

Abstract
Sepsis is a severe and complicated syndrome symbolized by dysregulation of host inflammatory responses and organ failure with high morbidity and mortality. The literature implies that autophagy is a crucial regulator of inflammation in sepsis. Here, we report that autophagy-related protein 7 (Atg7) is involved in inflammasome activation in Pseudomonas aeruginosa abdominal infection. Following intraperitoneal challenge with P. aeruginosa, atg7fl/fl mice showed impaired pathogen clearance, decreased survival, and widespread dissemination of bacteria into the blood and lung tissue compared to wild type mice. The septic atg7fl/fl mice also exhibited elevated neutrophil infiltration and severe lung injury. Loss of Atg7 resulted in increased production of IL-1β and pyroptosis, consistent with enhanced inflammasome activation. Furthermore, we demonstrated that P. aeruginosa flagellin is a chief trigger of inflammasome activation in the sepsis model. Collectively, our results provide insight into innate immunity and inflammasome activation in sepsis.
Introduction
Sepsis, associated with systemic inflammation in response to infection (1), is a major medical problem blamed for approximately 215,000 annual deaths in United States (2, 3). This disease sequentially progresses from systemic inflammatory response syndrome (SIRS), sepsis, severe sepsis, to septic shock (4). In sepsis, inflammation is required to defeat bacterial infection when it is finely adjusted to cause minimal tissue damage (5), while severe inflammatory response leads to organ dysfunction or risk for secondary infection especially in excessive cytokine-induced cytokine storm or SIRS (6). Although sepsis has been studied intensively over the last decades, the mechanism underlying the significant pathophysiological alterations has remained elusive.
A major feature of sepsis-induced hyper-response is inflammasome dysfunction (5, 7, 8). Growing evidence reveals that inflammasomes play a critical role in Pseudomonas aeruginosa infection (9, 10). An inflammasome is an intracellular multiprotein complex that mediates the activation of caspase-1, which promotes maturation and release of proinflammatory cytokines, such as IL-1β and IL-18, and thus leading to phagocyte pyroptosis (11). In sepsis, induced pyroptosis releases a number of inflammatory factors to cause plasma-membrane rupture and release of proinflammatory intracellular contents (12). Transition from autophagy to pyroptosis is reported to be critical for progression of bacterial infection (13). The active inflammasomes, especially the non-canonical inflammasome caspase-11 with activated caspase-1, may induce pyroptosis, while autophagy appears to negatively regulate pyroptosis during P. aeruginosa infection (14, 15).
P. aeruginosa is an opportunistic Gram-negative human pathogen and responsible for a broad range of infections in individuals with cystic fibrosis, immunodeficiency, ventilation-associated pneumonia, and skin integrity loss (severe burns and trauma) (9, 16). This bacterium has become increasingly resistant to various antibiotics, thus, further understanding of host-pathogen interaction may discover novel approaches to effectively preventing and treating its infection based on harnessing host processes, such as autophagy (17). Studies have recently demonstrated an important role for autophagy-related genes (atg) in host defense against pathogens including bacteria, parasites, and viruses (17–20). Autophagy may directly participate in elimination of pathogens, help deliver bacterial antigenic materials to the innate and adaptive immune system, and facilitate lymphocyte homeostasis (21). Autophagy-related protein 7 (Atg7) is an E1-like enzyme in the two ubiquitin-like conjugation systems essential for autophagosome biogenesis and is frequently implicated in innate immunity (22). For example, in HCV (hepatitis C virus)-infected hepatocytes, knockdown of Atg7 inhibited HCV growth by activating the interferon signaling pathway and inducing apoptosis (19). Atg7 also showed an antibacterial activity in host defense against S. typhimurium infection by mediating insulin signaling-mediated pathogen resistance (23), as well as pulmonary infection of Klebsiella pneumoniae (24). Because different infection models may involve distinct host response pathways, here we used Pseudomonas intraperitoneal injection to mimic a common clinical condition (peritoneal dialysis) afflicted by Pseudomonas infection (25). We hypothesize that Atg7 may be critical for inflammatory response to bacterial infection. Using a P. aeruginosa septic model, we investigated the role and underlying mechanism of Atg7 in host-pathogen interaction. Our results suggest that Atg7 may negatively regulate inflammasome activation as its deficiency resulted in uncontrolled activation of inflammasome in P. aeruginosa sepsis.
Materials and Methods
Mice
ER-Cre:atg7 fl/fl mice (thereafter atg7 fl/fl) were originally generated by Dr. Masaaki Komatsu at Tokyo Metropolitan Institute of Medical Science and our deficiency mice are a gift from Drs. Youwen He at Duke University. To generate atg7 fl/fl mice, exon 14 encoded regions were disrupted and the mice were bred with estrogen receptor cre mice. atg7 fl/fl mice and wild-type C57BL/6J mice (presumably expressing normal or WT levels of Atg7 in the lung) were bred in the same facility and injected with 0.1 mg/kg of tamoxifen (Sigma, St. Louis, MO) daily for 5 days prior to infection experiments to conditionally delete the target gene (26). Age- and sex-matched normal C57BL/6J mice were used as wild-type controls as the KO mice were based on C57BL/6J genetic background (after 7 generation backcrosses). Tamoxifen injection did not cause significant disturbance in the measured infection parameters (bacterial burdens, tissue damage, inflammation, and cytokine levels) in comparison with untreated controls. Animal care and experimental procedures were approved by the University of North Dakota institutional animal care and use committee (IACUC).
Cells
Alveolar macrophages (AMs) were isolated from bronchoalveolar lavage (BAL) (27, 28). MH-S cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). Bone marrow-derived macrophages (BMDM) were isolated from the femur of C57BL/6J wild-type (WT) mice and atg7fl/fl mice after challenged with tamoxifen (29), and cultured in DMEM+10% FBS+10 ng/ml GM-CSF (R&D systems, Minneapolis, MN, without tamoxifen) (12). AMs were isolated by BAL and maintained in RPMI 1640 (Invitrogen, Grand Island, NY) supplemented with 1% penicillin streptomycin glutamine and 10% fetal bovine serum (FBS).
Bacterial preparation and infection experiments
P. aeruginosa WT strain, PAO1 was kindly provided by Dr. S. Lory (Harvard Medical School). PA-Xen41 expressing bioluminescence was bought from Caliper Company (PerkinElmer, Waltham, MA). The fliC mutant of PAK strain was a gift from G. Pier (Channing Laboratory, Harvard Medical School, Boston, MA). PAO1 were grown for about 16 h in LB broth at 37°C with shaking (220 rpm). OD595 of the bacterial suspension was measured with a spectrophotometer and the bacteria were enumerated using 1 OD unit = 1×109 bacteria. The bacteria were centrifuged at 5,000g for 3 min and washed with PBS. Then mice were infected intraperitoneally with 5×106 colony forming units (CFU) of PAO1 after anesthetized by ketamine (45 mg/Kg). Mice were monitored for symptoms and euthanized when moribund to generate survival curves using Kaplan-Meier methods (30) and perform other assessments.
In vivo imaging
Mice were infected intraperitoneally with 1×107 CFU of PA-Xen41, at various time points after infection, the whole body of mice was imaged using IVIS XRII spectrum imaging system (PerkinElmer-Caliper) (31).
Inflammatory cell evaluation
Polymononuclear neutrophil (PMN) infiltration was measured by a Hema staining kit (Thermofisher Scientific, Waltham, MA) (32); and myeloperoxidase (MPO) activity in lungs was measured as previously described (30,33,34). Confocal microscopy for evaluating immune-stained cells was performed as previously described (31, 35).
Histological and immunohistochemistry analysis
Using H&E staining, tissue morphological damage was examined (30, 31). Lung samples underwent terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining with the DeadEnd™ Fluorometric TUNEL System (Promega kit).
Protein transfection
Flagellin (from P. aeruginosa) was purchased from InvivoGen (San Diego, CA). BMDMs were prestimulated with LPS (P. aeruginosa serotype 10, Sigma, 50 ng/ml) for 5 hours, then transfected with PAO1 flagellin by Profect P1 (EI Cajon, CA) for the indicated time points(36).
Cytotoxicity and cytokines
Cytotoxicity was measured with LDH cytotoxicity assay kit (Thermofisher Scientific). Cytokine concentrations were assessed by ELISA kits (eBioscience Company, San Diego, CA) (37).
RNA interference and labeling LC3
Cells were transfected with 50 nM siRNAs using LipofectAmine 2000 (Invitrogen), following manufacturer’s instruction. The siRNAs: ATG7 siRNA, sc-41448(m), ATG5 siRNA, sc-41446(m) and AMBRA1 (Beclin-1) siRNA, sc-141039 (m) were purchased from Santa Cruz Biotechnology. AM cells were translated with RFP-LC3 plasmid using LipofectAmine 2000 reagent following the manufacturer’s instructions (38).
Immunoblotting
Cells were lysed with T-PER Tissue Protein Extraction Reagent, and all lysates were quantified by Bio-Rad protein assay. The samples were separated by electrophoresis on 12% SDS-PAGE gels and transferred to PVDF transfer membranes. Proteins were detected by immunoblotting using primary Abs (all Abs from Santa Cruz Biotechnology) at a concentration of 1/1000 and were incubated overnight. Specific interaction was detected using corresponding secondary Abs, and detected using ECL reagents (33).
Confocal immunofluorescence imaging
MH-S cells were cultured onto glass cover slides in 24-well dishes. After washed 3 times by PBS, fixed with 4% paraformaldehyde (PFA) and permeabilized with 0.1% Triton X-100 for 15 min and blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature. Slides were incubated with ASC and caspase-1 antibodies (1:250 in 1% BSA in PBS) overnight at 4°C. The cells were stained with Alexa594-conjugated, FITC and Rhodamine red secondary antibody (molecular probes, diluted 1:1000) for 60 min at room temperature, Slides were visualized with Zeiss confocal microscope.
Statistical analysis
Data are presented as mean±SEM of the indicated times of experiments. Statistical analysis was done by Student t tests when appropriate (Data are normally distributed). A p value of < 0.05 was considered significant. Statistical analysis was performed using GraphPad Prism 6 (GraphPad Software).
Results
atg7fl/fl mice are susceptible to P. aeruginosa infection
To investigate whether Atg7 impacts immunity to P. aeruginosa infection, we challenged normal C57BL6J WT mice and ER-Cre:atg7 fl/fl mice (short as atg7 fl/fl) mice (both received tamoxifen injection) with PA-Xen 41 by i.p. injection (1×107 CFU per mouse) to investigate bacterial dissemination during infection. Results showed that atg7fl/fl mice exhibited wider dissemination of bioluminescence in the body than WT mice using an IVIS XRII imaging system (Figure 1A). We also examined survival using WT and atg7fl/fl mice after injecting 5×106 CFU PAO1. In additional control assays, ER-Cre:atg7 fl/fl mice that did not receive tamoxifen injection showed similar phenotype as normal control C57BL6J WT mice (bacterial burdens and survival). We observed that 25% of the atg7fl/fl mice survived compared to 75% of WT mice at 6 d, indicating that Atg7 may be required for host defense against P. aeruginosa infection in our sepsis model.
Figure 1.
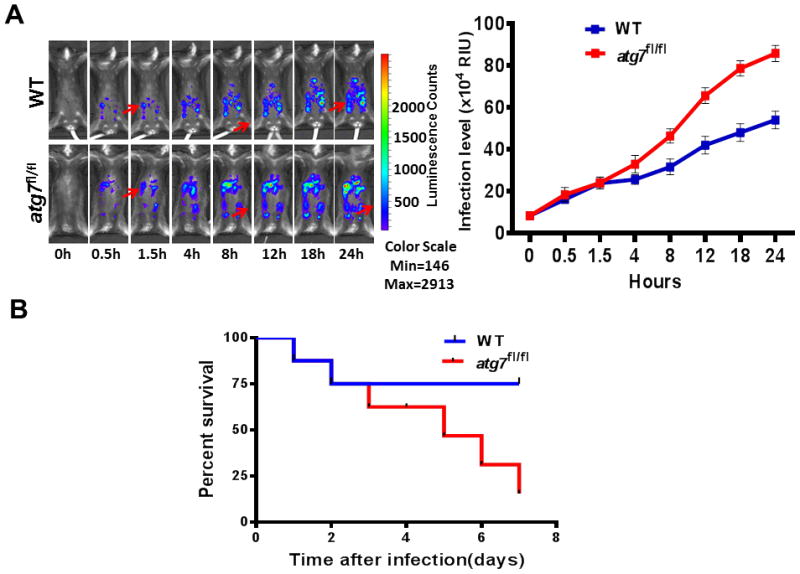
Infection progression of WT mice and atg7fl/fl mice after P. aeruginosa challenge. (A) Whole body imaging of infected WT mice and atg7fl/fl mice after i.p. injection of 1×107 CFU PA-Xen41, respectively. Representative images of WT and atg7fl/fl mice (n=8) at different time points using IVIS XRII imaging (arrows indicating spread of infection). Data are representative of three experiments. (B) WT mice and atg7fl/fl mice were infected by i.p. injection of 5×106 CFU of PAO1, survival was represented by Kaplan-Meier curves. (*P <0.05, n=8, log-rank test).
Atg7 deficiency aggravates bacterial dissemination, PMN penetration, and lung injury in P. aeruginosa sepsis
Bacterial dissemination is an important pathogenesis feature of sepsis syndrome (39). At 24 h post bacterial infection (psi), we evaluated the number of live bacteria in the blood of WT mice and atg7fl/fl mice, respectively. Atg7 loss resulted in profoundly elevated bacteremia (Figure 2A), and based on the hematogenous spread, bacteria were significantly disseminated to the lung tissue and BAL fluid (BALF) of septic atg7fl/fl mice (Figure 2B–C). Blood and BALF were Hema stained at the indicated time points to evaluate polymorphonuclear neutrophil (PMN) infiltration, which was increased in septic atg7fl/fl mice (Figure 2D–E). To corroborate the PMN filtration with subsequent tissue injury, we examined myeloperoxidase (MPO) activity as another measure of PMN presence in the lungs of septic WT and atg7fl/fl mice, which was significantly increased in septic atg7fl/fl mice at both 12 and 24 h psi (Figure 2F). We also assessed the extent of lung injury by H&E staining and found more severe histological alterations, indicating exacerbated lung injury in septic atg7fl/fl mice (Figure 2G). Thus, these data collectively demonstrated that depletion of Atg7 resulted in increased bacterial dissemination, PMN penetration, and lung injury in P. aeruginosa-induced sepsis.
Figure 2.
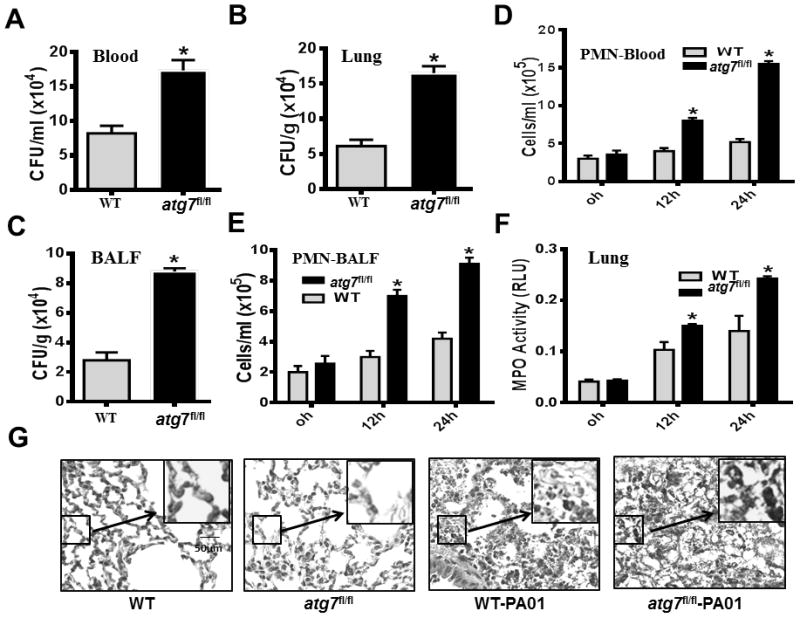
Disruption of Atg7 leads to severe bacterial dissemination, PMN penetration and oxidative lung injury in P. aeruginosa-induced sepsis. (A–C) Twenty-four hours after the onset of i.p. PAO1, blood, lung tissues and bronchoalveolar lavage fluid (BALF) of WT mice and atg7fl/fl mice were collected to detect bacterial dissemination by CFU assay. (*P<0.05; n=5, T tests.) (D–E) At 0, 12 and 24 hours after i.p. injection of PAO1, PMN infiltration levels in the blood and BALF were measured by Hema staining. (*P<0.05; n=5, T tests.) (F) MPO activity in lungs following i.p. PAO1 challenge in WT mice and atg7fl/fl mice. At 0, 12 and 24 hours after i.p. infection with PAO1, lung tissues were collected to determine MPO activity. (*P<0.05; n=5, T tests.) (G) Representative histological views of lungs of WT mice and atg7fl/fl mice after 24 hour i.p. infection with PAO1 by H&E staining (Inset shows the enlarged view). Data are shown as mean ± SEM of three independent experiments (*P<0.05; n=3 mice; T tests.)
Loss of Atg7 results in hampered autophagosome formation and increased IL-1β production upon P. aeruginosa infection
Autophagy is a conserved cellular mechanism and may play a role in host defense during bacterial infection (40). To explore this notion, we quantified the number of autophagosomes in individual cells by counting LC3 puncta. We found that atg7fl/fl MHS cells manifested reduced autophagosomes compared to WT cells (Figure 3A), indicating that Atg7 deficiency inhibits autophagosome formation. Given the importance of autophagy in interplay with inflammatory cytokines in regulating inflammatory responses, we intraperitoneally infected WT mice and atg7fl/fl mice with PAO1, and assessed inflammatory cytokines in BALF and blood. In septic atg7fl/fl mice, IL-1β levels in BALF and blood were significantly elevated compared to those of septic WT mice (Figure 3B–C). However, other proinflammatory cytokines (i.e., TNF-α and IL-6) were not apparently altered in the BALF (Figure 3D–E). In vitro data again showed significantly elevated IL-1β in Atg7 knockdown MH-S cells, while TNF-α showed no obvious change (Figure 3F–G), indicating that Atg7 deficiency specifically impacts IL-1β production upon P. aeruginosa infection.
Figure 3.
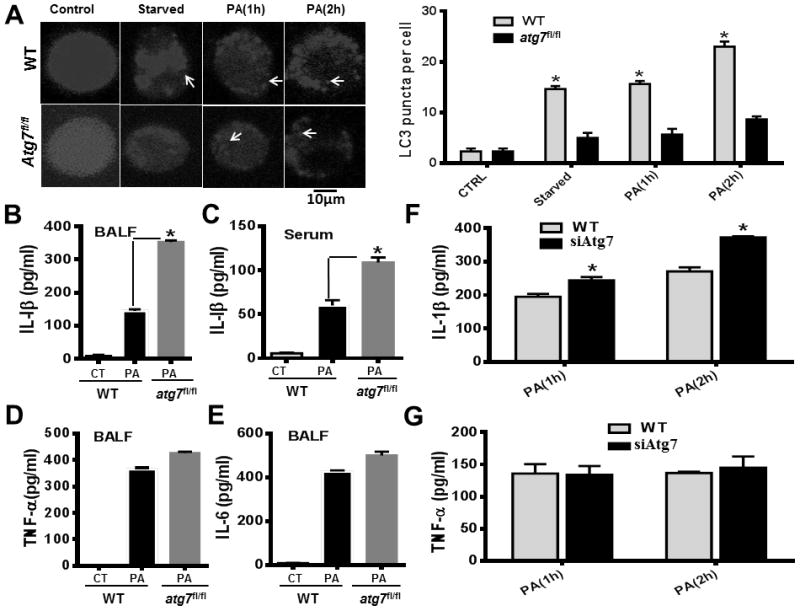
Hampered autophagosome formation and increased IL-1β in Atg7-deficient macrophages and septic atg7fl/fl mice (A) AM cells were transfected with RFP-LC3 plasmid for 24 hours. Then the cells were infected with PAO1 at MOI of 10:1 for the indicated time periods. The number of LC3 puncta (arrows) within each cell was determined by confocal laser scanning microscopy. Arrows show RFP-LC3 punctation. WT mice and atg7fl/fl mice (*P<0.05; n=5, T tests.) were infected with 5×106 CFU of PAO1 for 24 hours. (B) IL-1β (eBioscience, Cat #, 88-7013) production in BALF of WT mice and atg7fl/fl mice. (C) IL-1β levels in blood of WT mice and atg7fl/fl mice. (D) TNF-α (88-7324) concentrations in BALF of WT mice and atg7fl/fl mice. (E) IL-6 (88-7064) levels in BALF of WT mice and atg7fl/fl mice. (*P<0.05; n=5, T tests.). (F) IL-1β (88-7013) concentrations in siRNA-transfected and WT MH-S cells at different times. (G) TNF-α (88-7324) concentrations in siRNA-transfected and WT MH-S cells at different times. Cytokines were measured by ELISA. Data are representative of three independent experiments (*P<0.05; n=5, T tests.)
Atg7 deficiency increases inflammasome activation upon P. aeruginosa infection
Hypersecretion of IL-1β is a typical marker of inflammasome activation (41). We examined the effect of Atg7 knock-out on inflammasome activation by PAO1 infection using primary mouse BMDMs. Since IL-1β precursor is induced by activation of the NF-κB pathway in response to the stimuli of pathogen-associated molecular patterns (PAMPs) (42), we pre-stimulated BMDMs with LPS for 5 hours with different doses, and infected the cells with PAO1 for another 4 hours. In Atg7 knock-out conditions, we found that inflammasome activation was markedly increased as shown by hypersecretion of the active forms of both IL-1β and caspase-1 by western blotting (Figure 4A). Concordant with these observations, IL-1β processing was increased under Atg7 knock-out conditions upon P. aeruginosa infection by ELISA assay (Figure 4B), but TNF-α and IL-6 were not (Figure 4C–D). We further knocked down Atg5 or Beclin1 with siRNA silencing in BMDM, and examined the production of caspase-1 and IL-1β (Figure S1). We also observed that reduction in autophagy due to Atg7 knock-out induces inflammasome hyperactivation, which was in agreement with other reports from defects of critical autophagy factor Atg5 and Atg16L1 (43, 44).
FIGURE 4.
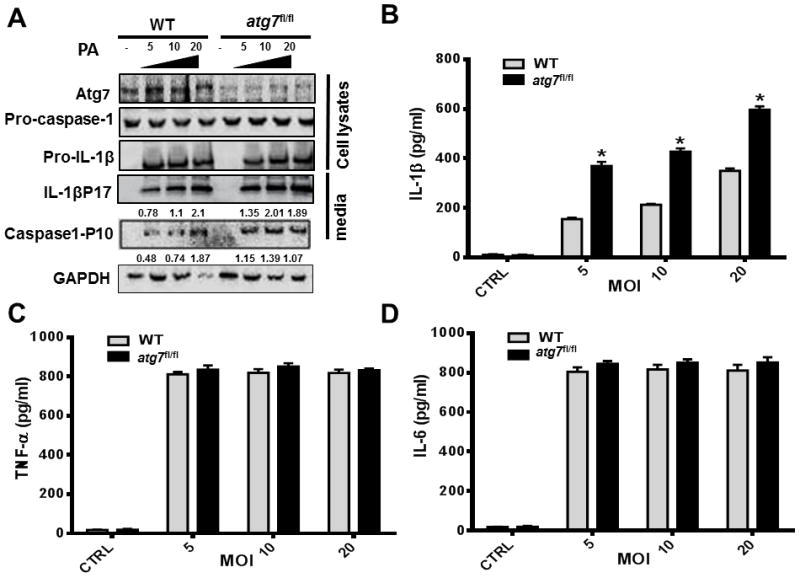
Atg7 deficiency hyperactivates inflammasome in macrophages upon P. aeruginosa infection. (A–B) Bone marrow-derived macrophages (BMDMs) were isolated from atg7fl/fl mice and C57BL/6J WT mice, then pre-stimulated with LPS for 5 hours. (A) Cells were infected with PAO1 at different MOIs. 4 hours later, cell lysates and cell culture media were harvested for western blot analysis. Atg7, pro-caspase-1, pro-IL-1, caspase-1 (P10), and IL-1β (P17) were evaluated by Western blotting. Densitometric analysis of caspase-1 (P10), and IL-1β (P17) by quantity one (B–D). Cell culture media were also assayed for IL-1β, TNF-α, IL-6 concentration by ELISA. (*P<0.05; n=3, T tests.)
Atg7 deficiency increases pyroptosis in macrophages upon P. aeruginosa infection
Hyperactivation of IL-1β signaling is associated with pyroptosis (41). Herein, we isolated AMs from WT and atg7fl/fl mice, and infected them with PAO1 at different MOI. Cell death was determined by lactate dehydrogenase (LDH) release (Figure 5A). In addition, to identify cell death relating to Atg7, we tested cell survival after infected with PAO1 and found that the Atg7 deficiency cells have lower viability (Figure 5B). To probe the cell death type, we performed western blotting and detected increased ASC and IL-18 in the lung of atg7fl/fl mice, indicating pyroptosis occurrence in the cells (Figure 5C). To verify the pyroptotic cell death occurred in MH-S cells, we examined caspase-1 expression and ASC oligomerization in response to LPS/ATP stimulation and P. aeruginosa infection by immunofluorescence. Both caspase-1 specks and ASC pyroptosomes were low in unstimulated controls, but were induced over time by LPS/ATP stimulation and P. aeruginosa infection in Atg7 siRNA-transfected MH-S cells (Figure 5D). These data showed that Atg7 deficiency increases pyroptosis in macrophages upon P. aeruginosa infection.
FIGURE 5.
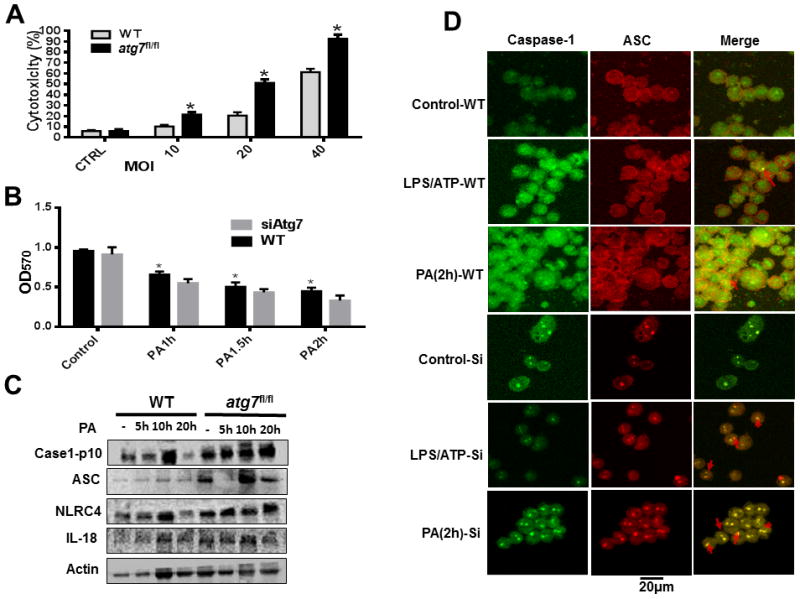
Exacerbated P. aeruginosa-induced pyroptosis in AM cells from atg7fl/fl mice and siRNA-transfected MH-S cell. (A) Alveolar macrophages of WT mice and atg7fl/fl mice were infected with PAO1 at indicated MOI for 4 hours, cytotoxicity was determined by LDH release. (*P<0.05; n=3, T tests.) (B) MTT assay of MHS cells infected by PAO1. (*P<0.05; n=10, T tests.) (C) WT and Atg7 deficiency mouse lungs were tested by western blotting after infected for 5h, 10h and 20h, respectively. Caspase-1, IL-18, NLRC4, and ASC were evaluated. (D) WT and Atg7 siRNA-transfected MH-S cells were infected with PAO1 (MOI=10:1) for 2 h or stimulated for 5 h with or without LPS (500 ng/ml) and ATP (5 mmol). Cells were stained with FITC and Rhodamine secondary antibodies, caspase-1 speck and ASC cells were analyzed by immunofluorescence. Data are representative of three independent experiments. (*P<0.05; n=3, T tests.)
Flagellin is a potential mediator of inflammasome hyperactivation in Atg7 deficiency by P. aeruginosa infection
To establish the mechanistic relationship between IL-1β production and pyroptosis in Atg7 depleted condition, we studied inflammasome activation phenotypes in AMs. We noted that in the absence of Atg7, there was an increase in the activation of the NLRC4 inflammasome following PAO1 infection with increased cleaved-IL-1β and caspase1P10 (Figure 6A). Inflammasome activation is critical for sepsis, and is associated with various forms of PAMPs potent for activating inflammasome (45). Flagellin, a conserved PAMP, has been shown to be the major stimulus for IL-1β production in acute P. aeruginosa pneumonia (46). To clarify whether flagellin is indeed involved in the inflammasome hyperactivation under Atg7-deficiency condition, BMDMs were transfected with PAO1 flagellin by Profect-P1. Atg7 depletion significantly elevated the expression of IL-1β and caspase-1 as determined by western blotting (Figure 6B). Similar effects were observed by ELISA assay (Figure 6C). However, TNF-α and IL-6 expression showed no apparent changes after Atg7 depletion (Figure 6D–E). We also performed H&E staining to assess the extent of lung injury and the results showed no significant histological alterations (Figure S2). Taken together, these findings suggest that Atg7 plays an essential role in the downregulation of inflammasome activation, and that flagellin may be a potential mediator of inflammasome hyperactivation in Atg7 deficient condition under P. aeruginosa infection.
Figure 6.
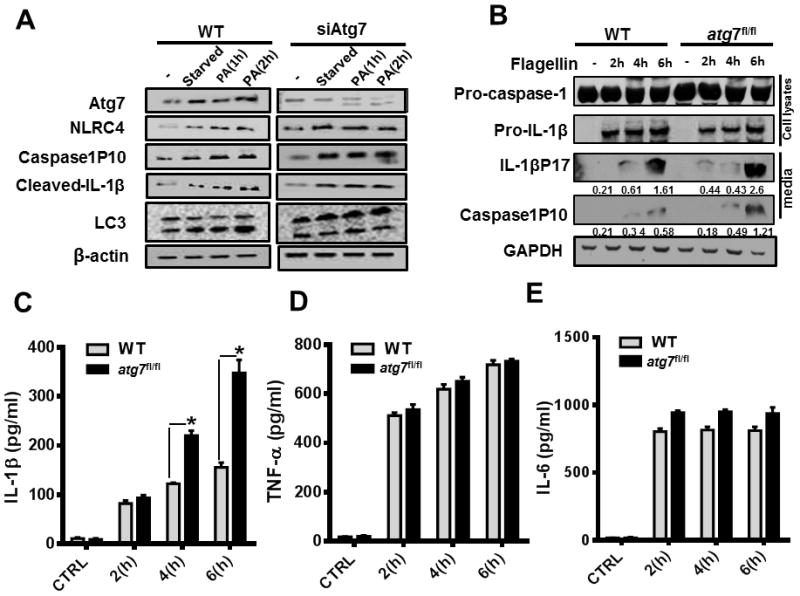
Flagellin mediates inflammasome hyperactivation in Atg7 deficient condition by P. aeruginosa infection. (A) MH-S and Atg7 siRNA-transfected MH-S cells were infected with PAO1 at different times (MOI=10:1). Western blotting of Atg7, caspase1P10, NLRC4, cleaved IL-1β, LC3 and β-actin was performed with no infection and 2 hour –starved controls. (B–C) Cells were transfected with PAO1 flagellin by Profect-P1 for indicated time points. Atg7, pro-caspase-1, pro-IL-1β, caspase-1 (P10), and IL-1β (P17) were evaluated by Western blotting. Densitometric analysis of caspase-1 (P10), and IL-1β (P17) by quantity one (B–D). Cell culture media were also assayed for IL-1β concentration by ELISA (bottom panel). (D–E) Cells were transfected with PAO1 flagellin by Profect P1 for the indicated time points. TNF-α and IL-6 levels in culture media were analyzed by ELISA. Data are shown as mean ± SEM of three independent experiments (*P<0.05; n=3, T tests.)
Discussion
This study demonstrates that Atg7 deficiency profoundly impacts the progression of P. aeruginosa-induced sepsis. Atg7-deficient septic mice exhibited exacerbated bacterial dissemination and decreased survival, along with severe lung injury and enhanced inflammatory response. Furthermore, the autophagy-deficient setting appears to aggravate P. aeruginosa or flagellin-mediated hyperactivation of macrophage inflammasome. These results presented in this study offer insight into Atg7 functional relevance to host defense and disease pathogenesis in bacterial sepsis. We analyzed the involvement of flagellin in initiating inflammasome activation in Atg7 deficiency. However, we cannot exclude the possibility that other bacterial structural and virulence factors including type 3 secretion systems (T3SS), which may participate in the complex pathogenesis responsible for inflammation (47–49). A recent report demonstrated that lipopolysaccharide (LPS) was delivered from gram-negative bacteria by outer membrane vesicles to the host cell cytosol and triggered caspase-11 activation (50). Thus, LPS may be another factor for inflammasome activation on P. aeruginosa-induced sepsis progression.
Sepsis is a complex disease involving inflammatory response that occurs during severe infection (6). Our understanding of autophagy relevance to sepsis indicates the critical role of Atg7 protein. Previous studies have shown that autophagy not only plays specific roles in functioning in a cell-autonomous manner to degrade intracellular pathogens (40), but also helps orchestrate the systemic immune response by serving as a regulator of innate immunity, adaptive immunity, and inflammation (51). In vitro study shows that silencing of autophagy-related genes leads to heavier bacterial burdens in macrophages (17, 23); and deletion of atg genes in plant and drosophila results in increased viral replication and increased mortality or more severe pathological phenotypes (52). Our in vivo data provide support for the hypothesis that in P. aeruginosa-induced sepsis progression, deficiency in Atg7 exacerbated infection outcomes. We found that Atg7-deficiency septic mice exhibited stronger dissemination of bacterial infection in the body and lower survival than WT septic mice. These observations seem to be consistent with previous studies (53). Furthermore, knockdown of Atg7 increased tissue injury and inflammatory responses, particularly IL-1β and pyroptosis by inflammasome activation. In addition, increasing studies explored the immune crosstalk between autophagy and inflammasome pathways in bacterial infection (54, 55), particularly the P. aeruginosa-induced sepsis progression. Extending the first report of P. aeruginosa-induced autophagy in alveolar cells, we have previously described the regulatory role of Atg7 in inflammatory response in Klebsiella pneumoniae pulmonary infections (24). We also found that Atg7 can augment immunity against P. aeruginosa in intranasal infection through activation of nitric oxide pathway (26) and enhancement of phagocytosis maturation (56). Note our previous studies were based on pulmonary models, different from the current sepsis model, which appears to be regulated through distinct cellular pathways following the same bacterial infection. Although intraperitoneal infection was used, surprisingly significant pulmonary infection (pneumonia) and lung injury were apparently induced. More recently, we observed that proinflammatory cytokine IL-17A(57) is related to P. aeruginosa-induced infection and inflammation. These studies suggest that infection-induced inflammatory response regulation is extremely important for infection control and the mechanism of the inflammatory regulation is very complex. Finally, Atg-7-mediated autophagy mechanisms involve many other diseases including acute liver injury (58, 59).
P. aeruginosa is a ubiquitous environmental bacterium and is amongst the top three causes of opportunistic human infections (60). The role of inflammasome pathways in P. aeruginosa pathogenesis is rather complex. In previous studies, IL-1 receptor type 1 gene-deficient (il-1r−/−) mice that were intranasally infected with P. aeruginosa showed attenuated outgrowth of bacteria in lungs, and exhibited decreased influx of neutrophils in bronchoalveolar lavage fluids (26). Similarly, inflammatory responses in the lung were lower in il-1r−/− mice than those in WT mice. Furthermore, treatment of WT mice with IL-1R antibodies also diminished outgrowth of P. aeruginosa when compared to WT mice (61). In addition, other studies have indicated involvement of the caspase-1/inflammasome signaling pathway in P. aeruginosa infection (62, 63). These studies demonstrated that P. aeruginosa infection activates caspase-1 in macrophages in an IPAF (NLRC4)-dependent manner (64). Strategies that block inflammasome activation, such as deletion of IPAF, reduction of IL-1β production, and inhibition of caspase-1 lead to enhanced bacterial clearance and alleviated pathology in P. aeruginosa pneumonia mice (9). These reports also suggest that some conserved PAMPs triggered inflammasome signaling; and as an example, flagellin, the basic structure protein that enables bacterial mobility, is a key stimulus for IL-1β production (9).
Recently, AIM2 (absent in melanoma 2) and NLRP3 (NLR family pyrin domain containing 3) inflammasomes are also reported to be activated during P. aeruginosa infection in Atg7 siRNA knockdown conditions (64, 65). These papers together with our study emphasize that multiple types of inflammasomes with diverse functions can be activated during P. aeruginosa infection. NLRC3 is recently shown in inhibiting tumorigenesis by suppressing mTOR and growth factor receptors (66), raising a possibility that some signaling regulators inhibit autophagy while others enhance it to tightly regulate inflammatory responses. The interplay between inflammasomes and autophagy varies greatly with pathogens, infection conditions, time points, host cells, animal models, etc. Hence, Atg7 deletion may contribute to elevated inflammatory responses and the underlying molecular mechanism in bacterial infection and sepsis progression merits further studies.
Autophagy has been considered crucial for inflammasome signaling. Following LPS stimulation, deficiency in Atg16L1 led to increased amounts of inflammatory cytokines (IL-1β and IL-18). Mice lacking Atg16L1 are highly susceptible to dextran sulphate sodium-induced acute colitis, which can be alleviated by injection of anti-IL-1β and IL-18 antibodies (44). Other groups have also reported that in atherosclerotic progression, defective autophagy is associated with proatherogenic inflammasome activation and autophagy dysfunction promotes atherosclerosis in part through inflammasome hyperactivation (43). Likewise in our present study, we observed overproduction of IL-1β and intensified pyroptosis in septic Atg7-deficient mice compared to septic WT mice. Particularly, transfection of flagellin into Atg7-deficient macrophages also led to inflammasome hyperactivation, suggesting that autophagy can regulate host immune response to flagellin through some unknown mechanisms. This study mostly focuses on the role of immune cells (AM and BMDM) in inflammasome activation while other cell types, such as epithelial cells, may also participate in this inflammatory process, which can be a future topic.
In summary, our data have characterized an important role of Atg7 in host defense against P. aeruginosa-induced sepsis in mice, revealing the crosstalk between autophagy and inflammasome signaling. Given the importance of inflammasome hyperactivation caused by Atg7 deficiency in the progression of P. aeruginosa sepsis, further mechanistic studies are warranted to investigate the molecular mechanism of autophagy dysfunction in inflammasome hyperactivation.
Supplementary Material
Acknowledgments
Funding: This project was supported by Flight Attendant Medical Research Institute (FAMRI, Grant #103007), NIH AI101973-01, AI109317-01A1, and AI097532-01A1 to MW and the University of North Dakota Core Facilities were supported by NIH grants INBRE P20GM103442, COBRE P30GM103329, and COBRE P20GM113123)
The authors thank Sarah Abrahamson and Steven Adkins of university of North Dakota imaging core for using microscopy and Flow Cytometry Core for measuring lymphocyte populations, respectively.
Footnotes
AUTHOR CONTRIBUTIONS
Q. P., C. G., and M. W. designed research and wrote the manuscript. C.G., Q. P., R. L., Y. L., S. T., and X. L. performed experiments. Y. W., L. L., X. D., H. L., F. M., and X. J. provided regents and suggestions.
Conflict of interest: none declared.
References
- 1.Bone RC, Sibbald WJ, Sprung CL. The ACCP-SCCM consensus conference on sepsis and organ failure. Chest. 1992;101:1481–1483. doi: 10.1378/chest.101.6.1481. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 4.Stubljar D, Skvarc M. Effective Strategies for Diagnosis of Systemic Inflammatory Response Syndrome (SIRS) due to Bacterial Infection in Surgical Patients. Infect Disord Drug Targets. 2015;15:53–56. doi: 10.2174/1871526515666150320161804. [DOI] [PubMed] [Google Scholar]
- 5.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 6.Cinel I, Dellinger RP. Advances in pathogenesis and management of sepsis. Curr Opin Infect Dis. 2007;20:345–352. doi: 10.1097/QCO.0b013e32818be70a. [DOI] [PubMed] [Google Scholar]
- 7.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 8.Bauernfeind F, Ablasser A, Bartok E, Kim S, Schmid-Burgk J, Cavlar T, Hornung V. Inflammasomes: current understanding and open questions. Cell Mol Life Sci. 2011;68:765–783. doi: 10.1007/s00018-010-0567-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest. 2013;123:1630–1637. doi: 10.1172/JCI66142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci USA. 2008;105:2562–2567. doi: 10.1073/pnas.0712183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vince JE, Silke J. The intersection of cell death and inflammasome activation. Cell Mol Life Sci. 2016 doi: 10.1007/s00018-016-2205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8:1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 13.Swanson MS, Molofsky AB. Autophagy and inflammatory cell death, partners of innate immunity. Autophagy. 2005;1:174–176. doi: 10.4161/auto.1.3.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki T, Nunez G. A role for Nod-like receptors in autophagy induced by Shigella infection. Autophagy. 2008;4:73–75. doi: 10.4161/auto.5101. [DOI] [PubMed] [Google Scholar]
- 15.Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49:331–338. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 16.Walter S, Gudowius P, Bosshammer J, Romling U, Weissbrodt H, Schurmann W, von der Hardt H, Tummler B. Epidemiology of chronic Pseudomonas aeruginosa infections in the airways of lung transplant recipients with cystic fibrosis. Thorax. 1997;52:318–321. doi: 10.1136/thx.52.4.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan K, Huang C, Fox J, Laturnus D, Carlson E, Zhang B, Yin Q, Gao H, Wu M. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci. 2012;125:507–515. doi: 10.1242/jcs.094573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cervantes S, Bunnik EM, Saraf A, Conner CM, Escalante A, Sardiu ME, Ponts N, Prudhomme J, Florens L, Le Roch KG. The multifunctional autophagy pathway in the human malaria parasite, Plasmodium falciparum. Autophagy. 2014;10:80–92. doi: 10.4161/auto.26743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology. 2011;53:406–414. doi: 10.1002/hep.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Gan CP, Zhang S, Zhou XK, Li XF, Wei YQ, Yang JL, Wu M. FIP200 is involved in murine pseudomonas infection by regulating HMGB1 intracellular translocation. Cell Physiol Biochem. 2014;33:1733–1744. doi: 10.1159/000362954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol. 2004;15:231–236. doi: 10.1016/j.semcdb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Jia K, Thomas C, Akbar M, Sun Q, Adams-Huet B, Gilpin C, Levine B. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc Natl Acad Sci USA. 2009;106:14564–14569. doi: 10.1073/pnas.0813319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye Y, Li X, Wang W, Ouedraogo KC, Li Y, Gan C, Tan S, Zhou X, Wu M. Atg7 deficiency impairs host defense against Klebsiella pneumoniae by impacting bacterial clearance, survival and inflammatory responses in mice. Am J Physiol Lung Cell Mol Physiol. 2014;307:L355–363. doi: 10.1152/ajplung.00046.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taber TE, Hegeman TF, York SM, Kinney RA, Webb DH. Treatment of Pseudomonas infections in peritoneal dialysis patients. Perit Dial Int. 1991;11:213–216. [PubMed] [Google Scholar]
- 26.Li X, Ye Y, Zhou X, Huang C, Wu M. Atg7 enhances host defense against infection via downregulation of superoxide but upregulation of nitric oxide. J Immunol. 2015;194:1112–1121. doi: 10.4049/jimmunol.1401958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu M, Hussain S, He YH, Pasula R, Smith PA, Martin WJ., 2nd Genetically engineered macrophages expressing IFN-gamma restore alveolar immune function in scid mice. Proc Natl Acad Sci USA. 2001;98:14589–14594. doi: 10.1073/pnas.251451498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wisniowski PE, Spech RW, Wu M, Doyle NA, Pasula R, Martin WJ., 2nd Vitronectin protects alveolar macrophages from silica toxicity. Am J Respir Crit Care Med. 2000;162:733–739. doi: 10.1164/ajrccm.162.2.9808015. [DOI] [PubMed] [Google Scholar]
- 29.Daubeuf F, Frossard N. Performing Bronchoalveolar Lavage in the Mouse. Curr Protoc Mouse Biol. 2012;2:167–175. doi: 10.1002/9780470942390.mo110201. [DOI] [PubMed] [Google Scholar]
- 30.Zhou X, Li X, Ye Y, Zhao K, Zhuang Y, Li Y, Wei Y, Wu M. MicroRNA-302b augments host defense to bacteria by regulating inflammatory responses via feedback to TLR/IRAK4 circuits. Nat Commun. 2014;5:3619. doi: 10.1038/ncomms4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Zhou X, Ye Y, Li Y, Li J, Privratsky B, Wu E, Gao H, Huang C, Wu M. Lyn regulates inflammatory responses in Klebsiella pneumoniae infection via the p38/NF-kappaB pathway. Eur J Immunol. 2014;44:763–773. doi: 10.1002/eji.201343972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li R, Tan S, Yu M, Jundt MC, Zhang S, Wu M. Annexin A2 Regulates Autophagy in Pseudomonas aeruginosa Infection through the Akt1-mTOR-ULK1/2 Signaling Pathway. J Immunol. 2015;195:3901–3911. doi: 10.4049/jimmunol.1500967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li G, Fox J, 3rd, Liu Z, Liu J, Gao GF, Jin Y, Gao H, Wu M. Lyn mitigates mouse airway remodeling by downregulating the TGF-beta3 isoform in house dust mite models. J Immunol. 2013;191:5359–5370. doi: 10.4049/jimmunol.1301596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan K, Huang C, Fox J, Gaid M, Weaver A, Li G, Singh BB, Gao H, Wu M. Elevated inflammatory response in caveolin-1-deficient mice with Pseudomonas aeruginosa infection is mediated by STAT3 protein and nuclear factor kappaB (NF-kappaB) J Biol Chem. 2011;286:21814–21825. doi: 10.1074/jbc.M111.237628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kannan S, Audet A, Huang H, Chen LJ, Wu M. Cholesterol-rich membrane rafts and Lyn are involved in phagocytosis during Pseudomonas aeruginosa infection. J Immunol. 2008;180:2396–2408. doi: 10.4049/jimmunol.180.4.2396. [DOI] [PubMed] [Google Scholar]
- 36.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 37.Yan J, Liu X, Wang Y, Jiang X, Liu H, Wang M, Zhu X, Wu M, Tien P. Enhancing the potency of HBV DNA vaccines using fusion genes of HBV-specific antigens and the N-terminal fragment of gp96. J Gene Med. 2007;9:107–121. doi: 10.1002/jgm.998. [DOI] [PubMed] [Google Scholar]
- 38.Wu M, Audet A, Cusic J, Seeger D, Cochran R, Ghribi O. Broad DNA repair responses in neural injury are associated with activation of the IL-6 pathway in cholesterol-fed rabbits. J Neurochem. 2009;111:1011–1021. doi: 10.1111/j.1471-4159.2009.06390.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 40.Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–5916. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 41.Sellin ME, Maslowski KM, Maloy KJ, Hardt WD. Inflammasomes of the intestinal epithelium. Trends Immunol. 2015;36:442–450. doi: 10.1016/j.it.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Jamilloux Y, Henry T. The inflammasomes: platforms of innate immunity. Med Sci (Paris) 2013;29:975–984. doi: 10.1051/medsci/20132911013. [DOI] [PubMed] [Google Scholar]
- 43.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 45.Kim YK, Shin JS, Nahm MH. NOD-Like Receptors in Infection, Immunity, and Diseases. Yonsei Med J. 2016;57:5–14. doi: 10.3349/ymj.2016.57.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai S, Batra S, Wakamatsu N, Pacher P, Jeyaseelan S. NLRC4 inflammasome-mediated production of IL-1beta modulates mucosal immunity in the lung against gram-negative bacterial infection. J Immunol. 2012;188:5623–5635. doi: 10.4049/jimmunol.1200195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kannan S, Pang H, Foster DC, Rao Z, Wu M. Human 8-oxoguanine DNA glycosylase increases resistance to hyperoxic cytotoxicity in lung epithelial cells and involvement with altered MAPK activity. Cell Death Differ. 2006;13:311–323. doi: 10.1038/sj.cdd.4401736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng X, Weerapana E, Ulanovskaya O, Sun F, Liang H, Ji Q, Ye Y, Fu Y, Zhou L, Li J, Zhang H, Wang C, Alvarez S, Hicks LM, Lan L, Wu M, Cravatt BF, He C. Proteome-wide quantification and characterization of oxidation-sensitive cysteines in pathogenic bacteria. Cell Host Microbe. 2013;13:358–370. doi: 10.1016/j.chom.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang H, Weaver A, Wu E, Li Y, Gao H, Fan W, Wu M. Lipid-based signaling modulates DNA repair response and survival against Klebsiella pneumoniae infection in host cells and in mice. Am J Respir Cell Mol Biol. 2013;49:798–807. doi: 10.1165/rcmb.2013-0069OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, Rathinam VA. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell. 2016;165:1106–1119. doi: 10.1016/j.cell.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lenz HD, Haller E, Melzer E, Kober K, Wurster K, Stahl M, Bassham DC, Vierstra RD, Parker JE, Bautor J, Molina A, Escudero V, Shindo T, van der Hoorn RA, Gust AA, Nurnberger T. Autophagy differentially controls plant basal immunity to biotrophic and necrotrophic pathogens. Plant J. 2011;66:818–830. doi: 10.1111/j.1365-313X.2011.04546.x. [DOI] [PubMed] [Google Scholar]
- 53.Figueiredo N, Chora A, Raquel H, Pejanovic N, Pereira P, Hartleben B, Neves-Costa A, Moita C, Pedroso D, Pinto A, Marques S, Faridi H, Costa P, Gozzelino R, Zhao JL, Soares MP, Gama-Carvalho M, Martinez J, Zhang Q, Doring G, Grompe M, Simas JP, Huber TB, Baltimore D, Gupta V, Green DR, Ferreira JA, Moita LF. Anthracyclines induce DNA damage response-mediated protection against severe sepsis. Immunity. 2013;39:874–884. doi: 10.1016/j.immuni.2013.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C, Nunez G. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:e111. doi: 10.1371/journal.ppat.0030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harris J, Hope JC, Lavelle EC. Autophagy and the immune response to TB. Transbound Emerg Dis. 2009;56:248–254. doi: 10.1111/j.1865-1682.2009.01069.x. [DOI] [PubMed] [Google Scholar]
- 56.Xing Y, Ge Y, Liu C, Zhang X, Jiang J, Wei Y. ER stress inducer tunicamycin suppresses the self-renewal of glioma-initiating cell partly through inhibiting Sox2 translation. Oncotarget. 2016 doi: 10.18632/oncotarget.8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He S, Li X, Li R, Fang L, Sun L, Wang Y, Wu M. Annexin A2 Modulates ROS and Impacts Inflammatory Response via IL-17 Signaling in Polymicrobial Sepsis Mice. PLoS Pathog. 2016;12:e1005743. doi: 10.1371/journal.ppat.1005743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhuang Y, Li Y, Li X, Xie Q, Wu M. Atg7 Knockdown Augments Concanavalin A-Induced Acute Hepatitis through an ROS-Mediated p38/MAPK Pathway. PLoS One. 2016;11:e0149754. doi: 10.1371/journal.pone.0149754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ren F, Zhang L, Zhang X, Shi H, Wen T, Bai L, Zheng S, Chen Y, Chen D, Li L, Duan Z. Inhibition of glycogen synthase kinase 3beta promotes autophagy to protect mice from acute liver failure mediated by peroxisome proliferator-activated receptor alpha. Cell Death Dis. 2016;7:e2151. doi: 10.1038/cddis.2016.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 61.Schultz MJ, Rijneveld AW, Florquin S, Edwards CK, Dinarello CA, van der Poll T. Role of interleukin-1 in the pulmonary immune response during Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol. 2002;282:L285–290. doi: 10.1152/ajplung.00461.2000. [DOI] [PubMed] [Google Scholar]
- 62.Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, Nunez G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol. 2007;37:3030–3039. doi: 10.1002/eji.200737532. [DOI] [PubMed] [Google Scholar]
- 63.Faure E, Mear JB, Faure K, Normand S, Couturier-Maillard A, Grandjean T, Balloy V, Ryffel B, Dessein R, Chignard M, Uyttenhove C, Guery B, Gosset P, Chamaillard M, Kipnis E. Pseudomonas aeruginosa type-3 secretion system dampens host defense by exploiting the NLRC4-coupled inflammasome. Am J Respir Crit Care Med. 2014;189:799–811. doi: 10.1164/rccm.201307-1358OC. [DOI] [PubMed] [Google Scholar]
- 64.Jabir MS, Hopkins L, Ritchie ND, Ullah I, Bayes HK, Li D, Tourlomousis P, Lupton A, Puleston D, Simon AK, Bryant C, Evans TJ. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy. 2015;11:166–182. doi: 10.4161/15548627.2014.981915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Deng Q, Wang Y, Zhang Y, Li M, Li D, Huang X, Wu Y, Pu J, Wu M. Pseudomonas aeruginosa Triggers Macrophage Autophagy To Escape Intracellular Killing by Activation of the NLRP3 Inflammasome. Infect Immun. 2015;84:56–66. doi: 10.1128/IAI.00945-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karki R, Man SM, Malireddi RK, Kesavardhana S, Zhu Q, Burton AR, Sharma BR, Qi X, Pelletier S, Vogel P, Rosenstiel P, Kanneganti TD. NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature. 2016;540:583–587. doi: 10.1038/nature20597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.