

See Pluchino and Peruzzotti-Jametti (doi:10.1093/aww266) for a scientific commentary on this article.
Macrophages have a critical role in remyelination. Psachoulia et al. show that IL4I1, a macrophage-secreted enzyme, promotes CNS remyelination by modulating T cell driven inflammation after focal demyelination in mice. Injection of recombinant IL4I1 protein reverses disease progression in a mouse model of multiple sclerosis, resulting in recovery from hindlimb paralysis.
Keywords: inflammation, remyelination, macrophages, interleukin-4 induced 1 (IL4I1), experimental autoimmune encephalomyelitis (EAE)
Abstract
See Pluchino and Peruzzotti-Jametti (doi:10.1093/aww266) for a scientific commentary on this article.
Myelin regeneration (remyelination) is a spontaneous process that occurs following central nervous system demyelination. However, for reasons that remain poorly understood, remyelination fails in the progressive phase of multiple sclerosis. Emerging evidence indicates that alternatively activated macrophages in central nervous system lesions are required for oligodendrocyte progenitor differentiation into remyelinating oligodendrocytes. Here, we show that an alternatively activated macrophage secreted enzyme, interleukin-four induced one (IL4I1), is upregulated at the onset of inflammation resolution and remyelination in mouse central nervous system lesions after lysolecithin-induced focal demyelination. Focal demyelination in mice lacking IL4I1 or interleukin 4 receptor alpha (IL4Rα) results in increased proinflammatory macrophage density, remyelination impairment, and axonal injury in central nervous system lesions. Conversely, recombinant IL4I1 administration into central nervous system lesions reduces proinflammatory macrophage density, enhances remyelination, and rescues remyelination impairment in IL4Rα deficient mice. We find that IL4I1 does not directly affect oligodendrocyte differentiation, but modulates inflammation by reducing interferon gamma and IL17 expression in lesioned central nervous system tissues, and in activated T cells from splenocyte cultures. Remarkably, intravenous injection of IL4I1 into mice with experimental autoimmune encephalomyelitis at disease onset significantly reversed disease severity, resulting in recovery from hindlimb paralysis. Analysis of post-mortem tissues reveals reduced axonal dystrophy in spinal cord, and decreased CD4+ T cell populations in spinal cord and spleen tissues. These results indicate that IL4I1 modulates inflammation by regulating T cell expansion, thereby permitting the formation of a favourable environment in the central nervous system tissue for remyelination. Therefore, IL4I1 is a potentially novel therapeutic for promoting central nervous system repair in multiple sclerosis.
Introduction
Multiple sclerosis is a chronic inflammatory disorder of the CNS that is characterized by demyelination, axonal injury, and progressive neurodegeneration (Compston and Coles, 2002; Lassmann et al., 2012; Dutta and Trapp, 2014). During the early stage of multiple sclerosis, most patients experience episodes of inflammatory attack and demyelination between intervals of inflammation-resolution and myelin regeneration (remyelination). Remyelination is a process by which oligodendrocyte precursor cells migrate to and proliferate in CNS lesions, and differentiate into oligodendrocytes for myelin replacement (Lassmann et al., 1997; Franklin and ffrench-Constant, 2008). This spontaneous regenerative response is critical for limiting the axonal dysfunction that otherwise occurs with myelin loss (Irvine and Blakemore, 2008; Huang and Franklin, 2012; Chamberlain et al., 2015). However, with disease progression, remyelination becomes increasingly impaired and eventually fails, leading to chronic axonal dystrophy and neurodegeneration that manifest in the accumulation of permanent disability (Dutta and Trapp, 2011; Franklin et al., 2012). Why regenerative decline occurs in multiple sclerosis is not fully understood. One possible explanation is that the balance between inflammation and its resolution, which is critical to controlling the different facets of oligodendrocyte lineage progression in the injured CNS, is altered or dysregulated in the chronic progressive stage of the disease (Franklin, 2002; Fitzner and Simons, 2010). The chronically inflamed landscape in the diseased CNS is likely to contribute to increasing oligodendrocyte precursor cell presence in lesions, but the failure to modulate inflammation may prevent them from differentiating into oligodendrocytes to restore myelin. Therefore, pharmacological strategies to modulate inflammation in the damaged CNS may enable myelin repair and prevent subsequent neurodegeneration in multiple sclerosis.
Macrophages display a spectrum of activation states (Murray and Wynn, 2011). Their range in function, which is often opposing, plays a critical role in myelin repair (Kotter et al., 2001; Miron and Franklin, 2014). In CNS injury, macrophages may be characterized functionally as proinflammatory, classically activated macrophages (CAM), or conversely as anti-inflammatory, alternatively activated macrophages (AAM) (Kigerl et al., 2009; Wynn et al., 2013; Miron and Franklin, 2014). The spatial and temporal distribution of CAM and AAM in the injured CNS is important for successful remyelination, such that enhanced CAM activity corresponds with oligodendrocyte precursor cell proliferation and death, whereas AAM activity corresponds with oligodendrocyte differentiation/remyelination (Schonberg et al., 2007; Miron et al., 2013). Differential macrophage distribution and corresponding activities may therefore dictate the inflammatory status of injured CNS tissues, and a disturbance in the balance of CAM and AAM activity in CNS lesions may contribute to remyelination decline (Miron and Franklin, 2014). Indeed, an imbalanced distribution towards CAM has been found to promote relapse in rodents with experimental autoimmune encephalomyelitis (EAE), and the adoptive transfer of AAM into mice with EAE is able to significantly improve clinical status (Weber et al., 2007; Mikita et al., 2011). These intriguing observations demonstrate the importance of CAM to AAM balance in regulating tissue repair, and suggest that AAM produce critical factors necessary to modulate inflammation and autoimmunity.
Here, we found that interleukin-4 induced one (IL4I1) is highly expressed during CNS remyelination in mice following lysolecithin-induced spinal cord demyelination. IL4I1 is a known secreted l-amino acid oxidase that oxidizes l-amino acids, namely l-phenylalanine, to corresponding α-ketoacids, hydrogen peroxide (H2O2), and ammonia (Chavan et al., 2002). It has been found to be expressed by immune cells, including macrophages, T cells, and B cells on stimulation by interleukin-4 (IL4), and has been shown to perform immunomodulatory functions in various tumours and bacterial infections (Chu and Paul, 1997; Copie-Bergman et al., 2003; Boulland et al., 2007; Lasoudris et al., 2011; Puiffe et al., 2013). However, the role of IL4I1 in CNS injury or repair has not been suggested. We found that IL4I1 expression is induced in AAM through IL4 receptor signalling, and that IL4I1 is necessary to modulate the inflammatory environment to enhance CNS remyelination and prevent axonal injury. Additionally, we found that IL4I1 significantly reduces interferon gamma (IFN-γ) and IL17 expression in CNS lesions and in activated CD4+ T cells from splenocytes, suggesting that the effect of IL4I1 on CD4+ T cells is critical to reduce proinflammatory macrophage activity for successful remyelination. Remarkably, intravenous injection of IL4I1 into mice with EAE significantly reduces disease severity and reverses the course of the disease. This suggests that IL4I1 has therapeutic potential for modulating the inflammation landscape in CNS lesions, thereby promoting tissue repair and preventing disease progression in multiple sclerosis.
Materials and methods
Mice
All experiments were performed in accordance with approved Institutional Animal Care and Use Committee (IACUC) protocols of Georgetown University. C57BL/6 mice were purchased from The Jackson Laboratory and Charles River. Il4i1−/− mice were purchased from MMRRC. Il4ra−/− mice were purchased from Taconic Farms.
Focal spinal cord demyelination
Focal demyelination was induced by injecting of 1.0% lysolecithin (Sigma-Aldrich) in saline into the spinal cord ventral funiculus of male or female wild-type C57BL/6, Il4i1−/−, or Il4ra−/− mice at 10–12 weeks old. For treated animals, 200 ng/ml of recombinant mouse IL4I1 (R&D Systems) was co-injected along with 1.0% lysolecithin into the ventral spinal cord. The animals (n = 3–5 in each group) were sacrificed at 3, 5, 10, 15 and 20 days after surgery for analysis.
Experimental autoimmune encephalomyelitis and recombinant IL4I1 therapeutic treatment
C57BL/6 female mice (Charles River) at age 9–10 weeks were acclimatized for 7 days prior to EAE. EAE was induced according to the Hooke Laboratories protocol (http://hookelabs.com/protocols/eaeAI_C57BL6.html). Briefly, mice were immunized by subcutaneous injection of an emulsion of MOG35-55 in complete Freund’s adjuvant (CFA) (Day 0), followed by administration of pertussis toxin (PTX) in phosphate-buffered saline (PBS) intraperitoneally, first on the day of immunization (Day 0), and then again the following day (Day 1). Pre-filled MOG35-55/CFA emulsion syringes and PTX were obtained from Hooke Laboratories (Cat. No: EK-2110). Each 1 ml syringe contained ∼1 mg MOG35-55/ml emulsion, ∼2–5 mg killed mycobacterium tuberculosis H37Ra/ml emulsion (all concentrations adjusted by lot for consistent EAE induction). Emulsion was administered subcutaneously at two sites, 0.1 ml/site (0.2 ml/mouse total). PTX was administered intraperitoneally at 0.13 ml/dose, and repeated 24 h later. Approximately 250 ng PTX/dose or 2.5 μg/ml for each of the two PTX administrations were used. The mice were scored blindly and daily from EAE Day 7 until at least EAE Day 28 according protocol from Hooke Laboratories. The scoring system used was as follows: 0.0 = no obvious changes in motor function; 0.5 = tip of tail is limp; 1.0 = limp tail; 1.5 = limp tail and hind leg inhibition; 2.0 = limp tail and weakness of hind legs or signs of head tilting; 2.5 = limp tail and dragging of hind legs or strong head tilting; 3.0 = limp tail and complete paralysis of hind legs or limp tail with paralysis of one front and one hind leg; 3.5 = limp tail and complete paralysis of hind legs plus mouse unable to right itself when placed on its side; 4.0 = limp tail, complete hind leg and partial front leg paralysis, mouse is minimally moving but appears alert and feeding; 4.5 = complete hind and partial front leg paralysis, no movement around the cage, mouse is not alert; 5.0 = mouse is found dead due to paralysis or mouse is euthanized due to severe paralysis.
For IL4I1 treatment studies, mice were housed in groups of five per cage and identified by ear notches. The mice that developed EAE were then randomly assigned into IL4I1 treated, or PBS (vehicle) treated group within each cage in a balanced manner to achieve groups with similar time of EAE onset and similar onset scores (as recommended by Hooke Laboratories). For treatment, 100 μl of recombinant mouse IL4I1 (1 μg/ml blood volume), or PBS was injected intravenously into the tail vein at clinical score 2.0–2.5. A second dose was injected after 3 days of rest. In other experiments, mice without any treatment were used as controls. Mice were anaesthetized briefly with isofluorane before IL4I1 or PBS injections to minimize stress, while untreated mice were not handled. The clinical scores and weight of mice were recorded daily until the end of experiment. Mice that had spontaneously recovered from EAE, or did not reach a score of 3.0 were not considered in the analysis of the therapeutic study. For each experiment, 8 to 10 mice from each group were analysed.
Cell cultures
RAW 264.7 cell lines were kindly provided by Dr Steven Singer (Georgetown University, Department of Biology) and were maintained in Dulbecco’s modified Eagle medium (DMEM) containing 10% foetal bovine serum (FBS). Cells were passaged every 2–3 days and were used until postnatal Day 15. Mouse primary microglia cultures were prepared from mixed glia shake-off as previously described, and kindly provided by Dr Kathleen Maguire-Zeiss, Georgetown University (Daniele et al., 2014). Briefly, mixed glia cultures were first prepared from postnatal Day 3–5 old mouse cortices and maintained in DMEM-F12 containing 10% FBS, 1% GlutaMax™ (GIBCO), and 100 U/ml penicillin/streptomycin (GIBCO) for 2 weeks. For microglia enrichment, mixed glia cultures in sealed T-75 flasks are rotated at 200 rpm and 37°C for 5 h before supernatants containing the microglia are collected and centrifuged. For plating, the microglia pellet was resuspended and cultured in 1 mM sodium pyruvate, 0.6% v/v glucose, 1 mM l-glutamine, 100 μg/ml penicillin/streptomycin, 5% v/v FBS. BV2 cells, also provided by Dr Kathleen Maguire-Zeiss, were maintained in media containing DMEM, 10% FBS, and 100 U/ml penicillin/streptomycin (GIBCO). For CAM and AAM polarization, primary microglia, BV2 cells, and RAW264.7 cells were treated with 100 ng/ml lipopolysaccharide or IL4 for 24 h. Primary oligodendrocyte precursor cell cultures were obtained from postnatal Day 3–5 mouse cortices using magnetic-activated cell sorting (MACS) with anti-O4 microbeads according to the manufacturers protocol (Miltenyi). Once purified, primary oligodendrocyte lineage cells were expanded in growth media (DMEM-F12 with N2, B27, penicillin/streptomycin, bovine serum albumin, PDGF and FGF) for 24 h and then differentiated in defined media (DMEM-F12, N2, B27, penicillin/streptomycin, insulin and T3) without PDGF and FGF for 48 h as previously described (Dincman et al., 2012). In vitro experiments were replicated twice for analysis.
RNA extraction, cDNA synthesis and quantitative reverse transcription polymerase chain reaction
For cells, total RNA was extracted by the RNeasy® Micro kit (QIAGEN). For tissue, total RNA was isolated using the TRIzol® Reagent protocol (Life Technologies) and homogenizer (Argos). All quantitative polymerase chain reaction (PCR) primers were purchased from Bio-Rad. SYBR® Green reverse transcriptase PCR was performed using the SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad) and analysed by the CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad). Results were normalized against peptidylprolyl isomerase A (Ppia) for spinal cord extracts, or Gapdh for cell cultures, and were expressed as mean ± standard error of the mean (SEM). Ppia and Gapdh are recommended normalization factors for gene expression studies (Gong et al., 2014).
In situ hybridization
Sense and antisense probes against IL4I1 were generated using primers against the mouse Il4i1 mRNA sequence (forward primer, 5’- CCAAGAGAGCTGAAGACAGCAG; reverse primer, 5’- GTAGCCCAGCTTTTCTGGCA). In situ hybridization was performed on 12 μm mouse spinal cord cryosections collected directly on SuperFrost®Plus slides (VWR International) as previously described (Huang et al., 2011).
Antibodies and cytokines
Lipopolysaccharide (Escherichia coli 0111:B4) was obtained from InvivoGen. Recombinant mouse IL4 and IL4I1 were obtained from R&D Systems. The following antibodies were used for immunohistochemistry. Primary antibodies: rat anti-CD11b (1:100; AbD Serotec), rabbit anti-Ym1 (1:100; StemCell Technologies), mouse anti-iNOS (1:50; BD Pharmingen), rabbit anti-Olig2 (1:300; Millipore), mouse anti-CC1 (1:300; Millipore), mouse anti-Nkx2.2 (1:100; DSHB), mouse anti-GFAP (1:400; Sigma), rat anti-Tenascin-C (1:100, Abcam), rabbit anti-NF200 (1:100; Sigma), mouse anti-SMI-32 (1:1000; Calbiochem), mouse anti-IST-9 (1:200; Abcam). Secondary antibodies: Alexa Fluor® 488 Goat Anti-Rabbit IgG (1:1000), Alexa Fluor® 488 Goat Anti-Rat IgG (1:500), Alexa Fluor® 594 Goat Anti-Mouse IgG (1:1000), Alexa Fluor® 594 Chicken Anti-Goat IgG (1:500) and Alexa Fluor® 594 Goat Anti-Rat IgG (1:500). Flow cytometry primary antibodies: PE/Cy7 anti-CD4 (BioLegend), Brilliant Violet 711 anti-T-bet (Biolegend), PE anti-RORγt (BD Pharmingen) and PerCP/Cy5.5 anti-Gata3 (BioLegend), anti-NOS2 PE (Santa Cruz Biotechnology) and anti-CD11b APC/Cy7 (Biolegend). LIVE/DEAD® Fixable Yellow Dead Cell Stain Kit (Invitrogen) was used to monitor cell death.
Immunohistochemistry
Mice were perfusion-fixed with 4% (w/v) paraformaldehyde (PFA; Sigma) in PBS. Spinal cord tissue was dissected and lightly postfixed in 4% PFA at room temperature. Tissue was cryoprotected in 20% (w/v) sucrose (Sigma) in PBS before freezing in O.C.T. on the surface of dry ice. Twelve micrometre spinal cord cryosections were collected directly on SuperFrost®Plus slides (VWR International) and were allowed to dry for 30 min before storing at −80°C. For in vitro experiments, cells were fixed with 4% (w/v) PFA for 10 min and then washed with PBS. Sections/cells were then incubated in blocking solution [0.1% (v/v) Triton™ X-100 and 10% FBS in PBS] for 1 h at room temperature. Primary and secondary antibodies were diluted in PBS blocking solution and applied to sections/cells overnight at 4°C. Tris-buffered saline (TBS) was substituted for PBS when immunolabelling with anti-CC1. For detection of Nkx2.2 and CC1, mouse-on-mouse antigen retrieval was performed before immunohistochemistry according to the manufacturer’s instructions (M.O.M.™ kit; Vector Laboratories). For immunohistochemistry of spinal cord sections from mice with EAE, roughly 1 mm of the lower lumbar spinal cord (where the most obvious pathology was) was collected from each mouse, and n = 3 sections (12 μm) were randomly chosen for immunostaining analysis.
Electron microscopy
Electron microscopy was performed on lysolecithin demyelinated mouse spinal cord sections. Lesioned mice at 10 days post-lesion (dpl) were transcardially perfused with a 0.9% NaCl solution for exsanguination followed by a 0.1 M Millong’s buffering solution containing 4% paraformaldehyde and 5% glutaraldehyde (pH 7.3). Whole animal carcasses were post-fixed in the same aldehyde fixative solution for 2 weeks at 4°C. Lumbar spinal cords were harvested and stored in 0.1 M cacodylate buffer (pH 7.4) overnight to remove excess aldehyde. The following day the samples were postfixed in 2% osmium tetroxide in 0.1 M cacodylate buffer for 1 h with constant agitation, rinsed in 0.1 M cacodylate buffer, dehydrated by serial dilutions of ethanol and infiltrated with and embedded in Poly/Bed® 812 resin (Polysciences). All samples were oriented for cross-section analysis. One-micrometre sections, stained with toluidine blue, were used to identify the lesioned site before collection for electron microscopy analysis. Following site identification, 90 nm sections were stained with a combination of lead citrate and uranyl acetate and used for ultrastructural evaluation. All images were collected using a JEOL JEM 1230 transmission electron microscope equipped with a Gatan Orius SC1000 side mount CCD camera.
G-ratio analysis
Axon and myelin circumference were measured using ImageJ (NIH). G-ratio was calculated as the axonal circumference (internal to the myelin layers) divided by the circumference of compact myelin (external to the myelin layers) at a given cross-section of the axon. Myelin tongues and non-compacted myelin were excluded from this calculation.
Flow cytometry
Spinal cords and spleens were collected from EAE mice at Day 35 in PBS and were mechanically dissociated. Tissues from two mice belonging to the same group were then combined and passed through 100 nm filter. Spleen samples were next incubated in red blood cell lysis buffer for 10–15 min in the dark and were then washed with PBS. Cells from spinal cords and spleens were then incubated with the anti-CD4 antibody and Live/Dead®. For intracellular staining, cells were incubated in Fix/Perm solution and Perm buffer (Biolegend) and were then incubated in primary antibodies. Single staining tissue samples were used for both tissue types. Cells were gated at the Georgetown Lombardi Comprehensive Cancer Center Flow Cytometry and Cell Sorting Shared Resource (FCSR).
Imaging and quantification
For quantification of immunohistochemical staining, cells were manually counted from low magnification (×10 for tissues; ×20 for cell cultures), non-overlapping images, using Adobe Photoshop. For tissues, quantification of cells was concentrated in the lesioned area, as defined by nuclear (DAPI) staining. Co-localization was determined either as a percentage (the ratio of cells expressing two markers divided by the number of cells expressing a single marker multiplied by 100) or per mm2 (the number of cells expressing one or two markers divided by the area in µ2 × 1 000 000). Corrected SMI-32 and NF200 fluorescence was calculated by measuring the integrated intensity minus the area of the selected region times the mean fluorescence of background reading. For all quantification a minimum of three sections from n = 3–5 mice was examined. The proportion or density of cells was determined per mouse. The average and standard error was then calculated for each group using Microsoft Excel.
Statistical analysis
All statistics were performed using GraphPad Prism 6 (La Jolla, CA, USA). Data are represented as mean ± SEM. For all other data significance was determined using two-tailed Student’s t-tests, or one- and two-way ANOVA with Tukey’s range test for post hoc analysis. EAE clinical score significance was determined using one-way Wilcoxon Rank-Sum Test. Exact P-values are stated where appropriate. Statistical significance is reported as not significant (n.s.), P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Results
Il4i1 is upregulated in central nervous system lesions during remyelination
Remyelination occurs spontaneously in the CNS after injury (Franklin and ffrench-Constant, 2008). It has been well established in rodents that following focal demyelination in the adult CNS, oligodendrocyte precursor cells migrate to the lesion, differentiate into mature oligodendrocytes and remyelinate axons within 1 month post-lesion (Blakemore and Franklin, 2008; Huang et al., 2011). During remyelination, innate immune cells, such as macrophages, are necessary to promote oligodendrocyte lineage cell progression (Kotter et al., 2001; Miron et al., 2013; Döring et al., 2015). To identify the immunomodulatory mechanisms that underlie the regulation of CNS remyelination, we analysed a previously published remyelination transcriptome from laser capture microdissected rat CNS lesions (Huang et al., 2011). Here, we found that Il4i1, a gene associated with immunomodulation, is upregulated during CNS remyelination (Fig. 1A and B). Il4i1 exhibits a distinctive and highly dynamic differential expression profile in CNS lesions during remyelination. We found that Il4i1 displays relatively low expression at 5 dpl, a time point that corresponds with proinflammatory activity and oligodendrocyte precursor cell recruitment to lesions, but is significantly upregulated at 14 dpl, a time point that corresponds with the reduction of proinflammatory activity and oligodendrocyte differentiation/remyelination (Fig. 1A and Supplementary Fig. 1). The differential expression of Il4i1 during remyelination is in opposition to the expression pattern of genes associated with proinflammatory activity, such as Mmp12 (Vos et al., 2003; Hansmann et al., 2012). This result indicates that Il4i1 is upregulated during inflammation resolution and at the onset of oligodendrocyte differentiation, suggesting that IL4I1 may be involved in modulating inflammation or promoting remyelination.
Figure 1.
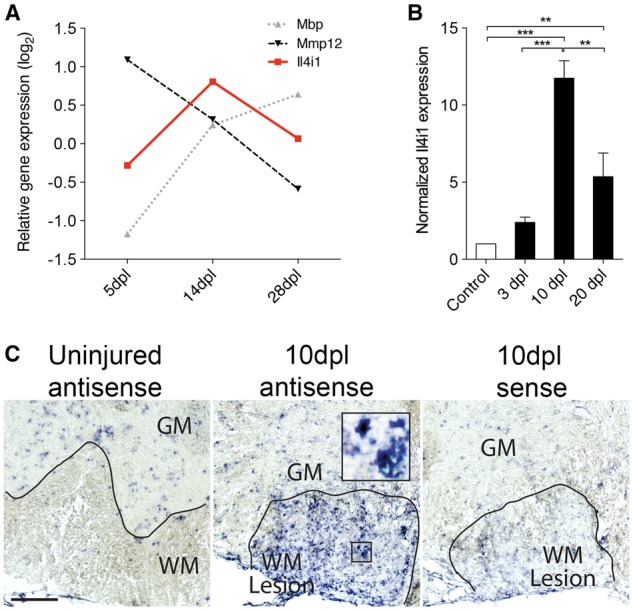
Il4i1 is expressed during remyelination in CNS lesions. (A) Graph showing the differential expression pattern of Il4i1 against Mmp12, and Mbp at 5, 14 and 28 dpl. The values were obtained from a previously published rat remyelination transcriptome (Huang et al., 2011). (B) qRT-PCR detection of Il4i1 in lesioned mouse spinal cord at 3, 10 and 20 dpl compared to control non-lesioned tissue (n = 3 per group). (C) In situ hybridization of Il4i1 in a non-lesioned and focally demyelinated mouse spinal cord sections at 10 dpl. Sense control staining shows faint labelling. GM = grey matter; WM = white matter. Scale bar = 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001; ANOVA followed by post hoc analysis.
To examine Il4i1 expression during remyelination in vivo, focal demyelination was performed by lysolecithin injection into the mouse spinal cord ventral funiculous, and analysed at 3, 10 and 20 dpl. These post-lesion time points correspond to the periods of oligodendrocyte precursor cell recruitment (3 dpl), oligodendrocyte differentiation (10 dpl), and remyelination completion (20 dpl) after demyelinating injury. Quantitative real time PCR (qRT-PCR) analysis of purified transcripts from spinal cord tissues revealed low Il4i1 expression in uninjured tissue and in lesioned tissue at 3 dpl. However, Il4i1 expression increased significantly at 10 dpl, and decreased by 20 dpl (Fig. 1B), corresponding with the pattern of Il4i1 expression in the rodent remyelination transcriptome. Moreover, in situ hybridization analysis of mouse spinal cord sections at 10 dpl showed that Il4i1 was highly expressed in lesions compared to uninjured white matter, and that Il4i1 was detected in cells whose morphology resembled foamy macrophages (Fig. 1C). A search in the mouse Brain RNA-Seq database (Zhang et al., 2014b) revealed that Il4i1 was enriched in microglia compared to other CNS cell populations in the cerebral cortex. Together, these findings suggest that Il4i1 is upregulated in CNS lesions and expressed in microglia/macrophages during CNS remyelination.
IL4I1 is induced in alternatively activated macrophages through IL4 receptor signalling
To determine if Il4i1 is induced upon alternative activation in microglia and macrophages, IL4 was added to primary microglia cultures, BV2 cells (immortalized murine microglia cell line), and RAW264.7 cells (monocyte derived macrophage cell line) for AAM activation, followed by qRT-PCR analysis after 24 h for Il4i1 expression. Control cells were either left untreated, or treated with lipopolysaccharide for CAM activation. We found that lipopolysaccharide had no significant effect on Il4i1 expression in primary microglia or RAW264.7 cells, and appeared to slightly reduce Il4i1 expression in BV2 cells compared to untreated cells (Fig. 2A–C). By contrast, IL4 addition significantly induced Il4i1 expression in primary microglia, BV2 cells, and RAW264.7 cells (Fig. 2A–C). These findings suggest that microglia-derived as well as monocyte-derived AAM express Il4i1 on IL4 stimulation, and supports a recent study showing that Il4i1 is upregulated in AAM (Yue et al., 2015).
Figure 2.

Il4i1 is upregulated in alternatively activated microglia and macrophages through IL-4 receptor signalling. qRT-PCR detection of Il4i1 in untreated, lipopolysaccharide (LPS)- and IL4-treated (A) microglia, (B) BV2 cells, and (C) RAW264.7 cell at 24 h after treatment (n = 3 per group). (D) qRT-PCR for Il4i1 expression in unlesioned wild-type (WT), lesioned wild-type and lesioned II4ra−/− spinal cord at 10 dpl (n = 3 per group). (E) In situ hybridization of Il4i1 in wild-type and II4ra−/− mice at 10 dpl. Spinal cord lesion is encircled. GM = grey matter, WM = white matter. Scale bar = 100 μm. Density of (F) oligodendrocyte precursor cells (PDGFRα+Olig2+) and (G) oligodendrocytes (CC1+Olig2+) from MACS purified primary oligodendrocyte precursor cell cultures at 3 days in vitro after treatment with vehicle (1XPBS) or recombinant IL4I1 for 24 h (n = 5 images per condition). All experimental results were replicated at least twice. **P < 0.01, ***P < 0.001, ****P < 0.0001; ANOVA followed by post hoc analysis.
To determine if Il4i1 expression depends on AAM activity during CNS remyelination in vivo, focal spinal cord demyelination was performed on IL4 receptor alpha knockout (Il4ra−/−) mice. These mice are deficient in both type 1 and type 2 IL4 receptor signalling, and therefore lack AAM activity and type 2 immunity (Wynn, 2015). qRT-PCR analysis of 10 dpl spinal cord tissues revealed that Il4i1 expression was severely reduced in Il4ra−/− mouse lesions (Fig. 2D). Moreover, in situ hybridization analysis revealed the absence of Il4i1 expression in CNS lesions in Il4ra−/− mice compared to wild-type (Fig. 2E). These results confirm that Il4i1 expression is dependent on IL4 receptor signalling in vivo, and that alternative activation of macrophages is required for Il4i1 expression in CNS lesions during remyelination.
AAM play a role in tissue remodelling and repair, and have recently been demonstrated to drive oligodendrocyte differentiation (Kigerl et al., 2009; Miron et al., 2013; Wynn et al., 2013). To determine if IL4I1 directly regulates oligodendrocyte differentiation, recombinant IL4I1 protein or vehicle (control) was added to oligodendrocyte lineage cells that were purified by MACS, and cultured in defined media. We found that IL4I1 addition did not affect the number of oligodendrocyte precursor cells or oligodendrocytes in vitro (Fig. 2F and G), indicating that IL4I1 does not directly affect oligodendrocyte lineage progression, and may regulate inflammation in CNS lesions.
IL4I1 modulates inflammation in central nervous system lesions
To determine if IL4I1 modulates inflammation during CNS remyelination, focal demyelination was performed on Il4i1 knockout (Il4i1−/−) mice. These mice do not display obvious developmental or behavioural abnormalities (according to MMRRC repository). The absence of Il4i1 expression in CNS lesion was confirmed by qRT-PCR and in situ hybridization (Supplementary Fig. 2A and B). To examine the distribution of proinflammatory macrophage subpopulations in lesions, immunostaining analysis for CD11b and iNOS co-labelling, corresponding to CAM, was performed on wild-type and Il4i1−/− mice at 5, 10 and 20 dpl. In wild-type lesions, we found that the relative density of CD11b+iNOS+ macrophages was high at 5 dpl, and reduced at 10 and 20 dpl (Fig. 3A and B). In Il4i1−/− lesions, as in the wild-type, we observed a relatively high density of CD11b+iNOS+ macrophages at 5 dpl. However, this density remained significantly elevated across all three post-lesion time points, unlike their wild-type counterparts (Fig. 3A and B). To assess the distribution of other proinflammatory cell populations, CNS lesions at 10 dpl were co-immunostained of tenascin-C and GFAP to identify reactive astrocytes, or intracellular fibronectin (IST-9) to identify meningeal fibroblasts. We found that mice deficient in Il4i1 displayed enhanced tenascin-C+ and IST-9+ staining in lesions, suggesting increased gliosis (Supplementary Fig. 3). These results suggest that inflammation remains unresolved in the absence of Il4i1 expression.
Figure 3.

IL4I1 modulates inflammation in CNS lesions. Quantification of macrophage subpopulations in lesions of wild-type, Il4i1−/− and IL4I1 treated mice at 5, 10 and 20 dpl. (A) CD11b+ iNOS+ cell quantification. (B) Immunostaining of iNOS (green), CD11b (red) and DAPI (blue) at 10 dpl. (C) CD11b+Ym1+ cell quantification. (D) Immunostaining of Ym1 (green), CD11b (red) and DAPI (blue) at 10 dpl. (E) Quantification of the ratio of iNOS+/Ym1+ cells in lesions. (F) Flow cytometry analysis of iNOS+CD11b+ cells in lesioned spinal cord of wild-type (WT) and IL4I1-treated mice at 10 dpl. For cell counts, n = 3–5 mice per group were used and n = 3 × 10 magnification images per mouse were analysed. Scale bar = 100 μm.
To examine the distribution of anti-inflammatory macrophages in lesions, immunostaining analysis for CD11b and Ym1 co-labelling, corresponding to AAM, was performed. We found that the relative density of CD11b+Ym1+ macrophages appeared to increase from 5 to 20 dpl in wild-type lesions; however, this change was not significant due to high variability across time points (Fig. 3C and D). In Il4i1−/− mice, we detected relatively fewer CD11b+Ym1+ macrophages in lesions compared to wild-type at 5 dpl, but found no significant difference between the two groups at the other post-lesion time points (Fig. 3C and D). These results suggest the absence of Il4i1 does not affect AAM distribution in lesions. We next analysed the ratio of iNOS/Ym1 in wild-type and Il4i1−/− lesions at 5, 10 and 20 dpl. The transition from high CAM/AAM to low CAM/AAM has been shown to be crucial for remyelination to proceed (Miron et al., 2013). In wild-type lesions, we observed a gradual and significant reduction of iNOS:Ym1 ratio from 5 to 20 dpl, suggesting a shift from acute inflammation to its resolution. By contrast, we found that the iNOS:Ym1 ratio in Il4i1−/− mice remained elevated at 20 dpl compared to wild-type, suggesting a failure to achieve inflammation resolution (Fig. 3E). These results show that Il4i1 is necessary to modulate inflammation during remyelination.
As IL4I1 is a secreted enzyme, we next asked if the administration of IL4I1 into CNS lesions is able to alter the state of inflammation in lesions. For IL4I1 delivery, we co-injected 200 ng/ml of recombinant mouse IL4I1 with lysolecithin into the spinal cord of wild-type mice and examined its effect on macrophage distribution in CNS lesions. We found that the density of CD11b+iNOS+ macrophages in IL4I1 treated lesions was high at 5 dpl, similar to that found in untreated lesions (Fig. 3A). However, IL4I1 treatment significantly reduced the density of CD11b+iNOS+ macrophages in lesions by 10 and 20 dpl (Fig. 3A and B). This reduction was confirmed by flow cytometry analysis of spinal cord tissues from untreated and IL4I1 treated mice (Fig. 3F). The observation that IL4I1 injection did not affect CD11b+iNOS+ initially at 5 dpl, but significantly reduced their density at 10 and 20 dpl, suggests that IL4I1 injection had a delayed effect on CAM distribution in lesions, and therefore may influence macrophage activity indirectly. We also determined the effect of IL4I1 injection on AAM distribution in lesions, and found that IL4I1 treatment did not significantly influence the density of CD11b+Ym1+ macrophages at 5 or 10 dpl compared to wild-type (Fig. 3C and D). However, we observed a significant decrease of CD11b+Ym1+ macrophages in IL4I1 treated lesions at 20 dpl compared to wild-type. This indicates a possible autoregulatory feedback mechanism in which IL4I1 may eventually reduce all activated macrophage subpopulations, or promote their clearance in lesions. An analysis of iNOS/Ym1 ratio revealed that IL4I1 treatment reduced proinflammatory activity in lesions at 5 dpl (Fig. 3E). Together, these results suggest that IL4I1 injection accelerates inflammation resolution.
IL4I1 promotes central nervous system remyelination and axonal integrity
To determine if IL4I1 is required for efficient remyelination, immunostaining analysis was performed on Il4i1−/− and wild-type mice at 5, 10 and 20 dpl. We did not detect a significant difference in the relative number of Nkx2.2+ oligodendrocyte precursor cells in CNS lesions of Il4i1−/− mice compared to wild-type in any of the post-lesion time points examined (Fig. 4A). We also did not detect a significant difference in the relative number of CC1+ oligodendrocytes in Il4i1−/− mice compared to wild-type at 5 or 10 dpl (Fig. 4B). However, a significant reduction of CC1+ oligodendrocytes was observed at 20dpl in Il4i1−/− lesions compared to wild-type (Fig. 4B). Interestingly, CC1+ oligodendrocytes were frequently observed in the periphery of the lesion at this time point (Fig. 4C), suggesting that oligodendrocytes within lesions had undergone cell death by 20 dpl. To examine the extent of remyelination, electron microscopy analysis was performed on CNS lesions at 10 dpl. In contrast to wild-type mouse lesions that exhibit many axons undergoing remyelination, we observed few remyelinating axons in Il4i1−/− mouse lesions (Fig. 4D). Moreover, g-ratio analysis revealed a significant reduction in the number of remyelinated axons in the lesions of Il4i1−/− mice compared to wild-type (Fig. 4E and F). Notably, we found that Il4i1−/− mice displayed prominent axonal dystrophy and Wallerian degeneration, which were infrequently observed in wild-type mice (Fig. 4G and H). Immunostaining analysis with SMI-32, an antibody that detects non-phosphorylated neurofilaments and marks dystrophic axons, showed significantly increased SMI-32 labelling in Il4i1−/− lesions compared to wild-type (Fig. 4I). These data indicate that Il4i1 deficiency results in significant remyelination impairment and axonal injury, and suggests that the failure to modulate inflammation results in a non-permissive environment for remyelination.
Figure 4.

IL4I1 regulates remyelination and preserves axonal integrity. Quantification of (A) Nkx2.2+Olig2+ and (B) CC1+Olig2+ cells per mm2 in lesions at 5, 10 and 20 dpl in wild-type (WT), Il4i1−/− and IL4I1-treated mice. (C) Immunostaining of Olig2 (green), CC1 (red) and DAPI (blue) in lesions at 10 dpl. Lesions are characterized as the cluster of DAPI+ nuclei in the spinal cord ventral funiculous. (D) Electron micrographs of lesion at 10 dpl show reduced remyelinated axons in Il4i1−/− lesions, and increased remyelinated axons in IL4I1 treated lesions compared to wild-type. (E) G-ratio analysis of remyelinated axons and corresponding axonal diameter. (F) Scatter plot showing the overall g-ratio in the mouse groups. (G) Electron micrographs of lesions in Il4i1−/− mice showing a dystrophic axon and an axon that has undergone Wallerian degeneration. (H) Immunostaining of SMI-32 (green), NF200 (red) and DAPI (blue) in spinal cord lesions at 10 dpl. Axonal dystrophy is detected by SMI-32+NF200+ co-labelling. (I) Graph of relative SMI-32 signal intensity normalized to NF200. For cell counts and fluorescence intensity, n = 3–5 mice per group were used and n = 3 ×10 magnification images per mouse were analysed. Scale bar = 100 μm for immunofluorescence images. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ANOVA followed by post hoc analysis.
To determine if the pharmacological administration of IL4I1 can enhance remyelination, we co-injected 200 ng/ml of recombinant mouse IL4I1 with lysolecithin into the spinal cord of wild-type mice. We found that IL4I1-treated mice displayed significantly increased numbers of Nkx2.2+ oligodendrocyte precursor cells in lesions at 5 dpl compared to untreated mice. However, by 20 dpl, the relative distribution of Nkx2.2+ oligodendrocyte precursor cells was reduced to levels comparable to those of the untreated mice (Fig. 4A). We also found that CC1+ oligodendrocyte number in lesions was significantly greater in IL4I1 treated mice compared to untreated mice in all three post-lesion time points examined (Fig. 4B and C). Analysis of CNS lesions by electron microscopy revealed that IL4I1 treated mice exhibited a higher proportion of axons undergoing remyelination as compared to untreated mice (Fig. 4D). G-ratio analysis confirmed that IL4I1 treatment significantly increased the number of remyelinated axons (Fig. 4E and F). These results indicate that IL4I1 administration enhances CNS remyelination.
IL4I1 rescues remyelination impairment in mice deficient in IL4 receptor signalling
Because IL4I1 is downstream of IL4 receptor signalling, and our findings suggest that IL4I1 plays role in inflammation modulation and remyelination, we next asked if remyelination is also impaired in Il4ra−/− mice. Immunostaining and analysis of CNS lesions at 10 dpl after lysolecithin-mediated demyelination showed that Il4ra−/− mice displayed significantly fewer CC1+ oligodendrocytes, elevated axonal dystrophy, and increased iNOS+CD11b+ macrophage number (Fig. 5A–C and Supplementary Fig. 4) in lesions compared to wild-type mice. Therefore, Il4ra−/− mice exhibit similar pathology in CNS lesions as that observed in the Il4i1−/− mice. To determine if IL4I1 administration can rescue remyelination impairment in Il4ra−/− mice, recombinant IL4I1 was co-injected with lysolecithin into the spinal cord of Il4ra−/− mice and the lesions were analysed at 10 dpl. We found significantly increased CC1+ oligodendrocyte density, reduced axonal dystrophy, and decreased iNOS+CD11b+ macrophages in the lesions of IL4I1 treated Il4ra−/− mice compared to untreated Il4ra−/− mice (Fig. 5A–C). These findings suggest that IL4I1 administration modulates inflammation, thereby promoting remyelination and preserving axonal integrity in mice with impaired AAM activity and type 2 immunity.
Figure 5.

IL4I1 injection rescues remyelination impairment in Il4ra−/− mice. Quantification of (A) CC1+Olig2+ cells, (B) SMI-32 signal intensity normalized to NF200, and (C) CD11b+iNOS+ macrophages in Il4ra−/− and Il4ra−/− treated mice at 10 dpl. *P < 0.05, **P < 0.01, ***P < 0.001; ANOVA followed by post hoc analysis.
IL4I1 modulates inflammation by reducing interferon gamma and IL17 expression
While our results indicated that IL4I1 modulates the inflammatory environment to promote CNS remyelination, its mechanism of action remained unclear. To determine if IL4I1 directly modulates the expression of the proinflammatory factor iNOS in innate immune cells, we treated purified astrocytes, microglia, and RAW264.7 cell cultures with recombinant IL4I1. qRT-PCR analysis showed that IL4I1 did not influence expression of Nos2, which encodes iNOS, in any of these cells in vitro (Supplementary Fig. 5). This observation suggests that IL4I1 regulates possibly other cell populations in CNS lesions in exerting its immunomodulatory effect. It has previously been demonstrated that IL4I1 regulates CD4+ T cell proliferation and interferon gamma (IFN-γ, encoded by IFNG) production in vitro (Lasoudris et al., 2011; Cousin et al., 2015). Because IFN-γ producing T cells are known to migrate to the CNS following blood–brain barrier breakdown and drive inflammation and oligodendrocyte destruction in the CNS (Ghasemlou et al., 2007; Ryu et al., 2015; Traka et al., 2016), we hypothesized that IL4I1 modulates CNS inflammation by regulating T cell activity. To determine if IL4I1 treatment affects T cell-associated cytokine production in CNS lesions, qRT-PCR analysis was performed on lysolecithin-demyelinated spinal cord tissues following recombinant IL4I1 or vehicle treatment. We found that IL4I1 treatment dramatically reduced Ifng and Il17 expression in CNS tissues at 10 dpl, and had no effect on Il4 expression (Fig. 6A), suggesting that IL4I1 injection modulates T-helper 1 (Th1) and Th17 cell activity in CNS lesions. To determine if IL4I1 directly regulates Ifng expression in CD4+ T cells, mouse splenocyte cultures were treated with ionomycin and phorbol myristate acetate (PMA) in the presence or absence of recombinant IL4I1 for 24 h. qRT-PCR analysis showed that IL4I1 addition significantly reduced Ifng expression in activated splenocytes (Fig. 6B). Moreover, IL4I1 treatment did not lead to apoptosis (Fig. 6C). This suggests that the reduction in Ifng resulted from the ability of IL4I1 to modulate T cell activity rather than an ability to decrease cell survival.
Figure 6.

IL4I1 modulates IFN-γ and IL-17 expression in CNS lesions and splenocytes. (A) Normalized expressions of Ifng, Il17 and Il4 in wild-type (WT, n = 4) and IL4I1-treated (n = 6) spinal cord lesions at 10 dpl by qRT-PCR. (B) Normalized expression of Ifng expression in splenocyte cultures treated with PMA+Ionomycin in the presence or absence of recombinant IL4I1 for 24 h (n = 3 per group) followed by qRT-PCR. (C) Cell death assay in splenocyte cultures treated with PMA+Ionomycin with or without recombinant IL4I1 for 24 h, and assessed by LIVE/DEAD® cell stain kit (n = 8 per group).
IL4I1 reverses experimental autoimmune encephalomyelitis-associated clinical symptoms by modulating CD4+ cell activation
To determine if IL4I1 can therapeutically modulate T cell activity in vivo, we examined the effect of recombinant IL4I1 administration in mice with EAE. EAE is an autoimmune-mediated inflammatory condition in the CNS that is characterized by demyelination and axonal injury (Kornek et al., 2000; Nikić et al., 2011). For the therapeutic study, mice that developed EAE were assigned either into IL4I1 treated, or PBS (vehicle) treated group (n = 8–10 per group). In a separate experiment, mice were assigned into IL4I1 treated, or untreated control group (n = 10 per group). For treatment, 100 μl of IL4I1 (1 μg/ml blood volume), or PBS was injected intravenously into mice with EAE—first dose at clinical score 2.0–2.5, when hindlimb weakness became apparent, and a second dose 4 days later. The mice were then monitored daily for disease severity until at least 28 days post EAE induction. We found that the untreated, and PBS-treated groups displayed the typical profile of disease progression, in which complete hindlimb paralysis (clinical score 3.0–3.5) appeared by 16–18 days post EAE induction, and persisted until the end of the experiment (Supplementary Video 1). We found that mice with IL4I1 treatment also displayed hindlimb paralysis, reaching the clinical score of 3.0. However, the clinical deficit was transient, as all of the mice subsequently recovered from paralysis (Fig. 7A and Supplementary Video 2). This result indicates that IL4I1 treatment reversed the course of the disease, leading to the reduction in disease severity.
Figure 7.

IL4I1 reverses clinical severity and preserves axons in mice with EAE. (A) Clinical scores of PBS-treated (n = 8) and IL4I1-treated (n = 10) mice for 28 days after EAE induction. Flow cytometry analysis of (B) spinal cord and (C) spleen from untreated and IL4I1 treated mice at 35 days after EAE induction showing the percentage and total number of gated T-bet+CD4+, Rorγt+CD4+ and Gata3+CD4+ cells (samples from n = 2 mice were combined for each group and analysed). (D) Immunostaining of SMI-32 (green), NF200 (red) and DAPI (blue) in spinal cord of untreated and IL4I1 treated mice with EAE at 35 days after EAE induction. Axonal dystrophy is detected by SMI-32+NF200+ co-labelling. (E) SMI-32 signal intensity normalized to NF200 in untreated, and IL4I1-treated mice showing reduced axonal dystrophy in IL4I1 treated mice (n = 3 per group). Scale bar = 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; one-way Wilcoxon Rank-Sum Test (EAE), two-tailed Student’s t-test (SMI-32 analysis).
Flow cytometry analysis of CD4+ T- cell populations in spinal cord tissues removed from the IL4I1 treated and untreated groups following completion of the EAE experiment revealed a reduction in T-bet+CD4+ (4.5-fold), RORγt+CD4+ (3.1-fold), and Gata3+CD4+ (2.7-fold) cells in the IL4I1-treated mice compared to control mice (Fig. 7B). Similarly, flow cytometry analysis of spleens removed from mice following completion of the EAE experiment showed a decrease in T-bet+CD4+ (2.8-fold), RORγt+CD4+ (1.7-fold), and Gata3+CD4+ (11.7-fold) cells in the IL4I1-treated mice compared to those without treatment (Fig. 7C). These data indicate that recombinant IL4I1 prevented Th1, Th17 and Th2 cell expansion in the CNS and spleen. To examine the extent of axonal injury, immunostaining with SMI-32 and NF200 was performed on spinal cord sections from IL4I1-treated and untreated mice at the end of the EAE experiment. We found that untreated mice with EAE displayed significant SMI-32+NF200+ co-labelling, which indicates increased axonal injury. By contrast, analysis of spinal cord sections from IL4I1-treated mice with EAE revealed reduced axonal injury compared to control mice (Fig. 7D and E). Together, these results suggest that recombinant IL4I1 is able to limit axonal damage by attenuating CD4+ T cell mediated inflammation.
Discussion
In multiple sclerosis, remyelination failure correlates with disease progression (Franklin, 2002; Hagemeier et al., 2012). We found that unresolved inflammation in the CNS results in remyelination failure and axonal injury. It has been demonstrated that the level of IL4, which stimulates AAM polarization, is significantly reduced or absent in clinically active and progressive multiple sclerosis (Calabresi et al., 1998; Clerici et al., 2001). Moreover, chronic multiple sclerosis lesions display deficient AAM, which may be a contributor of inflammation dysregulation in the CNS (Vogel et al., 2013). In EAE, AAM induction with IL4 is essential for controlling autoimmune inflammation in the CNS (Butovsky et al., 2006; Ponomarev et al., 2007). Moreover, AAM also provide regulatory factors, such as activin, to stimulate oligodendrocyte differentiation and remyelination (Miron et al., 2013). We found that AAM express IL4I1, and that IL4I1 is necessary to modulate inflammation and promote myelin repair. Deficiency in IL4I1 results in enhanced and/or unresolved inflammation in lesions, leading to exacerbated axonal injury and remyelination impairment. These results suggest that a major obstacle to remyelination success is the failure to resolve inflammation in the CNS.
Microglia interactions with T cells in demyelinating lesions have been suggested to play an important role in modifying the pathobiology of multiple sclerosis (Strachan-Whaley et al., 2014). CD4+ T cells exist in many discrete subtypes, and contribute to the development of autoimmunity and inflammation (O’Garra et al., 1997; Wang et al., 2015). These subtypes include Th1 and Th17 cells, which release proinflammatory cytokines, and Th2 and regulatory T cells (Treg) cells that induce immunosuppressive effects. Prominent infiltration of T cells into the CNS occurs following CNS injury or demyelination (Popovich et al., 1997; Ghasemlou et al., 2007; Ryu et al., 2015). Depending on their activities, T cells may be detrimental or protective to the CNS (Moalem et al., 1999; Fee et al., 2003; Walsh et al., 2015). It is possible that AAM are necessary to modulate T cell activity in the demyelinated CNS. Indeed, injection of either microglia-derived or monocyte-derived AAM into mice with EAE has been shown to reduce disease severity by suppressing T cell proliferation, and promoting Treg expansion (Weber et al., 2007; Zhang et al., 2014a). Our results suggest that IL4I1 is a critical AAM-derived factor that modulates T cell activity during CNS remyelination. Although we found that IL4I1 can be induced in both alternatively activated microglia and monocyte-derived macrophages, it is unknown if microglia-derived macrophages, monocyte-derived macrophages, or both are the source of IL4I1 in CNS lesions. It remains technically challenging to discriminate the origins of macrophages once they have taken on an amoeboid or foamy morphology in lysolecithin-induced CNS lesions.
IL4I1 has been shown to suppress antigen-specific T cell proliferation and cytokine secretion (Boulland et al., 2007; Cousin et al., 2015). We found that a single injection of recombinant IL4I1 into lysolecithin-demyelinated spinal cord significantly reduced inflammation and augmented remyelination. Interestingly, IL4I1 injection did not reduce CAM distribution in lesions until 10 dpl. Similarly, IL4I1 did not affect AAM distribution in lesions initially, but reduced their distribution at 20 dpl when remyelination is complete. These results suggest that IL4I1 administration accelerated inflammation resolution. As IL4I1 modulates T cell function, and does not appear to influence macrophage/microglia activity directly, it is possible that IL4I1 administration affected proinflammatory T cells that eventually resulted in the reduction of CAM and AAM macrophages in lesions. Exactly how IL4I1 modulates inflammation in the demyelinated CNS remains unclear. We found that IL4I1 injection reduced IFN-γ and IL17 expression in lysolecithin demyelinated spinal cords, thus supporting the role of IL4I1 in regulating T cell function. Although T cell infiltration into lysolecithin-mediated lesions has been thought to be rapid and transient, they may still have biological significance in injury (Bieber et al., 2003; Ghasemlou et al., 2007). One possible explanation for the observed reduction in IFNγ and IL-17 expression is that IL4I1 was co-injected with lysolecithin into the spinal cord (Day 0), thus was able to affect T cell function early in injury. It is possible that IL4I1 shifts the balance of CD4+ T cell activity in lesions from a Th1/Th17 state to an immunomodulatory Th2 state, and this may be necessary to allow remodelling of the inflammation landscape in the injured CNS for remyelination.
Finally, we found that therapeutic injection of IL4I1 into mice with EAE significantly reduced disease severity by decreasing CD4+ T cells in spinal cord and spleen. It remains unknown whether IL4I1 alters additional cell populations, or if it modulates cytokine signalling. Moreover, it remains to be determined if the effect of IL4I1 on inflammation in lysolecithin-demyelinated lesions and EAE are achieved through similar mechanisms. As IL4I1 is an l-amino acid oxidase, it is possible that IL4I1 modulates inflammation by depleting l-amino acids and/or by stimulating H2O2 production in lesions. Recently, IL4I1 has been shown to maintain the expression of Tob1, an antiproliferative protein, which is necessary to limit CD4+ T cell proliferation (Santarlasci et al., 2014). Furthermore, Tob1 deficiency in mice with EAE results in enhanced CNS inflammation (Schulze-Topphoff et al., 2013). In fact, individuals presenting with an initial demyelination episode who also display low Tob1 expression are more likely to progress to multiple sclerosis from clinically isolated syndrome than those with high Tob1 level (Corvol et al., 2008). Impaired IL4I1 expression or activity may therefore drive multiple sclerosis pathogenesis by reducing TOB1 expression in T cells. Interestingly the IL4I1 gene in humans has been mapped to chromosome 19q13.3-13.4, which is a region implicated in autoimmune disease susceptibility, including multiple sclerosis (Becker et al., 1998; Chavan et al., 2002), suggesting that dysregulated IL4I1 expression may correlate with multiple sclerosis susceptibility or pathogenesis.
In summary, we found that IL4I1 is a critical regulator of inflammation, and promotes inflammation resolution to permit spontaneous remyelination. Moreover, we found that therapeutic injection of recombinant IL4I1 significantly reduces disease severity, and improves behaviour in mice with EAE. This finding is particularly relevant in the context of multiple sclerosis therapy, as intravenous IL4I1 administrations may be able to reduce disease severity or modify the course of the disease in multiple sclerosis. IL4I1 is therefore a promising therapeutic compound for promoting CNS repair and preventing disease progression in multiple sclerosis.
Supplementary Material
Acknowledgements
We thank Stefano Daniele and Kathleen Maguire-Zeiss for assistance in primary microglia cultures, Alejandra Chipana for assistance in imaging, Aleksander Keselman and Steven Singer for flow cytometry assistance, Eric Schmitt (KU Leuven) for advice on statistical analysis, Rafal Olszewski and the Georgetown University Department of Comparative Medicine for mouse husbandry and handling, and members of the Huang lab for helpful discussion and suggestions on this project.
Funding
This project was supported, in part, by funding from the NIH-NINDS grant 1R21NS091890-01 (J.K.H.), National Multiple Sclerosis Society grant PP2159 (J.K.H.), Georgetown University Music for the Mind Award (J.K.H.), Georgetown University Partners in Research Award (J.K.H.), Georgetown College (J.K.H.), NIH-NINDS Training Award 5T32NS041218 (K.A.C), TurnFirst RDS Scholarship (S.E.D), and NIH-NCATS Training Award TL1TR001431 (S.E.D). Transmission electron microscopy was performed at the VCU – Dept. of Anatomy and Neurobiology Microscopy Facility, supported, in part, by funding from NIH-NINDS Center Core grant 5P30 NS047463 and NIH-NCI Cancer Center grant P30 CAO16059 (J.L.D).
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- AAM
alternatively activated macrophages
- CAM
classically activated macrophages
- dpl
days post-lesion
- EAE
experimental autoimmune encephalomyelitis
- qRT-PCR
quantitative real time polymerase chain reaction
References
- Becker KG, Simon RM, Bailey-Wilson JE, Freidlin B, Biddison WE, McFarland HF, et al. Clustering of non-major histocompatibility complex susceptibility candidate loci in human autoimmune diseases. Proc Natl Acad Sci USA 1998; 95: 9979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieber AJ, Kerr S, Rodriguez M. Efficient central nervous system remyelination requires T cells. Ann Neurol 2003; 53: 680–4. [DOI] [PubMed] [Google Scholar]
- Blakemore WF, Franklin RJM. Remyelination in experimental models of toxin-induced demyelination. Curr Top Microbiol Immunol 2008; 318: 193–212. [DOI] [PubMed] [Google Scholar]
- Boulland M-L, Marquet J, Molinier-Frenkel V, Möller P, Guiter C, Lasoudris F, et al. Human IL4I1 is a secreted L-phenylalanine oxidase expressed by mature dendritic cells that inhibits T-lymphocyte proliferation. Blood 2007; 110: 220–7. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Landa G, Kunis G, Ziv Y, Avidan H, Greenberg N, et al. Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J Clin Invest 2006; 116: 905–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi PA, Tranquill LR, McFarland HF, Cowan EP. Cytokine gene expression in cells derived from CSF of multiple sclerosis patients. J Neuroimmunol 1998; 89: 198–205. [DOI] [PubMed] [Google Scholar]
- Chamberlain KA, Nanescu SE, Psachoulia K, Huang JK. Oligodendrocyte regeneration: its significance in myelin replacement and neuroprotection in multiple sclerosis. Neuropharmacology 2015. pii: S0028-3908(15)30133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavan SS, Tian W, Hsueh K, Jawaheer D, Gregersen PK, Chu CC. Characterization of the human homolog of the IL-4 induced gene-1 (Fig1). Biochim Biophys Acta 2002; 1576: 70–80. [DOI] [PubMed] [Google Scholar]
- Chu CC, Paul WE. Fig1, an interleukin 4-induced mouse B cell gene isolated by cDNA representational difference analysis. Proc Natl Acad Sci USA 1997; 94: 2507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Saresella M, Trabattoni D, Speciale L, Fossati S, Ruzzante S, et al. Single-cell analysis of cytokine production shows different immune profiles in multiple sclerosis patients with active or quiescent disease. J Neuroimmunol 2001; 121: 88–101. [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet 2002; 359: 1221–31. [DOI] [PubMed] [Google Scholar]
- Copie-Bergman C, Boulland M-L, Dehoulle C, Möller P, Farcet J-P, Dyer MJS, et al. Interleukin 4-induced gene 1 is activated in primary mediastinal large B-cell lymphoma. Blood 2003; 101: 2756–61. [DOI] [PubMed] [Google Scholar]
- Corvol J-C, Pelletier D, Henry RG, Caillier SJ, Wang J, Pappas D, et al. Abrogation of T cell quiescence characterizes patients at high risk for multiple sclerosis after the initial neurological event. Proc Natl Acad Sci USA 2008; 105: 11839–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin C, Aubatin A, Le Gouvello S, Apetoh L, Castellano F, Molinier-Frenkel V. The immunosuppressive enzyme IL4I1 promotes FoxP3(+) regulatory T lymphocyte differentiation. Eur J Immunol 2015; 45: 1772–82. [DOI] [PubMed] [Google Scholar]
- Daniele SG, Edwards AA, Maguire-Zeiss KA. Isolation of cortical microglia with preserved immunophenotype and functionality from murine neonates. J Vis Exp 2014; 83: e51005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dincman TA, Beare JE, Ohri SS, Whittemore SR. Isolation of cortical mouse oligodendrocyte precursor cells. J Neurosci Methods 2012; 209: 219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döring A, Sloka S, Lau L, Mishra M, van Minnen J, Zhang X, et al. Stimulation of monocytes, macrophages, and microglia by amphotericin B and macrophage colony-stimulating factor promotes remyelination. J Neurosci 2015; 35: 1136–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Trapp BD. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog Neurobiol 2011; 93: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Trapp BD. Relapsing and progressive forms of multiple sclerosis: insights from pathology. Curr Opin Neurol 2014; 27: 271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fee D, Crumbaugh A, Jacques T, Herdrich B, Sewell D, Auerbach D, et al. Activated/effector CD4+ T cells exacerbate acute damage in the central nervous system following traumatic injury. J Neuroimmunol 2003; 136: 54–66. [DOI] [PubMed] [Google Scholar]
- Fitzner D, Simons M. Chronic progressive multiple sclerosis - pathogenesis of neurodegeneration and therapeutic strategies. Curr Neuropharmacol 2010; 8: 305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJM. Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci 2002; 3: 705–14. [DOI] [PubMed] [Google Scholar]
- Franklin RJM, ffrench-Constant C, Edgar JM, Smith KJ. Neuroprotection and repair in multiple sclerosis. Nat Rev Neurol 2012; 8: 624–34. [DOI] [PubMed] [Google Scholar]
- Franklin RJM, ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci 2008; 9: 839–55. [DOI] [PubMed] [Google Scholar]
- Ghasemlou N, Jeong SY, Lacroix S, David S. T cells contribute to lysophosphatidylcholine-induced macrophage activation and demyelination in the CNS. Glia 2007; 55: 294–302. [DOI] [PubMed] [Google Scholar]
- Gong Z-K, Wang S-J, Huang Y-Q, Zhao R-Q, Zhu Q-F, Lin W-Z. Identification and validation of suitable reference genes for RT-qPCR analysis in mouse testis development. Mol Genet Genomics 2014; 289: 1157–69. [DOI] [PubMed] [Google Scholar]
- Hagemeier K, Brück W, Kuhlmann T. Multiple sclerosis - remyelination failure as a cause of disease progression. Histol Histopathol 2012; 27: 277–87. [DOI] [PubMed] [Google Scholar]
- Hansmann F, Herder V, Kalkuhl A, Haist V, Zhang N, Schaudien D, et al. Matrix metalloproteinase-12 deficiency ameliorates the clinical course and demyelination in Theiler’s murine encephalomyelitis. Acta Neuropathol 2012; 124: 127–42. [DOI] [PubMed] [Google Scholar]
- Huang JK, Franklin RJM. Current status of myelin replacement therapies in multiple sclerosis. Prog Brain Res 2012; 201: 219–31. [DOI] [PubMed] [Google Scholar]
- Huang JK, Jarjour AA, Nait Oumesmar B, Kerninon C, Williams A, Krezel W, et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci 2011; 14: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine KA, Blakemore WF. Remyelination protects axons from demyelination-associated axon degeneration. Brain 2008; 131: 1464–77. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 2009; 29: 13435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol 2000; 157: 267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter MR, Setzu A, Sim FJ, Van Rooijen N, Franklin RJ. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia 2001; 35: 204–12. [DOI] [PubMed] [Google Scholar]
- Lasoudris F, Cousin C, Prevost-Blondel A, Martin-Garcia N, Abd-Alsamad I, Ortonne N, et al. IL4I1: an inhibitor of the CD8+ antitumor T cell response in vivo. Eur J Immunol 2011; 41: 1629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H, Brück W, Lucchinetti C, Rodriguez M. Remyelination in multiple sclerosis. Mult Scler 1997; 3: 133–6. [DOI] [PubMed] [Google Scholar]
- Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 2012; 8: 647–56. [DOI] [PubMed] [Google Scholar]
- Mikita J, Dubourdieu-Cassagno N, Deloire MS, Vekris A, Biran M, Raffard G, et al. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult Scler 2011; 17: 2–15. [DOI] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao J-W, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 2013; 16: 1211–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Franklin RJM. Macrophages and CNS remyelination. J Neurochem 2014; 130:165–71. [DOI] [PubMed] [Google Scholar]
- Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 1999; 5: 49–55. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol 2011; 89: 557–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 2011; 17: 495–9. [DOI] [PubMed] [Google Scholar]
- O’Garra A, Steinman L, Gijbels K. CD4+ T cell subsets in autoimmunity. Curr Opin Immunol 1997; 9: 872–83. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci 2007; 27: 10714–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol 1997; 377: 443–64. [DOI] [PubMed] [Google Scholar]
- Puiffe M-L, Lachaise I, Molinier-Frenkel V, Castellano F. Antibacterial properties of the mammalian L-amino acid oxidase IL4I1. PloS One 2013; 8: e54589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu JK, Petersen MA, Murray SG, Baeten KM, Meyer-Franke A, Chan JP, et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun 2015; 6: 8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarlasci V, Maggi L, Mazzoni A, Capone M, Querci V, Rossi MC, et al. IL-4-induced gene 1 maintains high Tob1 expression that contributes to TCR unresponsiveness in human T helper 17 cells. Eur J Immunol 2014; 44: 654–61. [DOI] [PubMed] [Google Scholar]
- Schonberg DL, Popovich PG, McTigue DM. Oligodendrocyte generation is differentially influenced by toll-like receptor (TLR) 2 and TLR4-mediated intraspinal macrophage activation. J Neuropathol Exp Neurol 2007; 66: 1124–35. [DOI] [PubMed] [Google Scholar]
- Schulze-Topphoff U, Casazza S, Varrin-Doyer M, Pekarek K, Sobel RA, Hauser SL, et al. Tob1 plays a critical role in the activation of encephalitogenic T cells in CNS autoimmunity. J Exp Med 2013; 210: 1301–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan-Whaley M, Rivest S, Yong VW. Interactions between microglia and T cells in multiple sclerosis pathobiology. J Interferon Cytokine Res 2014; 34: 615–22. [DOI] [PubMed] [Google Scholar]
- Traka M, Podojil JR, McCarthy DP, Miller SD, Popko B. Oligodendrocyte death results in immune-mediated CNS demyelination. Nat Neurosci 2016; 19: 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel DYS, Vereyken EJF, Glim JE, Heijnen PDAM, Moeton M, van der Valk P, et al. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflammation 2013; 10: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos CMP, van Haastert ES, de Groot CJA, van der Valk P, de Vries HE. Matrix metalloproteinase-12 is expressed in phagocytotic macrophages in active multiple sclerosis lesions. J Neuroimmunol 2003; 138: 106–14. [DOI] [PubMed] [Google Scholar]
- Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J Clin Invest 2015; 125: 2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Collins M, Kuchroo VK. Effector T cell differentiation: are master regulators of effector T cells still the masters? Curr Opin Immunol 2015; 37: 6–10. [DOI] [PubMed] [Google Scholar]
- Weber MS, Prod’homme T, Youssef S, Dunn SE, Rundle CD, Lee L, et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med 2007; 13: 935–43. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol 2015; 15: 271–82. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature 2013; 496: 445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Huang W, Liang J, Guo J, Ji J, Yao Y, et al. IL4I1 is a novel regulator of M2 macrophage polarization that can inhibit T cell activation via L-tryptophan and Arginine depletion and IL-10 production. PloS One 2015; 10: e0142979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X-M, Lund H, Mia S, Parsa R, Harris RA. Adoptive transfer of cytokine-induced immunomodulatory adult microglia attenuates experimental autoimmune encephalomyelitis in DBA/1 mice. Glia 2014a; 62: 804–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 2014b; 34: 11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.