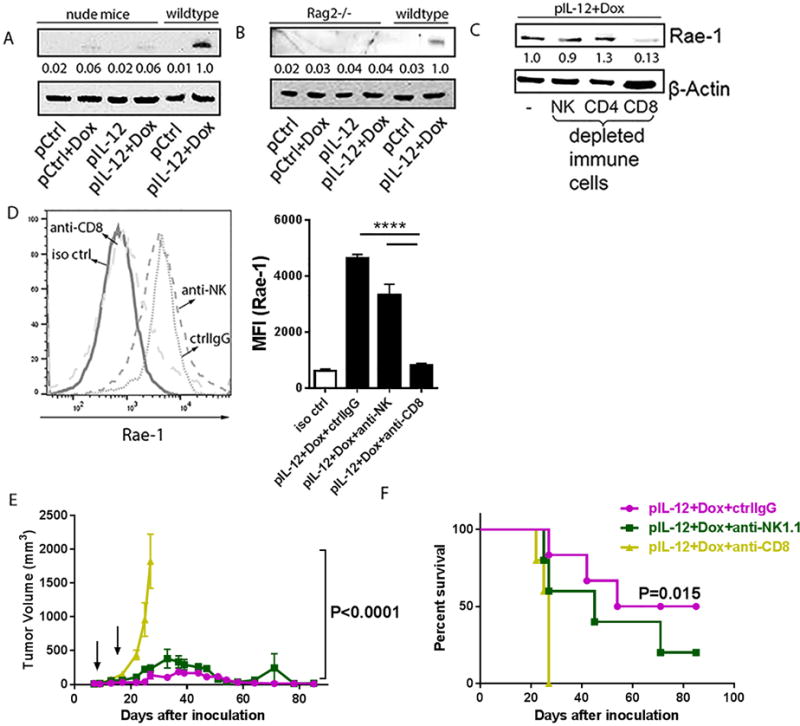
(A, B) Immunoblots of Rae-1 in 4T-1 tumor samples from nude mice (n=3) or LLC tumor samples from Rag2−/− (n = 3) that were subject to one of the four standard treatments. (C) Immunoblots of Rae-1 in LLC tumor samples from mice (n = 3) that received IL12 plus doxorubicin plus control IgG or one of the immune cell–depleting antibodies as shown. (D) Flow cytometry analyses of Rae-1 in LLC tumors subject to the same treatments as (C). (E, F) Tumor volume (E) and survival time (F) of LLC tumor bearing mice (n = 5) subject to the same treatments as (C). Intensity quantification (intensity of Rae-1/intensity of β-Actin) shown under each blot represents the mean intensity of three repeated experiments. Bar graphs (D) show the mean fluorescence intensity (MFI) of Rae-1 as means ± SEM (n = 3). Results are representative of three repeated experiments.
To verify that activated CD8+ T cells were the only critical subpopulation of immune cells for Rae-1 restoration, we cocultured immune cells and tumor cells in vitro to simulate the tumor microenvironment. Wild-type IFNγ+/+ splenocytes, or IFNγ−/− splenocytes isolated from IFNγ knockout mice, were incubated with control IgG, or blocking antibodies to IL12 or IL12Rβ2, for 2 hours, and then were stimulated with IL12 recombinant protein (10 ng/ml) for 6 hours. CT26 or K7M3 tumor cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) and directly treated with IL12 (10 ng/ml) or IFNγ (10 ng/ml), or cocultured with naïve or stimulated splenocytes (1:5 tumor cell-to-splenocyte ratio) for 24 hours. Rae-1 expression was then assessed on the tumor cell surface. We found that IL12 or IFNγ recombinant protein alone did not affect Rae-1 expression on the cell surface. In contrast, only stimulated, but not naïve, splenocytes restored Rae-1 on the tumor cell surface. Blocking either IL12 or IL12 receptor with cognate antibodies completely abolished Rae-1 upregulation, and IFNγ−/− splenocytes also significantly impaired Rae-1 induction (Fig. 5A, 5B). The isolated wild-type splenocytes were then depleted of, or enriched with, different subpopulations of immune cells (NK cells, CD4+ T cells, CD8+ T cells; Supplementary Fig. S9). CFSE-labeled tumor cells were co-incubated with splenocytes after depletion (1:5 tumor cell-to-splenocyte ratio) (Fig. 5C, 5D) or enrichment of immune cells (1:1 tumor cell-to-immune cell ratio) (Fig. 5E, 5F). Only the CD8+ T cell–depleted splenocytes could not restore Rae-1 expression on tumor cells, whereas the CD8+ T cell–enriched splenocytes significantly restored Rae-1 in both tumor cell lines. Thus IL12 acted on CD8+ T cells to produce inflammatory cytokines, such as IFNγ, that played critical roles in CD8+ T cell–mediated Rae-1 upregulation on tumor cells. Taken together, the in vivo and in vitro results suggest a CD8+ T cell–mediated mechanism for Rae-1 restoration by IL12 plus doxorubicin.
Fig. 5. In vitro model of activated CD8+ T cell-mediated Rae-1 restoration.
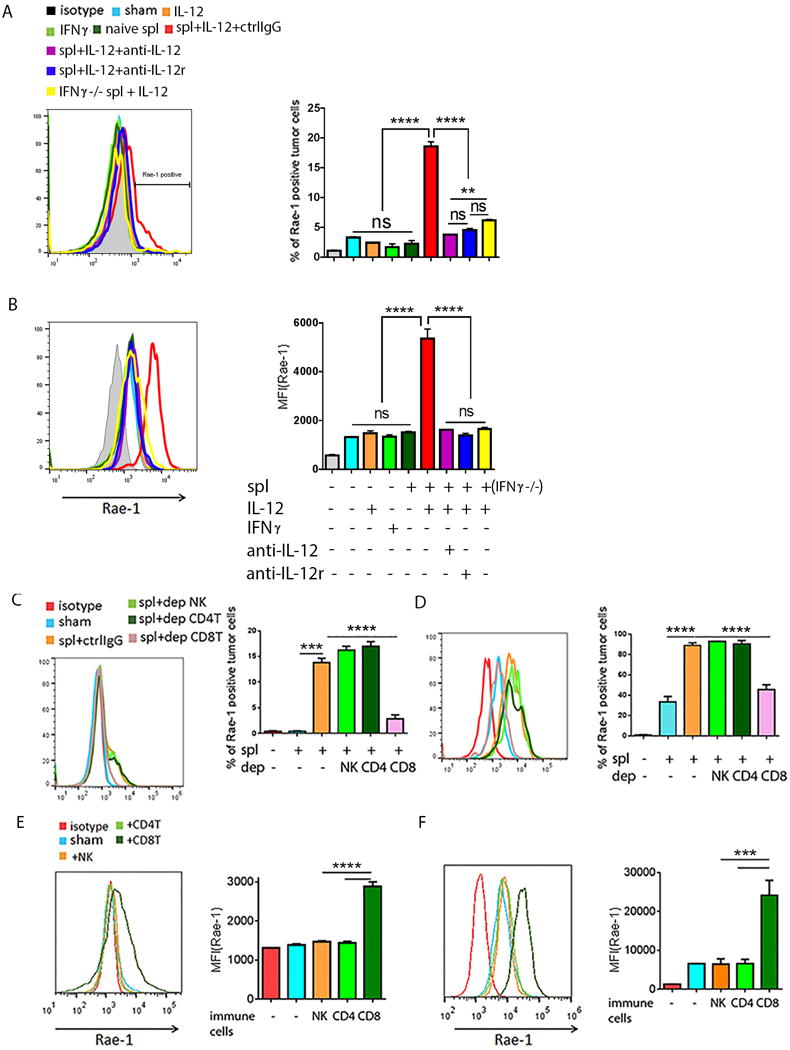
(A, B) Rae-1 on CT26 (A) or K7M3 (B) tumor cells after treated with IL12 (10 ng/ml) or IFNγ (10 ng/ml) recombination protein, or co-incubation with naïve splenocytes or stimulated spl. (C, D) Rae-1 on CT26 tumor cells (C) and K7M3 tumor cells (D) after co-incubation with activated splenocytes lacking the indicated lymphocyte subpopulation through antibody depletion. spl+ctrlIgG: splenocytes plus control IgG; spl+dep NK: splenocytes in which NK cells were depleted; spl+dep CD4T: splenocytes in which the CD4+ T cells were depleted; spl+dep CD8T: splenocytes in which CD8+ T cells were depleted. (E, F) Rae-1 on CT26 (E) and K7M3 (F) tumor cell surfaces after incubation with splenocytes enriched in immune cell subpopulations from wild-type mice as indicated. Bar graphs show percentage of Rae-1+ cells or mean fluorescence intensity (MFI) of Rae-1 as means ± SEM. Results are representative of three repeated experiments.
The above results showed that CD8+ T cells must be present in the tumor microenvironment and engaged with tumor cells to induce NKG2D ligands on tumor cells. Because cytotoxic CD8+ T cells release Th1 cytokines (e.g., interferons, tumor necrosis factors, interleukins) in tumor microenvironment, and such inflammatory signals may recruit histone acetyltransferases to regulate gene expression (22–24), we hypothesized that the presence of activated CD8+ T cells within the tumor could facilitate HAT-mediated upregulation of NKG2D ligands. To test this hypothesis, CT26 and K7M3 cells were treated with the HDAC inhibitors trichostatin A (TSA; 0.3 μM) or sodium butyrate (SB; 1 mM) for 24 hours. Rae-1 expression was significantly upregulated, suggesting the potential impact of acyltransferases on Rae-1 regulation (Fig. 6A, 6B).
Fig. 6. GCN5 and PCAF account for CD8+ T cell-mediated Rae-1 restoration.
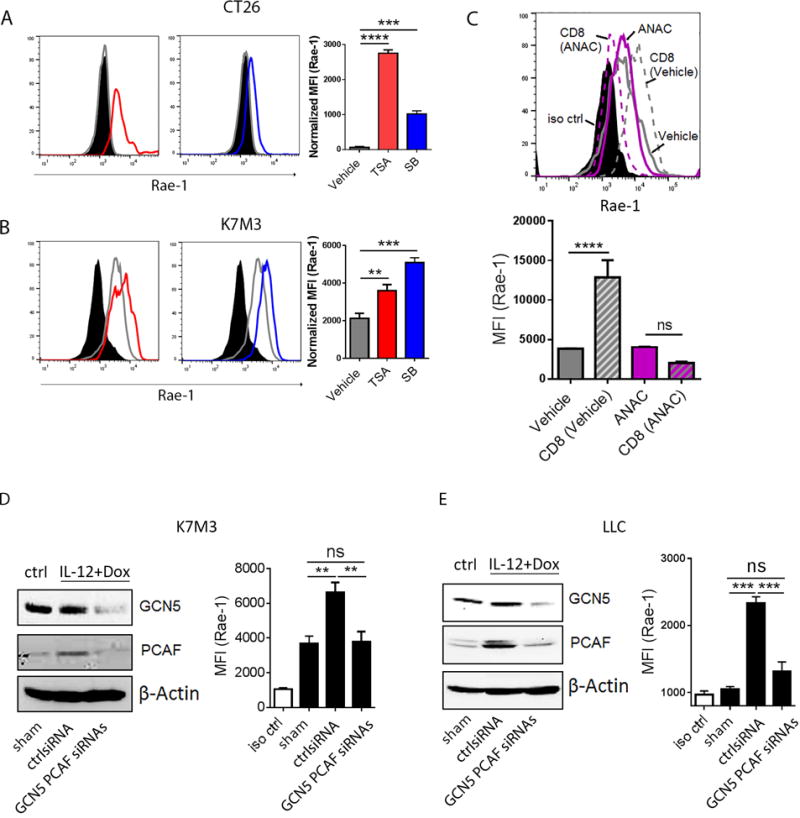
(A, B) HDAC inhibitors induced Rae-1 on CT26 and K7M3 tumor cells. CT26 (A) and K7M3 (B) cells were treated with vehicle control, TSA (0.3 μM) or SB (1 mM) for 24 hours. Rae-1 was detected by using flow cytometry. Black filled, isotype control; grey line, vehicle control; red line, TSA treated; blue line, SB treated. (C) HAT inhibitor anacardic acid (ANAC) abolished CD8+ T cell mediated Rae-1 restoration. K7M3 cells were pre-treated with vehicle control or ANAC (15 μM) for 3 hours, and subsequently co-incubated with CD8+ T cells at a ratio of 1:1 for 24 hours. Rae-1 expression on tumor cells was determined by flow cytometry. Black filled, isotype control; grey line, vehicle control; grey dash, vehicle and CD8+ T cells; purple line, ANAC; purple dash, ANAC and CD8+ T cells. (D, E) Loss of GCN5 and PCAF abrogated CD8+ T cell mediated Rae-1 restoration. K7M3 (D) and LLC (E) tumor-bearing mice (n = 5) were subjected to intratumoral administrations of control siRNA or GCN5 and PCAF siRNAs plus electroporation twice weekly, starting on day 7 after tumor inoculation. All the mice were treated with IL12 plus doxorubicin as described in Figs. 1 and 2. Four days after the second treatment, tumors were dissociated to test GCN5 and PCAF by immunoblots, and Rae-1 by flow cytometry. Bar graphs show the mean fluorescence intensity (MFI) of Rae-1 as means ± SEM. Results are representative of three repeated experiments.
To determine whether acyltransferases are involved in the induction of Rae-1 on tumor cells by activated CD8+ T cells, K7M3 cells were pretreated with vehicle or acetyltransferase inhibitor anacardic acid (ANAC) (15 μM) for 3 hours, and then cocultured with stimulated CD8+ T cells for 24 hours (Fig. 6C). Of note, Rae-1 induction by engagement with CD8+ T cells was completely abolished by pretreatment with anacardic acid, implying tthat acetyltransferases play a crucial role in CD8+ T cell–induced Rae-1 expression.
Given that anacardic acid functions as a noncompetitive HAT inhibitor, interfering with a broad spectrum of HATs, we next identified the specific HATs involved in our system. Rae-1 is activated by E2F transcription factors (25), and the E2F family is directly regulated by the GCN5/PCAF HAT complex (26–28). We therefore “knocked down” (KD) GCN5 and PCAF from K7M3 (Fig. 6D) and LLC (Fig. 6E) tumors using intratumoral siRNA electroporation, and treated control and KD tumor–bearing mice with IL12 plus doxorubicin. Untreated tumor-bearing mice were used as a control. The efficiency of GCN5 and PCAF knockdown was validated by immunoblots (Fig. 6D–6E). Loss of GCN5 and PCAF abrogated Rae-1 upregulation by IL12 plus doxorubicin (Fig. 6D–6E). Taken together, we demonstrated that GCN5 and PCAF are the major regulators to induce Rae-1 after IL12 plus doxorubicin treatment, in a CD8+ T cell–dependent manner.
Discussion
NKG2D ligands are often downregulated in established tumors. NKG2D ligand mRNA is detectable in both human neuroblastoma cell lines and primary tumors, but the protein has been found only in the cell lines (29). As tumor malignancy increases, the expression of NKG2D ligands in tumors decreases (30). In human cancer patients, expression of NKG2D ligands is associated with good prognoses in many types of cancers (10,31–33). NKG2D–NKG2D ligand intereations are critical to immune surveillance, especially at early stages of tumor development, which was demonstrated in NKG2D−/− transgenic mice (34).
The main challenge is the restoration of this NKG2D surveillance. One approach could be the genetic transfection of tumor cells, which inhibits tumor development. Because the genetic engineering of every single tumor cell to express NKG2D ligands in vivo is not feasible, it is important to explore therapeutic approaches that can induce NKG2D ligands in different types of solid tumors in vivo. Khallouf et al. found that IFNα plus 5-fluorouracil induced NKG2D ligands in pancreatic tumor cells in vitro (35), but they never clarified whether this combination was effective in vivo.
In the present study, IL12 electroporation plus a low dose of doxorubicin administered only twice, at a 10-day interval, induced tumor-specific NKG2D ligand expression across all types of tested tumors in animal models of both transplanted and spontaneous tumors. These treatments also boosted NKG2D immune surveillance in a human xenograft solid tumor model. Although NKG2D ligand expression on tumor-associated myeloid cells desensitizes NKG2D+ immune cells (36), IL12 plus doxorubicin–mediated NKGD immune surveillance was not affected, because Rae-1 induction was limited to GFP+ tumor cells, and not detected on any other cell types (GFP−) in the tumors. This implies that the induction of NKG2D ligands observed in this study may not downregulate NKG2D receptors. Instead, NKG2D ligand induction by IL12 plus doxorubicin was associated with induction of NKG2D receptors on CD8+ T cells and increased the accumulation of NKG2D+ CD8+ T cells in tumors (37). The success of sustained NKG2D ligand induction by IL12 plus doxorubicin suggested this approach may be effective in a clinical setting to treat solid tumors.
As shown in this study, several chemotherapeutic agents induced Rae-1 in vitro, but none of them induced sustained Rae-1 expression in the growing solid tumors in vivo. We emphasized solid tumors because only a limited amount of chemotherapeutic agents can penetrate solid tumors, whereas the same chemotherapeutic drugs can easily induce NKG2D ligands on blood tumor cells in vivo (38). Other chemotherapy agents, when combined with IL12, were less effective than doxorubicin for inducing NKG2D ligands. However, the combination of electroporated IL12 plus doxorubicin may not be the best possible combination, and the approach needs to be further optimized.
The observed Rae-1 induction required mature CD8+ T cells. This finding was supported by evidence from our in vivo studies of CD8+ T-cell or NK-cell depletion and Rag2−/− mice, and our in vitro studies in a CD8+ T cell and tumor cell coculture model. Given that IFNγ−/−splenocytes could not restore Rae-1 expression on tumor cells after coculture in vitro, inflammatory cytokines like IFNγ were required for CD8+ T cell–mediated Rae-1 upregulation on tumor cells. One previous study indicated that in MCA sarcoma cell lines, IFNγ may downregulate H60 and Mult to some extent, but not regulate Rae-1 expression (39). Here we found that Rae-1 expression on CT26 and K7M3 cells was not changed by treatment with IFNγ, which confirms that Rae-1 expression was not regulated by IFNγ alone. Another report found that human NKG2D ligands MICA and ULBP2 were downregulated in certain melanoma cell lines in vitro by incubation with a high concentration (200 U/ml) of IFNγ for a long period (96 hours)(40), conditions very different from those in our treatment model. Our IL12 plus doxorubicin treatment induced activated immune cells to produce only about 50 pg of IFNγ per milligram of total protein in tumors (21).
Effector CD8+ T cells that infiltrate tumors release Th1 cytokines to generate an inflammatory context that may modulate the effect of HDACs and HATs within tumor cells (24,41). It is well-established that most NKG2D ligands can be upregulated by HDAC inhibitors, and CBP/p300 as an essential histone acetyltransferase (HAT) that can upregulate mouse and human NKG2D ligands on a panel of tumor cells (42). To unveil the underlying mechanism of the CD8+ T cell–dependent upregulation of Rae-1 expression, we impaired HAT activity in tumor cells before their stimulation by CD8+ T cells, which inhibited the induction of Rae-1, suggesting that HAT activity was crucial for Rae-1 induction by our treatment. Jung et al. and Soriani et al. showed that NKG2D ligands can be regulated by E2F family of transcriptional factors (25,43), which often requires the GCN5/PCAF complex as co-activators. When we knocked down GCN5 and PCAF expression from our K7M3 and LLC tumor models, Rae-1 induction by IL12 plus doxorubicin was impaired, suggesting that Rae-1 induction was regulated by GCN5 and PCAF.
Nevertheless, the signaling pathway through which the GCN5/PCAF complex is recruited in response to inflammatory signals remains to be defined. Nuclear factor-κB (NF-κB) cooperates with GCN5/PCAF complex to regulate the expression of multiple genes (44–46), and NF-κB also serves as a regulator of NKG2D ligands (47). Hence, it is possible that inflammatory cytokines activate NF-κB, which in turn recruits GCN5/PCAF to upregulate NKG2D ligands. This hypothesis needs to be thoroughly validated.
In summary, this study revealed that (i) NKG2D ligands could be specifically induced only on tumor cells, using a panel of mouse and human solid tumors; (ii) this induction required the engagement of CD8+ T cells; and (iii) the regulation of Rae-1 was through the HAT GCN5/PCAF complex. NKG2D ligand induction by IL12 plus doxorubicin was associated with induction of NKG2D receptor on CD8+ T cells and the increased accumulation of NKG2D+ CD8+ T cells in tumors (37). This approach has the potential to boost NKG2D tumor immune surveillance and therefore may have therapeutic potential.
Supplementary Material
Acknowledgments
The authors thank Ms. Donna Reynolds for preparing the tumor sections.
Grant support: This study was supported by the National Institutes of Health grant R01 CA200574 and by MD Anderson Cancer Center’s Genetically Engineered Mouse facility which are partially supported by the NIH/NCI under award number P30CA016672.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Bindea G, Mlecnik B, Fridman WH, Pages F, Galon J. Natural immunity to cancer in humans. Curr Opin Immunol. 2010;22:215–22. doi: 10.1016/j.coi.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 3.Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol. 2000;1:119–26. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- 4.Strid J, Roberts SJ, Filler RB, Lewis JM, Kwong BY, Schpero W, et al. Acute upregulation of an NKG2D ligand promotes rapid reorganization of a local immune compartment with pleiotropic effects on carcinogenesis. Nat Immunol. 2008;9:146–54. doi: 10.1038/ni1556. [DOI] [PubMed] [Google Scholar]
- 5.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–9. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 6.Routes JM, Ryan S, Morris K, Takaki R, Cerwenka A, Lanier LL. Adenovirus serotype 5 E1A sensitizes tumor cells to NKG2D-dependent NK cell lysis and tumor rejection. J Exp Med. 2005;202:1477–82. doi: 10.1084/jem.20050240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayakawa Y, Kelly JM, Westwood JA, Darcy PK, Diefenbach A, Raulet D, et al. Cutting edge: tumor rejection mediated by NKG2D receptor-ligand interaction is dependent upon perforin. J Immunol. 2002;169:5377–81. doi: 10.4049/jimmunol.169.10.5377. [DOI] [PubMed] [Google Scholar]
- 8.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413:165–71. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11521–6. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGilvray RW, Eagle RA, Watson NF, Al-Attar A, Ball G, Jafferji I, et al. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res. 2009;15:6993–7002. doi: 10.1158/1078-0432.CCR-09-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Sullivan T, Dunn GP, Lacoursiere DY, Schreiber RD, Bui JD. Cancer immunoediting of the NK group 2D ligand H60a. J Immunol. 2011;187:3538–45. doi: 10.4049/jimmunol.1100413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vales-Gomez M, Chisholm SE, Cassady-Cain RL, Roda-Navarro P, Reyburn HT. Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. Cancer Res. 2008;68:1546–54. doi: 10.1158/0008-5472.CAN-07-2973. [DOI] [PubMed] [Google Scholar]
- 13.Jinushi M, Vanneman M, Munshi NC, Tai YT, Prabhala RH, Ritz J, et al. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1285–90. doi: 10.1073/pnas.0711293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diermayr S, Himmelreich H, Durovic B, Mathys-Schneeberger A, Siegler U, Langenkamp U, et al. NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood. 2008;111:1428–36. doi: 10.1182/blood-2007-07-101311. [DOI] [PubMed] [Google Scholar]
- 15.Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005;65:6321–9. doi: 10.1158/0008-5472.CAN-04-4252. [DOI] [PubMed] [Google Scholar]
- 16.Tieng V, Le Bouguenec C, du Merle L, Bertheau P, Desreumaux P, Janin A, et al. Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc Natl Acad Sci U S A. 2002;99:2977–82. doi: 10.1073/pnas.032668099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–90. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmudde M, Braun A, Pende D, Sonnemann J, Klier U, Beck JF, et al. Histone deacetylase inhibitors sensitize tumour cells for cytotoxic effects of natural killer cells. Cancer Lett. 2008;272:110–21. doi: 10.1016/j.canlet.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 19.Lopez-Soto A, Folgueras AR, Seto E, Gonzalez S. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: potential implications for the immunosurveillance of cancer. Oncogene. 2009;28:2370–82. doi: 10.1038/onc.2009.117. [DOI] [PubMed] [Google Scholar]
- 20.Forget MA, Malu S, Liu H, Toth C, Maiti S, Kale C, et al. Activation and propagation of tumor-infiltrating lymphocytes on clinical-grade designer artificial antigen-presenting cells for adoptive immunotherapy of melanoma. Journal of immunotherapy. 2014;37:448–60. doi: 10.1097/CJI.0000000000000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu S, Waguespack M, Barker SA, Li S. Doxorubicin directs the accumulation of interleukin-12 induced IFN gamma into tumors for enhancing STAT1 dependent antitumor effect. Clin Cancer Res. 2007;13:4252–60. doi: 10.1158/1078-0432.CCR-06-2894. [DOI] [PubMed] [Google Scholar]
- 22.Selvi BR, Mohankrishna DV, Ostwal YB, Kundu TK. Small molecule modulators of histone acetylation and methylation: a disease perspective. Biochim Biophys Acta. 2010;1799:810–28. doi: 10.1016/j.bbagrm.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Clarke DL, Clifford RL, Jindarat S, Proud D, Pang L, Belvisi M, et al. TNFalpha and IFNgamma synergistically enhance transcriptional activation of CXCL10 in human airway smooth muscle cells via STAT-1, NF-kappaB, and the transcriptional coactivator CREB-binding protein. J Biol Chem. 2010;285:29101–10. doi: 10.1074/jbc.M109.0999952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh AK, Yuan W, Mori Y, Chen S, Varga J. Antagonistic regulation of type I collagen gene expression by interferon-gamma and transforming growth factor-beta. Integration at the level of p300/CBP transcriptional coactivators. J Biol Chem. 2001;276:11041–8. doi: 10.1074/jbc.M004709200. [DOI] [PubMed] [Google Scholar]
- 25.Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J Exp Med. 2012;209:2409–22. doi: 10.1084/jem.20120565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagy Z, Tora L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene. 2007;26:5341–57. doi: 10.1038/sj.onc.1210604. [DOI] [PubMed] [Google Scholar]
- 27.Lang SE, McMahon SB, Cole MD, Hearing P. E2F transcriptional activation requires TRRAP and GCN5 cofactors. J Biol Chem. 2001;276:32627–34. doi: 10.1074/jbc.M102067200. [DOI] [PubMed] [Google Scholar]
- 28.Guo R, Chen J, Mitchell DL, Johnson DG. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res. 2011;39:1390–7. doi: 10.1093/nar/gkq983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia. 2004;6:558–68. doi: 10.1593/neo.04316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, et al. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain: a journal of neurology. 2006;129:2416–25. doi: 10.1093/brain/awl205. [DOI] [PubMed] [Google Scholar]
- 31.Cho H, Chung JY, Kim S, Braunschweig T, Kang TH, Kim J, et al. MICA/B and ULBP1 NKG2D ligands are independent predictors of good prognosis in cervical cancer. BMC cancer. 2014;14:957. doi: 10.1186/1471-2407-14-957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamimura H, Yamagiwa S, Tsuchiya A, Takamura M, Matsuda Y, Ohkoshi S, et al. Reduced NKG2D ligand expression in hepatocellular carcinoma correlates with early recurrence. Journal of hepatology. 2012;56:381–8. doi: 10.1016/j.jhep.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 33.de Kruijf EM, Sajet A, van Nes JG, Putter H, Smit VT, Eagle RA, et al. NKG2D ligand tumor expression and association with clinical outcome in early breast cancer patients: an observational study. BMC cancer. 2012;12:24. doi: 10.1186/1471-2407-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. 2008;28:571–80. doi: 10.1016/j.immuni.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khallouf H, Marten A, Serba S, Teichgraber V, Buchler MW, Jager D, et al. 5-Fluorouracil and interferon-alpha immunochemotherapy enhances immunogenicity of murine pancreatic cancer through upregulation of NKG2D ligands and MHC class I. Journal of immunotherapy. 2012;35:245–53. doi: 10.1097/CJI.0b013e31824b3a76. [DOI] [PubMed] [Google Scholar]
- 36.Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, et al. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science. 2015;348:136–9. doi: 10.1126/science.1258867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu J, Zhu S, Xia X, Zhang L, Kleinerman ES, Li S. CD8+T cell-specific induction of NKG2D receptor by doxorubicin plus interleukin-12 and its contribution to CD8+T cell accumulation in tumors. Molecular cancer. 2014;13:34. doi: 10.1186/1476-4598-13-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113:3503–11. doi: 10.1182/blood-2008-08-173914. [DOI] [PubMed] [Google Scholar]
- 39.Bui JD, Carayannopoulos LN, Lanier LL, Yokoyama WM, Schreiber RD. IFN-dependent down-regulation of the NKG2D ligand H60 on tumors. J Immunol. 2006;176:905–13. doi: 10.4049/jimmunol.176.2.905. [DOI] [PubMed] [Google Scholar]
- 40.Schwinn N, Vokhminova D, Sucker A, Textor S, Striegel S, Moll I, et al. Interferon-gamma down-regulates NKG2D ligand expression and impairs the NKG2D-mediated cytolysis of MHC class I-deficient melanoma by natural killer cells. International journal of cancer Journal international du cancer. 2009;124:1594–604. doi: 10.1002/ijc.24098. [DOI] [PubMed] [Google Scholar]
- 41.Nusinzon I, Horvath CM. Positive and negative regulation of the innate antiviral response and beta interferon gene expression by deacetylation. Molecular and cellular biology. 2006;26:3106–13. doi: 10.1128/MCB.26.8.3106-3113.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sauer M, Schuldner M, Hoffmann N, Cetintas A, Reiners KS, Shatnyeva O, et al. CBP/p300 acetyltransferases regulate the expression of NKG2D ligands on tumor cells. Oncogene. 2016 doi: 10.1038/onc.2016.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soriani A, Iannitto ML, Ricci B, Fionda C, Malgarini G, Morrone S, et al. Reactive oxygen species- and DNA damage response-dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1. J Immunol. 2014;193:950–60. doi: 10.4049/jimmunol.1400271. [DOI] [PubMed] [Google Scholar]
- 44.Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, et al. Transcriptional activation by NF-kappaB requires multiple coactivators. Mol Cell Biol. 1999;19:6367–78. doi: 10.1128/mcb.19.9.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhatt D, Ghosh S. Regulation of the NF-kappaB-Mediated Transcription of Inflammatory Genes. Front Immunol. 2014;5:71. doi: 10.3389/fimmu.2014.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim N, Sun HY, Youn MY, Yoo JY. IL-1beta-specific recruitment of GCN5 histone acetyltransferase induces the release of PAF1 from chromatin for the de-repression of inflammatory response genes. Nucleic Acids Res. 2013;41:4495–506. doi: 10.1093/nar/gkt156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin D, Lavender H, Soilleux EJ, O’Callaghan CA. NF-kappaB regulates MICA gene transcription in endothelial cell through a genetically inhibitable control site. J Biol Chem. 2012;287:4299–310. doi: 10.1074/jbc.M111.282152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.