

Abstract
Purpose of review
Endothelial dysfunction is an early feature of vascular disease induced by cardiovascular risk factors (CRF). In growing populations with obesity, diabetes, hypertension and heart failure, mineralocorticoid receptor (MR) antagonism improves endothelial function. This review summarizes recent advances in our understanding of the specific role of endothelial cell (EC) MR in vascular function in health and disease.
Recent findings
Using transgenic mice with MR expression specifically modulated in ECs, recent studies support the emerging concept that while EC-MR may be protective in health, in the presence of CRFs, EC-MR contributes to endothelial dysfunction and progression of vascular disease. Proposed mechanisms include a role for EC-MR in decreased nitric oxide production and bioavailability, increased vascular oxidative stress, regulation of epithelial sodium channels that enhance vascular stiffness, and increased EC adhesion molecules promoting inflammation. The role of EC-MR may also depend on the sex, race, or vascular bed involved.
Summary
Recent advances support the idea that EC-MR is a mediator of the switch from vascular health to disease in response to CRFs. Further investigation of the molecular mechanism is underway to identify therapeutic interventions that will limit the detrimental effects of EC-MR in patients at cardiovascular risk.
Keywords: Mineralocorticoid Receptor, Endothelial Function, Cardiovascular Disease
Introduction
The vasculature is lined with endothelial cells (EC) which dynamically regulate vascular tone, stiffness, inflammation, and thrombotic potential in health and disease. Through these functions, the endothelium contributes to regulation of organ blood flow, blood pressure (BP), and vascular integrity. Endothelial function is influenced by paracrine and circulating factors regulating the production and activity of endothelium-derived vasoactive and growth factors, adhesion molecules that mediate leucocyte-EC interaction, and regulators of blood coagulation. In the healthy vasculature, the endothelium is anti-inflammatory, anti-thrombotic and promotes vasodilation. In the presence of cardiovascular risk factors, the endothelium becomes pro-inflammatory and pro-thrombotic and is characterized by impaired endothelium-dependent relaxation, a marker of endothelial dysfunction that predicts cardiovascular risk[1,2]. The mechanism by which the healthy endothelium becomes dysfunctional in response to cardiovascular risk factors is not completely understood.
Endothelial dysfunction occurs in response to common cardiovascular risk factors including obesity, metabolic syndrome, hypertension and heart failure. These conditions are all also associated with increased levels of the BP-regulating hormone aldosterone and activation of its mineralocorticoid receptor (MR)[3]. In patients with these conditions, MR antagonism improves endothelial function[4,5,6**]. In addition to its role in controlling renal salt and water homeostasis, MR is also expressed in the vascular endothelium[7,8]. Recent reports that perivascular tissue is a paracrine source of MR ligands in obesity [9] and hyperadrenergic states [10] further supports the potential for direct vascular MR activation to contribute to vascular disease in high risk patients. However, the specific role of EC-MR in vascular health and disease has been controversial until recently[8]. The development of transgenic mice in which MR expression can be specifically modulated in ECs has recently advanced our understanding of the role of EC-MR in maintaining a healthy vasculature and its contribution to cardiovascular disease. This review summarizes our understanding of the role of endothelial MR in vascular function and disease with an emphasis on data published in the past year.
Physiological versus pathological role of MR in endothelial function: still a paradox?
Early studies investigating the role of EC-MR in endothelial function produced conflicting findings[reviewed in 3,11]. Aldosterone can induce either vasoconstriction or vasodilation in isolated vessels [reviewed in 8,12]. Rapid vasodilator action of aldosterone was described to be dependent on MR activation of PI3-kinase/Akt signaling, enhancing the production of the endothelium-derived vasodilator nitric oxide (NO) via phosphorylation of endothelial NO synthase (eNOS, Figure 1)[12,13]. However, when tested in vitro, EC-MR activation decreased eNOS activity is some studies[14,15] while it enhanced eNOS-derived NO production in others[16].
Figure 1. Proposed model for the role of EC-MR in endothelial function.
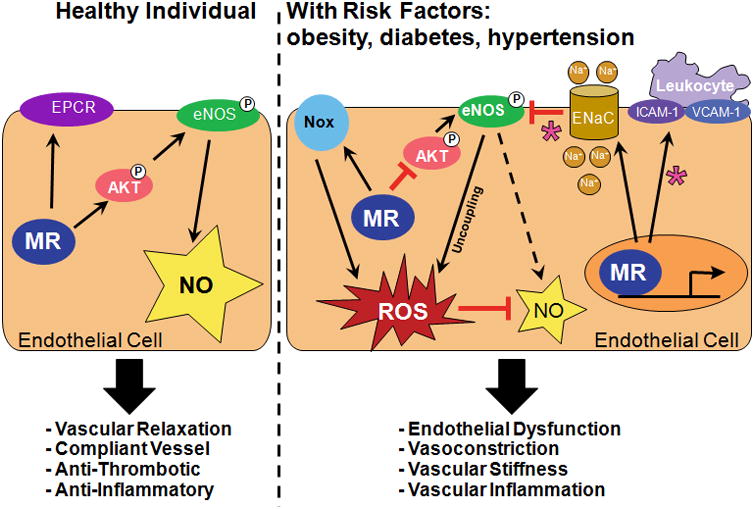
In the healthy vasculature, EC-MR exerts a protective role on endothelial function. In the presence of cardiovascular risk factors such as obesity, diabetes, or hypertension, EC-MR contributes to endothelial dysfunction through NADPH oxidase (Nox) activation, eNOS uncoupling, increased ENaC expression, and ICAM1/VCAM1-mediated inflammation. Asterisks indicate EC-MR mechanisms differentially regulated in females compared to males. EC=endothelial cells, MR=mineralocorticoid receptor, eNOS=endothelial nitric oxide synthase, ENaC=epithelial sodium channel, EPCR=endothelial cell protein C receptor, ICAM-1=intercellular adhesion molecule-1, NO=nitric oxide, P=serine phosphorylation, ROS=reactive oxygen species, VCAM1= vascular cell adhesion molecule-1.
Clinical studies also originally appeared contradictory. Several studies reveal a protective role of MR on endothelial function in healthy populations. Acute and chronic MR activation in healthy men improved endothelium-dependent relaxation and enhanced NO bioactivity[17]. Accordingly, short-term MR antagonism with eplerenone impaired endothelial function in healthy older adults, as measured by brachial artery flow-mediated dilation (FMD), in association with reduced eNOS activity as determined by decreased eNOS Ser1177-phosphorylation[18*]. In another study, MR inhibition improved FMD; however, the degree of improvement correlated with increased adiposity and fasting glucose, with no effect on vasorelaxation in non-obese patients[19]. This apparent contradiction is consistent with the emerging concept that although EC-MR may be protective in a healthy population, in the presence of cardiovascular risk factors (e.g. obesity, metabolic syndrome), EC-MR becomes a contributing factor to endothelial dysfunction. This is consistent with previous studies demonstrating that MR antagonism improves endothelial function in heart failure patients [4], and with a more recent reports showing that spironolactone improved coronary flow reserve (CFR) in patients with diabetes [6**] and enhanced EPC-derived NO production in patients with hypertension due to hyperaldosteronism[20**].
Overall, clinical studies reveal that MR inhibition in patients with cardiovascular disease or risk factors improves endothelial function. However, these studies cannot differentiate between direct effects of EC-MR or secondary effects on the endothelium mediated by MR in other tissues. To address this, genetic mouse models have been developed with EC-specific MR deletion. The first mouse model used the Tie2 promoter driving Cre recombinase expression resulting in MR deletion from EC as well as leukocytes (due to Tie2 expression in bone marrow–derived cells). In this model, Schafer et al. [21] found normal endothelium-dependent relaxation in aortae of healthy EC-MR-KO mice, while Rickard et al. [22] found vasodilation to be reduced in the aorta and mesenteric arteries, with no change in BP or vasoconstriction (Table 1). Because MR activation promotes macrophage activation and T lymphocyte differentiation [29], MR deletion in leukocytes could have independently influence endothelial function in this model.
Table 1. Comparison of Transgenic Mouse Models Used to Determine the Role of EC-MR in Blood Pressure and Vascular Function.
| Model | MR Phenotype | Sex | Blood Pressure | Vascular Phenotype | Molecular Mechanism | Ref. |
|---|---|---|---|---|---|---|
| Tie2-Cre/Floxed MR | MR deletion from endothelial cells and bone marrow-derived cells | male | Not measured | MR deletion did not affect basal aortic relaxation responses, but prevented HFD-induced endothelial dysfunction. | EC-MR contributes to increased COX-1 and p22phox subunit of NADPH oxidase in aorta. | [21] |
| male | No genotype effect at baseline or after DOCA/salt treatment | Reduced endothelium-dependent relaxation to Ach in aorta and MA of MR-KO at baseline; MR-KO prevented Aldo-induced endothelial dysfunction in aorta, but not in MA. | Not addressed | [22] | ||
| male | No genotype effect at baseline or after Aldo treatment. | EC-MR deletion prevented Aldo-induced superoxide production in cerebral arteries. | Not addressed | [23*] | ||
| VE-Cad-Cre/Floxed MR | MR deletion specifically from endothelial cells | male | No effect of EC-MR deletion at baseline or after AngII or Aldo± salt treatments to induce hypertension. | EC-MR does not contribute to MA or coronary vasodilation in healthy vessels; EC-MR deletion prevents endothelial dysfunction in MA of hypertensive mice; In coronary vessels, EC-MR contributes to increased vasoconstriction in response to AngII-dependent hypertension. | EC-MR deletion improved the NO/COX and EDH components of vasodilation in MA of AngII-induced hypertensive mice. | [24*] |
| female | No effect of EC-MR deletion at baseline or after WD feeding to induce obesity. | EC-MR deficiency prevented WD-induced aortic fibrosis, stiffness and endothelial dysfunction. | EC-MR contributes to ENaC activation, eNOS inhibition, oxidative stress, and inflammation after WD-induced obesity. | [25*,26] | ||
| VE-Cad-tetOFF/tetO-hMR | Tet-inducible human MR overexpression in endothelial cells | male | Increased at baseline | Delayed thrombosis in absence of additional risk factors. | MR-mediated up-regulation of protein C receptor expression in EC. | [27] |
| male | Increased at baseline | Normal vascular wall morphology and endothelial function. Increased MA myogenic tone and contraction to agonists. | Not addressed | [28] |
Ach: acetylcholine; Aldo: aldosterone; AngII: angiotensin II; EC: endothelial cell; EDH: endothelium-derived hyperpolarization; ENaC: epithelial sodium channel; eNOS: endothelial nitric oxide synthase; HFD: high-fat diet; KO: knockout; MR: mineralocorticoid receptor; MA: mesenteric arteries; VE-Cad: VE-cadherin; WD: Western diet.
To address this controversy, a truly EC-MR specific knockout mouse model (VE-cadherin promoter driven Cre recombinase) was recently developed with intact leukocyte MR[24*]. In this model, EC-MR deletion did not contribute to BP or to endothelial-dependent relaxation of coronary or mesenteric arteries in healthy animals. However, mesenteric arteries from male EC-MR knockout (EC-MR-KO) mice were protected from endothelial dysfunction following exposure to angiotensin II-induced hypertension[24*]. Similarly, in female mice, EC-MR deletion protected from aortic endothelial dysfunction caused by Western diet-induced obesity[25*]. This is consistent with data in the Tie2-Cre/EC-MR-KO model showing that aortic endothelial function was unchanged in healthy EC-MR-KO mice, but these mice were protected from endothelial dysfunction caused by high fat diet-induced obesity[21](Table 1).
These clinical and animal studies support an emerging paradigm in which basal EC-MR activity may contribute to normal vasodilation without determining vascular tone or BP, but that EC-MR contributes to the extent of endothelial dysfunction in the setting of cardiovascular risk factors such as hypertension, obesity, and diabetes (Figure 1).
Impact of Sex, Race and Vascular Bed Differences in the role of MR in Endothelial Function
Obesity is associated with increased plasma aldosterone levels[30] likely due to production of aldosterone-releasing factors, including leptin, by adipocytes[31**]. Interestingly, in a mouse model of Western diet-induced obesity, females developed higher plasma aldosterone levels compared to males [26,32] perhaps due to estrogen regulation of adrenal aldosterone production [33]. Consistent with this, improvement of endothelial function with MR blockade was more prominent in female compared to male hyperleptinemic mice[34*]. Female EC-MR-KO mice were also protected from increased endothelial stiffness and endothelial dysfunction induced by Western diet, further supporting a direct role for EC-MR in obesity-induced endothelial dysfunction[25*]. These studies suggest sex differences in the role of EC-MR in endothelial dysfunction, although the molecular mechanisms are still being explored. In human ECs in vitro, activated estrogen receptor (ER) has been shown to inhibit the transcriptional activity of MR [35], providing another potential mechanism for sex differences in the role of MR in EC function. Human coronary EC have been shown to express the cortisol-inactivating enzyme 11βHSD-2 supporting that aldosterone can act as a ligand for EC-MR ([7] and reviewed in [8]). However, whether 11βHSD-2 expression and/or function is modified by risk factors or other biological variables is not known and hence the potential for corticosteroids to activate EC-MR in vivo under different circumstances cannot be ruled out.
In contrast to the protective role of MR activation in brachial artery FMD observed in previous studies of healthy populations [18*,36], normotensive African-Americans developed endothelial microvascular dysfunction (evaluated by digital pulse arterial tonometry) in response to acute aldosterone administration and this was prevented by spironolactone[37*]. It was suggested that the endothelial dysfunction in healthy African-Americans may be due to decreased vascular G6PD activity in this population. Aldosterone has previously been shown to suppress G6PD expression in ECs [15], supporting a potential direct role for EC-MR in the mechanism. This detrimental effect of acute aldosterone administration seen in healthy African-Americans was not observed in brachial artery FMD in a predominantly Caucasian male population [17]. However, since these studies examined microvessel dysfunction in African-Americans and large vessels in the Caucasian populations, further studies are needed to clarify whether there are indeed racial differences in the vascular response to aldosterone or alternatively, a higher susceptibility of resistance microvessels to endothelial dysfunction in response to EC-MR activation when compared to large conduit vessels like the aorta or brachial arteries.
Indeed, clinical and animal data support differences in the role of EC-MR in conduit compared to microvessel function. Garg et al. [6**] observed that MR antagonism in type 2 diabetics improved CFR, an indicator of coronary microvascular function. The coronary benefits contrast with studies showing no improvement in forearm FMD with MR blockade in patients with diabetes[38,39], metabolic syndrome[40], or coronary artery disease[41]. These differences in the role of MR may be due to mechanistic differences in regulation of the coronary versus peripheral vasculature. Indeed, the role of EC-MR in hypertension-induced vascular dysfunction in mice differed in the coronary compared to the mesenteric microvasculature in the EC-specific MR-KO mouse model[24*] (Table 1). Further studies are needed to determine the mechanism for the distinct response to risk factors in the endothelium of different vascular beds and the role of EC-MR in mediating these differences.
New Insights into Mechanisms for the Role of EC-MR in Endothelial Function
Overall, clinical and experimental data support that MR blockade with spironolactone or eplerenone improves endothelial function in humans and animal models with high cardiovascular risk [reviewed in 3,11]. Tissue specific KO mice suggest a role for EC-MR in this protective effect (Table 1). While our understanding is still limited, the available data regarding the molecular mechanisms by which EC-MR contributes to endothelial dysfunction in the setting of cardiovascular disease are summarized in the following sections and depicted in Figure 1.
EC-MR Contributes to Vascular Oxidative Stress and Impaired NO Bioavailability
In the healthy endothelium, NO is produced by eNOS and diffuses to the underlying smooth muscle cells, where it mediates vasodilation. NO also acts locally on ECs to prevent inflammation and thrombosis. In the setting of cardiovascular disease risk factors NO production and bioavailability are impaired. Production of NO may decrease due to a decline in eNOS expression or activity, as indicated by phosphorylation of Ser1177. eNOS functions as a dimer and in the setting of vascular disease may also become uncoupled due to a decrease in the eNOS cofactor BH4 resulting in superoxide anion generation by eNOS rather than NO production. Superoxide reacts with NO to further decrease its bioavailability. Cardiovascular risk factors are associated with increased vascular oxidative stress due to reactive oxygen species produced by uncoupled eNOS and other enzymes, including NADPH and mitochondrial oxidases.
One mechanism involved in the protective role of MR blockade in endothelial function is increased NO bioavailability due to reduction in vascular oxidative stress and increased NO production[10,42,43,44]. Decreased eNOS uncoupling has been demonstrated in humans [20**] and rats [10] after spironolactone treatment. This effect was associated with increased eNOS dimerization, expression of the chaperone HSP-90 and availability of BH4 resulting in reduced superoxide generation and increased NO production. Spironolactone has also been shown to increase vascular superoxide dismutase and catalase expression, important anti-oxidants, and to reduce expression of p47phox, the regulatory subunit of NADPH oxidase[20**,43]. The specific role of EC-MR was recently demonstrated in vivo as EC-MR deletion prevented the aldosterone-induced increase in superoxide production in cerebral arteries [23*] and resulted in enhanced eNOS Ser1177 phosphorylation and improved endothelial function in aortae of Western-diet fed mice[25*]. Thus, human and animal studies support the concept that EC-MR contributes to vascular dysfunction in response to cardiovascular risk factors by enhancing vascular oxidative stress and impairing NO function.
EC-MR Modulates Endothelial Stiffness by Regulating the Epithelial Sodium Channel (ENaC)
The epithelial sodium channel (ENaC), a classical target of MR in the kidney, has more recently been identified in vascular ECs where it is also MR-regulated and mediates endothelial sodium transport thereby contributing to vascular stiffness[45]. Aldosterone increases expression of ENaC in human ECs in association with increased EC stiffness[46,47]. Enhanced EC stiffness alters shear forces in the vasculature resulting in impaired NO production[48]. In the Western diet-induced obesity model, aortic stiffening and decreased NO production in female mice was blocked by spironolactone[42]. In this model, EC-specific MR deletion prevented aortic stiffening and this beneficial effect was associated with attenuation of endothelial ENaC promoter activity and gene expression and restoration of eNOS phosphorylation[25*]. These data support that regulation of ENaC by EC-MR may be an additional mediator of endothelial dysfunction in response to cardiovascular risk factors including obesity.
Contributions of EC-MR to Vascular Thrombosis and Inflammation
When ECs lining the vasculature become damaged by cardiac risk factors, they also become dysfunctional in their anti-inflammatory and anti-thrombotic functions thereby contributing to development of atherosclerosis and its complications, myocardial infarction and stroke. Overexpression of MR in EC in mice or MR activation with aldosterone enhanced endothelial protein C receptor (EPCR) expression via a transcriptional mechanism, decreasing thrombin generation and attenuating vascular thrombosis[27]. These data suggest that MR activation could be antithrombotic in the endothelium. However, this contrasts with previous studies demonstrating that aldosterone increases thrombosis in experimental models of vascular injury in mice[49,50]. Once again these seemingly conflicting findings are consistent with the hypothesis that EC-MR has anti-thrombotic properties in healthy vessels, perhaps through increased NO bioavailability and EPCR, while in the presence of vascular injury or combined with risk factors, EC-MR may promote vascular thrombosis. Further studies are needed to test this hypothesis and explore potential mechanisms.
Substantial data support a role for EC-MR in vascular inflammation. In human coronary ECs, activation of MR by aldosterone increases transcription of the intercellular adhesion molecule-1 (ICAM-1) and promotes leucocyte adhesion[7]. This effect of EC-MR on ICAM-1 transcription is inhibited by estrogen activation of the ER[35]. This interaction between MR and ER in ECs may provide a mechanism for sex differences in the role of EC-MR in vascular dysfunction and inflammation. In Western diet-induced obese female mice, EC-MR deletion reduced aortic and myocardial oxidative stress, macrophage polarization to the M1 pro-inflammatory phenotype, and inflammatory cytokine levels while increasing expression of the anti-inflammatory cytokine IL-10[25*,26]. In contrast with the anti-inflammatory effects of EC-MR deletion in obese female mice, male EC-MR-KO mice were not protected from enhanced ICAM-1 expression, inflammatory cytokines, and T-cell recruitment in response to pressure overload-induced heart failure[51]. EC-MR deficiency also did not ameliorate the mineralocorticoid/salt-induced renal inflammation and kidney injury in male mice[52**]. Interestingly, obese female mice lost the protective effect of the endothelial ER-α seen in healthy females[53*]. Therefore, similar to endothelial dysfunction, there are important sex differences in the pro-inflammatory effects of EC-MR activation that require further investigation.
Conclusion
In summary, MR in EC may be vasculoprotective in normal physiology but ample data supports that in the setting of cardiovascular risk factors, including obesity, diabetes, and hypertension, EC-MR activation contributes to endothelial damage. Potential mechanisms include decreased NO bioavailability due to decreased NO production by eNOS and increased oxidative stress via eNOS uncoupling and NADPH oxidase activation and genomic regulation of ENaC and ICAM1 to increase vascular stiffness and inflammation, respectively. There are important differences in the role of EC-MR in vascular dysfunction in males compared to females, conduit versus resistance vessels, and in distinct vascular beds. The detailed molecular mechanisms are only beginning to be elucidated and thus further study is needed to determine the ideal therapeutic interventions to limit the detrimental effects of EC-MR activation in patients at risk for cardiovascular disease.
Key points.
In the healthy vasculature, EC-MR exerts a minimal or protective impact on endothelial function.
In the setting of cardiovascular risk factors, EC-MR contributes to endothelial dysfunction, vascular stiffness, vascular inflammation and thrombosis.
Mechanisms involved in EC-MR-induced vascular dysfunction include eNOS uncoupling, NADPH oxidase activation, ENaC-mediated endothelial stiffening, and ICAM-1-mediated vascular inflammation.
Differences in vascular health, sex, race, vessel size, and vascular bed might explain the controversial role of EC-MR in vascular health and disease.
Acknowledgments
none
Financial support: This work was supported by grants from the National Institute of Health (HL095590) and the American Heart Association (EIA 18290005) to IZJ and Sao Paulo Research Foundation (FAPESP 14/26192-6) to APD.
Footnotes
Disclosure: None.
Conflicts of interest: None.
References
- 1.Félétou M, Köhler R, Vanhoutte PM. Endothelium-derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets. Curr Hypertens Rep. 2010;12:267–275. doi: 10.1007/s11906-010-0118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daiber A, Steven S, Weber A, et al. Targeting vascular (endothelial) dysfunction. Br J Pharmacol. 2016 May 17; doi: 10.1111/bph.13517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lother A, Hein L. Vascular Mineralocorticoid Receptors: Linking Risk Factors, Hypertension, and Heart Disease. Hypertension. 2016;68:6–10. doi: 10.1161/HYPERTENSIONAHA.116.07418. [DOI] [PubMed] [Google Scholar]
- 4.Farquharson CA, Struthers AD. Spironolactone increases nitric oxide bioactivity, improves endothelial vasodilator dysfunction, and suppresses vascular angiotensin I/angiotensin II conversion in patients with chronic heart failure. Circulation. 2000;101:594–597. doi: 10.1161/01.cir.101.6.594. [DOI] [PubMed] [Google Scholar]
- 5.Thum T, Schmitter K, Fleissner F, et al. Impairment of endothelial progenitor cell function and vascularization capacity by aldosterone in mice and humans. Eur Heart J. 2011;32:1275–1286. doi: 10.1093/eurheartj/ehq254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **6.Garg R, Rao AD, Baimas-George M, et al. Mineralocorticoid receptor blockade improves coronary microvascular function in individuals with type 2 diabetes. Diabetes. 2015;64:236–242. doi: 10.2337/db14-0670. This clinical study suggests a role for MR as a mediator of microvascular coronary dysfunction in obese and diabetic patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caprio M, Newfell BG, la Sala A, et al. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ Res. 2008;102:1359–1367. doi: 10.1161/CIRCRESAHA.108.174235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCurley A, Jaffe IZ. Mineralocorticoid receptors in vascular function and disease. Mol Cell Endocrinol. 2012;350:256–265. doi: 10.1016/j.mce.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Briones AM, Nguyen Dinh Cat A, Callera GE, et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension. 2012;59:1069–1078. doi: 10.1161/HYPERTENSIONAHA.111.190223. [DOI] [PubMed] [Google Scholar]
- 10.Victorio JA, Clerici SP, Palacios R, et al. Spironolactone Prevents Endothelial Nitric Oxide Synthase Uncoupling and Vascular Dysfunction Induced by β-Adrenergic Overstimulation: Role of Perivascular Adipose Tissue. Hypertension. 2016;68:726–735. doi: 10.1161/HYPERTENSIONAHA.116.07911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moss ME, Jaffe IZ. Mineralocorticoid Receptors in the Pathophysiology of Vascular Inflammation and Atherosclerosis. Front Endocrinol (Lausanne) 2015;6:153. doi: 10.3389/fendo.2015.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skøtt O, Uhrenholt TR, Schjerning J, et al. Rapid actions of aldosterone in vascular health and disease--friend or foe? Pharmacol Ther. 2006;111:495–507. doi: 10.1016/j.pharmthera.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 13.Uhrenholt TR, Schjerning J, Hansen PB, et al. Rapid inhibition of vasoconstriction in renal afferent arterioles by aldosterone. Circ Res. 2003;93:1258–1266. doi: 10.1161/01.RES.0000106135.02935.E1. [DOI] [PubMed] [Google Scholar]
- 14.Nagata D, Takahashi M, Sawai K, et al. Molecular mechanism of the inhibitory effect of aldosterone on endothelial NO synthase activity. Hypertension. 2006;48:165–171. doi: 10.1161/01.HYP.0000226054.53527.bb. [DOI] [PubMed] [Google Scholar]
- 15.Leopold JA, Dam A, Maron BA, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–197. doi: 10.1038/nm1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mutoh A, Isshiki M, Fujita T. Aldosterone enhances ligand-stimulated nitric oxide production in endothelial cells. Hypertens Res. 2008;31:1811–1820. doi: 10.1291/hypres.31.1811. [DOI] [PubMed] [Google Scholar]
- 17.Nietlispach F, Julius B, Schindler R, et al. Influence of acute and chronic mineralocorticoid excess on endothelial function in healthy men. Hypertension. 2007;50:82–88. doi: 10.1161/HYPERTENSIONAHA.107.088955. [DOI] [PubMed] [Google Scholar]
- *18.Hwang MH, Yoo JK, Luttrell M, et al. Acute effect of mineralocorticoid receptor antagonism on vascular function in healthy older adults. Exp Gerontol. 2016;73:86–94. doi: 10.1016/j.exger.2015.11.017. This study supports a beneficial role for basal MR activity in endothelial function of healthy older adults without influencing blood pressure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang MH, Yoo JK, Luttrell M, et al. Mineralocorticoid receptors modulate vascular endothelial function in human obesity. Clin Sci (Lond) 2013;125:513–520. doi: 10.1042/CS20130200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **20.Chen L, Ding ML, Wu F, et al. Impaired Endothelial Repair Capacity of Early Endothelial Progenitor Cells in Hypertensive Patients With Primary Hyperaldosteronemia: Role of 5,6,7,8-Tetrahydrobiopterin Oxidation and Endothelial Nitric Oxide Synthase Uncoupling. Hypertension. 2016;67:430–439. doi: 10.1161/HYPERTENSIONAHA.115.06597. This study showed that treatment with the MR antagonist spironolactone restores the impaired endothelial repair capacity and NO production of early Endothelial Progenitor Cells in patients with primary hyperaldosteronemia. [DOI] [PubMed] [Google Scholar]
- 21.Schäfer N, Lohmann C, Winnik S, et al. Endothelial mineralocorticoid receptor activation mediates endothelial dysfunction in diet-induced obesity. Eur Heart J. 2013;34:3515–3524. doi: 10.1093/eurheartj/eht095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rickard AJ, Morgan J, Chrissobolis S, et al. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension. 2014;63:1033–1040. doi: 10.1161/HYPERTENSIONAHA.113.01803. [DOI] [PubMed] [Google Scholar]
- *23.Dinh QN, Young MJ, Evans MA, et al. Aldosterone-induced oxidative stress and inflammation in the brain are mediated by the endothelial cell mineralocorticoid receptor. Brain Res. 2016;1637:146–153. doi: 10.1016/j.brainres.2016.02.034. This is the first demonstration of a specific role for endothelial cell MR activation in mediating oxidative stress in cerebral arteries. [DOI] [PubMed] [Google Scholar]
- *24.Mueller KB, Bender SB, Hong K, et al. Endothelial mineralocorticoid receptors differentially contribute to coronary and mesenteric vascular function without modulating blood pressure. Hypertension. 2015;66:988–997. doi: 10.1161/HYPERTENSIONAHA.115.06172. This article describes the first mouse model with specific deletion of MR only in endothelial cells and shows that effects of EC-MR on vascular function depend on the presence of hypertension and differ in distinct vascular beds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *25.Jia G, Habibi J, Aroor AR, et al. Endothelial mineralocorticoid receptor mediates diet-induced aortic stiffness in females. Circ Res. 2016;118:935–943. doi: 10.1161/CIRCRESAHA.115.308269. This manuscript highlighted the importance of EC-MR-induced ENaC expression in mediating aortic stiffness in females. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jia G, Habibi J, DeMarco VG, et al. Endothelial mineralocorticoid receptor deletion prevents diet-induced cardiac diastolic dysfunction in females. Hypertension. 2015;66:1159–1167. doi: 10.1161/HYPERTENSIONAHA.115.06015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagrange J, Li Z, Fassot C, et al. Endothelial mineralocorticoid receptor activation enhances endothelial protein C receptor and decreases vascular thrombosis in mice. FASEB J. 2014;28:2062–2072. doi: 10.1096/fj.13-238188. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen Dinh Cat A, Griol-Charhbili V, Loufrani L, et al. The endothelial mineralocorticoid receptor regulates vasoconstrictor tone and blood pressure. FASEB J. 2010;24:2454–2463. doi: 10.1096/fj.09-147926. [DOI] [PubMed] [Google Scholar]
- 29.Bene NC, Alcaide P, Wortis HH, Jaffe IZ. Mineralocorticoid receptors in immune cells: emerging role in cardiovascular disease. Steroids. 2014;91:38–45. doi: 10.1016/j.steroids.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bentley-Lewis R, Adler GK, Perlstein T, et al. Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J Clin Endocrinol Metab. 2007;92:4472–4475. doi: 10.1210/jc.2007-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **31.Huby AC, Antonova G, Groenendyk J, et al. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation. 2015;132:2134–2145. doi: 10.1161/CIRCULATIONAHA.115.018226. This study identified that leptin may be one of the long sought after “adipocyte-derived aldosterone-releasing factors”. [DOI] [PubMed] [Google Scholar]
- 32.Jia G, Aroor AR, Martinez-Lemus LA, Sowers JR. Overnutrition, mTOR signaling, and cardiovascular diseases. Am J Physiol Regul Integr Comp Physiol. 2014;307:R1198–1206. doi: 10.1152/ajpregu.00262.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caroccia B, Seccia TM, Campos AG, et al. GPER-1 and estrogen receptor-β ligands modulate aldosterone synthesis. Endocrinology. 2014;155:4296–4304. doi: 10.1210/en.2014-1416. [DOI] [PubMed] [Google Scholar]
- *34.Huby AC, Otvos L, Belin de Chantemèle EJ. Leptin induces hypertension and endothelial dysfunction via aldosterone-dependent mechanisms in obese female mice. Hypertension. 2016;67:1020–1028. doi: 10.1161/HYPERTENSIONAHA.115.06642. This study highlights sex differences in the role of endothelial MR in leptin-induced endothelial dysfunction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrett Mueller K, Lu Q, Mohammad NN, et al. Estrogen receptor inhibits mineralocorticoid receptor transcriptional regulatory function. Endocrinology. 2014;155:4461–4472. doi: 10.1210/en.2014-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toda N, Nakanishi S, Tanabe S. Aldosterone affects blood flow and vascular tone regulated by endothelium-derived NO: therapeutic implications. Br J Pharmacol. 2013;168:519–533. doi: 10.1111/j.1476-5381.2012.02194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *37.Mohandas A, Suboc TB, Wang J, et al. Mineralocorticoid exposure and receptor activity modulate microvascular endothelial function in African Americans with and without hypertension. Vasc Med. 2015;20:401–408. doi: 10.1177/1358863X15584753. This study suggests enhanced responsiveness of African-American population to endothelial microvascular dysfunction in response to MR activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies JI, Band M, Morris A, Struthers AD. Spironolactone impairs endothelial function and heart rate variability in patients with type 2 diabetes. Diabetologia. 2004;47:1687–1694. doi: 10.1007/s00125-004-1510-8. [DOI] [PubMed] [Google Scholar]
- 39.Swaminathan K, Davies J, George J, et al. Spironolactone for poorly controlled hypertension in type 2 diabetes: conflicting effects on blood pressure, endothelial function, glycaemic control and hormonal profiles. Diabetologia. 2008;51:762–768. doi: 10.1007/s00125-008-0972-5. [DOI] [PubMed] [Google Scholar]
- 40.Hwang MH, Yoo JK, Luttrell M, et al. Effect of selective mineralocorticoid receptor blockade on flow-mediated dilation and insulin resistance in older adults with metabolic syndrome. Metab Syndr Relat Disord. 2015;13:356–361. doi: 10.1089/met.2015.0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sudano I, Naegele M, Roas S, et al. Vascular effects of eplerenone in coronary artery disease with preserved ejection fraction: a double-blind, randomized, placebo-controlled trial. Clin Cardiol. 2016;39:285–290. doi: 10.1002/clc.22528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeMarco VG, Habibi J, Jia G, et al. Low-Dose Mineralocorticoid Receptor Blockade Prevents Western Diet-Induced Arterial Stiffening in Female Mice. Hypertension. 2015;66:99–107. doi: 10.1161/HYPERTENSIONAHA.115.05674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silva MA, Bruder-Nascimento T, Cau SB, et al. Spironolactone treatment attenuates vascular dysfunction in type 2 diabetic mice by decreasing oxidative stress and restoring NO/GC signaling. Front Physiol. 2015;6:269. doi: 10.3389/fphys.2015.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kratz MT, Schirmer SH, Baumhäkel M, Böhm M. Improvement of endothelial function in a murine model of mild cholesterol-induced atherosclerosis by mineralocorticoid antagonism. Atherosclerosis. 2016;251:291–298. doi: 10.1016/j.atherosclerosis.2016.06.018. [DOI] [PubMed] [Google Scholar]
- 45.DuPont JJ, Hill MA, Bender SB, et al. Aldosterone and vascular mineralocorticoid receptors: regulators of ion channels beyond the kidney. Hypertension. 2014;63:632–637. doi: 10.1161/HYPERTENSIONAHA.113.01273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kusche-Vihrog K, Sobczak K, Bangel N, et al. Aldosterone and amiloride alter ENaC abundance in vascular endothelium. Pflugers Arch. 2008;455:849–857. doi: 10.1007/s00424-007-0341-0. [DOI] [PubMed] [Google Scholar]
- 47.Drüppel V, Kusche-Vihrog K, Grossmann C, et al. Long-term application of the aldosterone antagonist spironolactone prevents stiff endothelial cell syndrome. FASEB J. 2013;27:3652–3659. doi: 10.1096/fj.13-228312. [DOI] [PubMed] [Google Scholar]
- 48.Kusche-Vihrog K, Jeggle P, Oberleithner H. The role of ENaC in vascular endothelium. Pflugers Arch. 2014;466:851–859. doi: 10.1007/s00424-013-1356-3. [DOI] [PubMed] [Google Scholar]
- 49.Bodary PF, Sambaziotis C, Wickenheiser KJ, et al. Aldosterone promotes thrombosis formation after arterial injury in mice. Arterioscler Thromb Vasc Biol. 2006;26:233. doi: 10.1161/01.ATV.0000195782.07637.44. [DOI] [PubMed] [Google Scholar]
- 50.Gromotowicz A, Szemraj J, Stankiewicz A, et al. Study of the mechanisms of aldosterone prothrombotic effect in rats. J Renin Angiotensin Aldosterone Syst. 2011;12:430–439. doi: 10.1177/1470320310397405. [DOI] [PubMed] [Google Scholar]
- 51.Salvador AM, Nevers T, Velázquez F, et al. Intercellular Adhesion Molecule 1 Regulates Left Ventricular Leukocyte Infiltration, Cardiac Remodeling, and Function in Pressure Overload-Induced Heart Failure. J Am Heart Assoc. 2016;5:e003126. doi: 10.1161/JAHA.115.003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **52.Lother A, Fürst D, Bergemann S, et al. Deoxycorticosterone Acetate/Salt-Induced Cardiac But Not Renal Injury Is Mediated By Endothelial Mineralocorticoid Receptors Independently From Blood Pressure. Hypertension. 2016;67:130–138. doi: 10.1161/HYPERTENSIONAHA.115.06530. This article suggests no role for endothelial cell MR in the mineracorticoid/salt-induced kidney injury in male mice. [DOI] [PubMed] [Google Scholar]
- *53.Manrique C, Lastra G, Ramirez-Perez FI, et al. Endothelial Estrogen Receptor-α Does Not Protect Against Vascular Stiffness Induced by Western Diet in Female Mice. Endocrinology. 2016;157:1590–1600. doi: 10.1210/en.2015-1681. This study indicates loss of the protective effect in females when endothelial ER-α is deleted in obese female mice. [DOI] [PMC free article] [PubMed] [Google Scholar]