

Abstract
Reliance on glutamine has long been considered a hallmark of cancer cell metabolism. However, some recent studies have challenged this notion in vivo, prompting a need for further clarifications on the role of glutamine metabolism in cancer. We find that there is ample evidence of an essential role for glutamine in tumors and that a variety of factors, including tissue type, the underlying cancer genetics, the tumor microenvironment and other variables such as diet and host physiology collectively influence the role of glutamine in cancer. Thus the requirements for glutamine in cancer are overall highly heterogeneous. In this review, we discuss the implications both for basic science and for targeting glutamine metabolism in cancer therapy.
Keywords: Glutamine Metabolism, Cancer Metabolism, Glutaminase, TCA Cycle Anaplerosis
Metabolic reprogramming in cancer
Cancer cells undergo a reprogramming of metabolism in order to maintain bioenergetics, redox status, cell signaling and biosynthesis, in what is often a poorly vascularized, nutrient-deprived microenvironment [1–4]. To supply biosynthetic pathways with precursors, the uptake and catabolism of certain nutrients are upregulated in tumor cells. In particular, the ‘Warburg Effect’ occurs in many human tumors, such that positron emission tomography (PET) using the glucose analog 18F-flurodeoxyglucose is widely used for imaging tumors in the clinic [1, 5]. Another metabolic characteristic of many cancer cells is a dependence on an exogenous supply of glutamine, despite this being a non-essential amino acid (NEAA) that mammalian cells can synthesize de novo. Glutamine serves as an important source of reduced nitrogen for biosynthetic reactions, and as a source of carbon to replenish the tricarboxylic acid (TCA) cycle, produce glutathione, serve as a precursor to nucleotides and lipid synthesis via reductive carboxylation (Figure 1) [1, 2, 6, 7]. Indeed, an inhibitor of the mitochondrial enzyme glutaminase, which converts glutamine to glutamate, a precursor of the TCA cycle intermediate α-ketoglutarate (α-KG), is currently being evaluated in clinical trials for treatment of a range of malignancies [8–12].
Figure 1. The Glutamine Metabolic Footprint in Cancer.
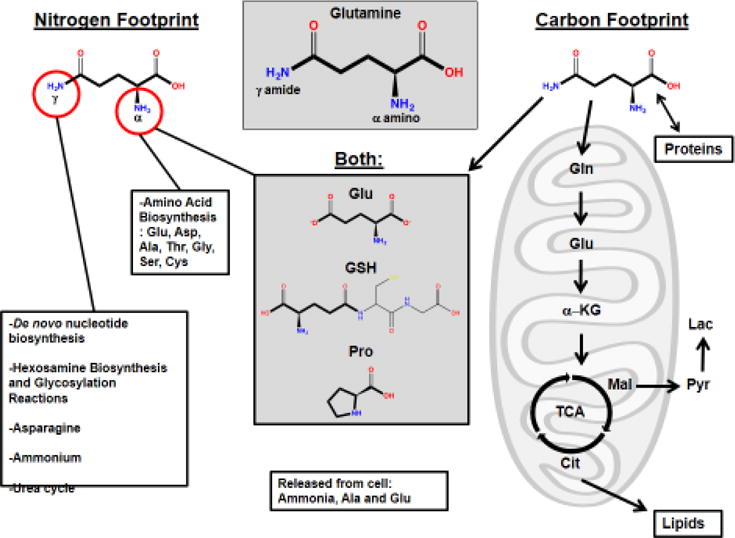
Glutamine has a five-carbon backbone and two nitrogen atoms (α and γ) to donate. Glutamine’s metabolic footprint goes well beyond TCA cycle anaplerosis. Gln: Glutamine, Glu: Glutamate, GSH: Glutathione, Asp: Aspartate, Ala: Alanine, Thr: Threonine, Gly: Glycine, Ser: Serine, Cys: Cysteine, Pro: Proline, Mal: Malate, Pyr: Pyruvate, Lac: Lactate, OAA: Oxaloacetate, Cit: Citrate, TCA: Citric acid cycle, α-KG: alpha-ketoglutarate.
Several recent studies have led to new insights in our understanding of the role of glutamine in cancer. As examples, one study found that the requirements for glutamine undergo changes upon transition from monolayer culture to anchorage-independent culture [7], and another study found that glutamine catabolism was not required for tumorigenesis in vivo in some mouse models, which led to the conclusion that glutamine metabolism may not have a role in cancer [13]. Nevertheless, numerous other studies have provided compelling evidence that a requirement for glutamine catabolism indeed exists in many in vivo settings [10, 14–21]. In this review, we discuss these seemingly contradictory recent findings in detail and explain the factors underlying the heterogeneity of glutamine metabolism in cancer.
Culture conditions and model systems influence glutamine metabolism
The ability to culture cell lines derived from human tumors has, over the past 60 years, provided researchers with a powerful tool for studying cancer biology. A common characteristic of mammalian cell lines grown in culture, noted by Harry Eagle in the 1950s [22], is a dependence on an abundant exogenous supply of the NEAA glutamine. After glucose, glutamine is the most rapidly consumed nutrient by many human cancer cell lines grown in culture [23, 24]. However, glutamine requirements are very heterogeneous among different cancer cell lines, ranging from those that are glutamine auxotrophs, to those that can survive and proliferate in the absence of an exogenous glutamine supply [18, 25]. Recent studies have demonstrated that the tissue of origin, the underlying genetics, and the microenvironment, can all impact cancer cell metabolism, including utilization of glutamine. Importantly, tumor microenvironment conditions can be modeled ex vivo, as the constituents of the culture media can be customized and other essential variables that determine metabolic requirements such as oxygen levels can be controlled [26, 27].
In monolayer culture, the non-small cell lung cancer (NSCLC) cell line H460 shows abundant uptake of glutamine, which is primarily utilized for TCA cycle anaplerosis. However, when the same cells are grown under anchorage-independent conditions as tumor spheroids, glutamine oxidation is suppressed and instead reductive glutamine metabolism via cytosolic isocitrate dehydrogenase 1 (IDH1) occurs [7]. The isocitrate/citrate generated by IDH1 then enters the mitochondria and undergoes oxidative metabolism via IDH2, generating mitochondrial NADPH to mitigate the elevated reactive oxygen species (ROS) levels that occur during anchorage-independent growth.
Distinct metabolic alterations take place when NSCLC cells are transitioned from an ex vivo to an in vivo environment. When cells derived from mutant KRAS-driven NSCLC tumors are grown in culture, glutamine supplies the TCA cycle with carbon, and inhibition of the mitochondrial enzyme glutaminase (GLS) suppresses cell proliferation [13]. However, this glutamine-dependent phenotype is lost when the same cells are grown as lung tumors in mice. Instead, TCA cycle intermediates in the tumors are derived primarily from glucose via glycolysis and the enzyme pyruvate carboxylase (PC), and tumors are resistant to glutaminase inhibition or to CRISPR/Cas9-mediated GLS deletion [13]. At first glance, these findings may seem surprising and contradictory to previous reports regarding the importance of glutamine metabolism in cancer cells both ex vivo and in vivo (see Table 1). However, they are consistent with earlier studies showing that some NSCLC tumors in human patients rely on glucose-derived carbon and PC activity, and show upregulated PC gene expression [28]. Indeed, PC has been identified as an essential factor in cancer cells that display glutamine-independent growth [29]. Nevertheless, many tumors rely on glutamine-mediated TCA cycle anaplerosis in vivo with concordance of glutamine dependence ex vivo and in vivo. (see Table 1), as discussed in detail below [9, 10, 12, 14, 15, 30] (Figure 2). Thus, recent observations in certain NSCLC mouse tumors cannot be generalized to other cancers.
Table 1.
Glutaminase Inhibition across different cancer types ex vivo and in vivo
| Tumor type | GLS inhibition | Experimental Models | Refs. | |
|---|---|---|---|---|
| ex vivo | in vivo | |||
| NSCLC (Lung) | Sensitive | Resistant | GEMs and xenografts | [13, 28, 34] |
| GBM | Resistant | Resistant | Patient derived xenografts | [33, 35] |
| Kidney | Sensitive | Transgenic MYC Mice | [14] | |
| Liver | Sensitive | Transgenic mice | [15] | |
| Breast (luminal) | Resistant | [17] | ||
| Breast (Basal) | Sensitive | Sensitive | xenografts | [10, 17] |
| T cell ALL | Sensitive | Sensitive | NOTCH1-induced T-ALL mice | [16] |
| Prostate | Sensitive (+metformin) | Sensitive (+metformin) | Transgenic (TRAMP) mice | [19] |
| Pancreas | Sensitive, sensitizes cells to β-lap treatment | Sensitive, sensitizes cells to β-lap treatment | KRAS induced PDACxenografts, β-lapachone (β-lap) | [18, 20] |
| Fibrosarcoma | Sensitive (+IKKβ) | Sensitive (+IKKβ) | xenografts, IκB kinase β (IKKβ) | [21] |
Figure 2. TCA cycle anaplerotic fluxes affect glutaminase inhibition efficacy.
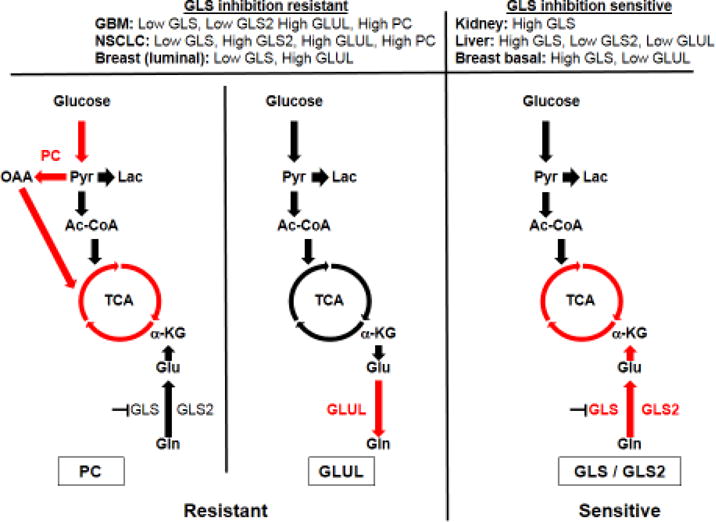
There are two main anaplerotic fluxes that can feed the citric acid cycle, a glutamine flux via glutaminase (GLS and/or GLS2) and a glucose flux via pyruvate carboxylase (PC). Glutamine synthetase (GLUL) is also an important enzyme for this process as it can synthesize glutamine from glutamate and thus allow cells to survive in glutamine-depleted conditions. The expression levels of these enzymes vary according to tissue type and can greatly affect their sensitivity to glutaminase inhibition. GBM: Glioblastoma, NSCLC: non-small cell lung carcinoma, Pyr: Pyruvate, Lac: Lactate, Cit: Citrate, TCA: Citric acid cycle, α-KG: alpha-ketoglutarate.
Metabolic phenotype varies with cancer subtype and microenvironment
Different mammalian organs exhibit distinct modes of glutamine metabolism. For example, the kidney mediates net glutamine catabolism, generating ammonia for pH homeostasis and supplying carbon for renal gluconeogenesis [31], whereas lung, skeletal muscle, and adipose tissues exhibit net de novo glutamine synthesis via the enzyme glutamine synthetase (GLUL) [32]. Similarly, human tumors show a range of metabolic phenotypes that vary with the tissue of origin, the cancer subtype, and the tumor microenvironment.
Although mammalian cells can synthesize glutamine de novo using GLUL [33], some cancer cells do not express high levels of GLUL and instead are dependent on an exogenous supply of glutamine, which can be catabolized in mitochondria via GLS. There is strong evidence that GLS plays an important role in the development of a range of cancers in vivo. While data in The Cancer Genome Atlas (TCGA) indicate that the GLS transcript is downregulated relative to surrounding healthy tissue in NSCLC, consistent with the studies described above, GLS mRNA levels are frequently upregulated in several other human malignancies (Figure 2). These include tumors of the colon, esophagus, liver, stomach, thyroid, as well as head and neck cancer. In conditional transgenic mouse models, overexpression of the MYC proto-oncogene in kidney or liver results in the formation of tumors in which GLS levels are highly upregulated relative to the surrounding healthy tissue [14, 15]. In both of these animal models, inhibition of GLS impedes tumor progression, and deletion of one GLS allele in the liver model significantly delays early tumor progression.
In contrast, glutamine synthesis is upregulated in some cancers. As outlined above, GLS is dispensable for growth of NSCLC tumors in vivo [13], and NSCLC tumors can actually accumulate newly-synthesized glutamine [34]. Similarly, some human glioblastoma (GBM) tumors do not significantly catabolize glutamine via GLS and the TCA cycle, but instead accumulate large pools of glutamine, synthesized by GLUL from glucose-derived carbon [35]. This glutamine feeds de novo purine biosynthesis, and renders GBM cells self-sufficient for glutamine requirements [33]. Consistent with this metabolic phenotype, GLUL and PC are expressed in most GBM tumors, whereas GLS expression is downregulated relative to surrounding brain tissue [33, 35].
Even among tumors that arise in a specific organ, different cancer subtypes can show distinct patterns of glutamine metabolism. Luminal breast cancers frequently exhibit high GLUL and low GLS expression, whereas the opposite is true of basal breast cancers [17]. Matching these expression patterns, most luminal breast cancer cells can be cultured in glutamine-free media, whereas basal cells are highly sensitive to glutamine withdrawal and to inhibition of GLS, both in cell culture and when grown as xenograft tumors in vivo [10, 17]. Metabolic heterogeneity can also arise between different regions of the same tumor. For example, highly perfused regions of NSCLC tumors oxidize diverse nutrients to fuel the TCA cycle, whereas less perfused regions primarily utilize glucose-derived carbon [36].
Thus, some tumors that arise in some tissues are typically dependent on glutamine anaplerosis, whereas NSCLC and GBM more frequently rely on pyruvate anaplerosis to maintain TCA cycle flux (Figure 2). A recent study using the mouse breast cancer cell line 4T1, which metastasizes to the lung with nearly 100% penetrance within a time frame of a day, sheds some light on the factors that influence choice of anaplerotic substrate. In contrast to primary breast tumors, lung metastases were found to rely on PC for TCA cycle anaplerosis, indicating that the tissue microenvironment might favor one metabolic phenotype over another [37]. Supporting this notion, the pyruvate/glutamine ratio is approximately 3-fold higher in the interstitial fluid of the lungs than in blood plasma, and the pyruvate concentration and expression level of PC in breast cancer lung metastases are elevated relative to primary breast tumors [37]. When cultured ex vivo, cell lines established from the lung metastases reverted to low levels of PC-dependent anaplerosis, similar to the parental cell line, again illustrating the effect of environment on metabolic phenotype [38]. Nevertheless, cell lines established from 4T1 metastases in different organs do retain some distinctions in their gene expression and metabolic profiles, indicating that adaptation to the microenvironmental nutrient supply does not fully explain the metabolic reprogramming that occurs during metastasis [38]. Another factor that may alter metabolism in the tumor microenvironment is the state of the immune compartment in the tumor. Recent reports have shown that cancer cells can compete with T cells for glucose within the tumor microenvironment and the resulting glucose limitation in T cells suppresses anti-tumor immunity [39, 40]. Given that glutamine metabolism is an important requirement for the metabolism of immune cells (i.e. Immunometabolism) [41–43] a similar effect may occur due to competition for glutamine within the microenvironment.
The impact of oncogenes on glutamine metabolism
Tumors that arise in different organs, but from the same genetic lesion can also have distinct phenotypes for glutamine metabolism. One study demonstrated that MYC-induced liver tumors exhibit elevated glutamine catabolism, with increased GLS expression and suppressed GLUL expression relative to surrounding tissue [15, 34]. In contrast, MYC-induced NSCLC tumors exhibit increased expression of GLUL, and accumulate glutamine [34]. c-Myc can regulate the expression of both GLS and GLUL through mechanisms involving suppression of miRNA-23a/b in the case of GLS, or upregulation of thymine DNA glycosylase, which leads to demethylation of, and increased expression from, the GLUL gene promoter [44, 45]. These mechanisms serve as examples to potentially explain, albeit non-exhaustively, why c-Myc has contrasting effects on glutamine metabolism in different cellular contexts. Tumors arising within the same tissue, but driven by different oncogenes, can also be metabolically divergent. In contrast to MFC-induced liver tumors, MET-induced liver tumors lose GLS expression, and overexpress GLUL and accumulate glutamine [34]. Furthermore, there are many other factors that can regulate glutamine metabolism such as the activity c-Jun [46], Rb [47, 48], PGC-1/ERRα [49], GLUL acetylation levels [50] and others [4, 6]. Thus, the metabolic phenotype of a given cancer seems to be determined by three key parameters: the tissue of origin, the underlying genetic factors driving tumorigenesis, and the microenvironment.
Tumor glutamine supply
Glutamine can be imported from the microenvironment by the solute carrier (SLC) group of transporters, including members of the SLC1, SLC6, SLC7, SLC36, and SLC38 families. The Na+/amino acid exchanger SLC1A5 and the unidirectional Na+/Cl−/amino acid symporter SLC6A14 are both regulated by c-Myc, and are overexpressed in several cancers [51]. Certain tumors such as pancreatic ductal adenocarcinoma (PDAC) are typically poorly vascularized, and consequently do not have an abundant serum supply of glutamine [52]. PDAC cells instead can in some cases use macropinocytosis to engulf extracellular proteins, which are then degraded in lysosomes to release glutamine and other amino acids [52–55]. An alternative route for cancer cell glutamine supply involves delivery of amino acids via extracellular vesicles shed by neighboring cells into the tumor microenvironment. Both PDAC and prostate cancer cells can grow in nutrient-deprived conditions when supplied with amino acids by exosomes shed by cancer-associated fibroblasts (CAFs) [56]. Additionally, recent work using ovarian carcinoma mouse models uncovered a reliance of cancer cells on stromal CAFs to maintain growth when glutamine is scarce. These fibroblasts boost glutamine synthesis from atypical sources to feed adjacent cancer cells [57]. Nevertheless, tumors that utilize macropinocytosis or uptake extracellular vesicles to acquire glutamine may still require glutaminase and exhibit sensitivity to its inhibition [18, 58].
The metabolic fate of glutamate in cancer
The first step of glutamine catabolism is its conversion to glutamate, which is catalyzed by cytosolic glutamine amidotransferases or by mitochondrial glutaminases. Glutamine-derived glutamate has diverse fates in proliferating cells, including consumption during protein synthesis, supplying nitrogen for transamination reactions, secretion from the cell in exchange for other nutrients, incorporation into the antioxidant tripeptide glutathione, and conversion into α-KG for TCA cycle anaplerosis. Formation of α-KG is catalyzed by the glutamate dehydrogenases (GLUD1/2), which release ammonia as a byproduct, or by transaminases, which transfer the amine-nitrogen of glutamate onto an α-keto acid to generate another amino acid. Some breast cancer cells are known to catabolize glutamate primarily via transaminases, thus conserving the amine nitrogen [59]. Alanine transaminase 2 (GPT2), in particular, is critical for α-KG generation and therefore for glutamine/glutamate-mediated TCA cycle anaplerosis in colon cancer cells [60, 61]. In contrast to proliferative cells, transaminase expression is low in quiescent cells, and instead GLUD expression is induced [62].
Another major fate of glutamine-derived glutamate is efflux from the cell, and glutamine consumption is closely mirrored by glutamate release across the NCI-60 panel of cell lines [23]. The transporter SLC7A11 mediates exchange of intracellular glutamate for extracellular cystine, which, once inside the cell, is reduced to cysteine, the rate-limiting amino acid for glutathione biosynthesis [51]. Expression of the SLC7A11 gene is induced by c-Myc, and is upregulated in a number of cancers [22]. In addition to the important role of cystine import for redox homeostasis, in some tumors the secreted glutamate can stimulate proliferation by acting on metabotropic and ionotropic glutamate receptors [51].
Epigenetics and signaling
Underlying some of the connections between tumor tissue-of-origin, microenvironment, and metabolic phenotype, is the tumor epigenetic landscape (see Text Box 1). Many epigenetic modifications and processes are regulated by glutamine-derived metabolites including α-KG, which is a cofactor for Jumonji-domain-containing histone demethylases [63]. In a variety of xenograft tumors, the poorly vascularized tumor core shows a selective deficiency of glutamine relative to other amino acids, and a corresponding depletion of glutamine-derived α-KG [26]. This results in inhibition of α-KG-dependent histone demethylation of H3K27 loci by a Jumonji-domain containing histone demethylase. Consequently, the tumor core exhibits pronounced histone hypermethylation, leading to suppressed expression of differentiation-related genes and cancer cell dedifferentiation.
Text Box 1. Glutamine’s effect on cancer epigenetics and posttranslational modifications.
The connection between metabolism and epigenetics has been better appreciated in recent years [63, 86]. Given the importance of glutamine in various crucial metabolic pathways, it is not surprising that it is also an important link between metabolism and various epigenetic and post-translational marks. This role is best illustrated in glutamine’s importance to the TCA cycle and specifically to α-KG levels, as this metabolite can act as a co-activator of Jumonji-C domain containing histone demethylases and TET demethylases. Additionally, downstream metabolites such as succinate, fumarate and the oncometabolite 2-hydroxyglutarate (2-HG), which arises from mutated isocitrate dehydrogenase 1/2 (IDH1/2) in gliomas and acute myeloid leukemias, can all inhibit these same demethylases [87]. Furthermore, glutamine can contribute to the intracellular acetyl-CoA pool as well as to NAD/NADH levels, suggesting a potential role in regulating histone and protein acetylation levels and patterns. Moreover, it has also been shown that UDP-glucosamine (UDP-GlcNAc), used for N-acetylglucosamination (GlcNAcylation) of histones and proteins and can regulate gene expression [88], can be synthesized from glutamine and can also be modulated by it through hexosamine biosynthesis via mTORC2 [89, 90].
Additionally, glutathione (GSH) can inhibit the activity of S-adenosyl methionine synthetase (MAT1A), a key enzyme in the synthesis of S-adenosyl methionine (SAM), the main methyl donor in the cell [91]. It has recently been reported that histone H3 can be S-glutathionated on Cyc110, which can cause changes in nucleosome stability and alter chromatin structure. Interestingly this modification was observed to increase in proliferating cells relative to quiescent cells [92]. Together, this indicates a critical role for glutamine metabolism in regulating and controlling a wide array of epigenetic and PTM marks.
Histone hypermethylation can be induced in V600EBRAF melanoma cells by withdrawing glutamine, and the consequent changes in gene expression lead to resistance to BRAF inhibitor treatment [26]. This mechanistic link between glutamine levels and gene-regulatory chromatin changes has important implications for the development of targeted cancer treatments. Inhibition of GLS is currently being evaluated in clinical trials (see below), and might be expected to trigger a histone hypermethylation phenotype that can lead to drug resistance following depletion of glutamine-derived α-KG. Although this suggests a possible mode of resistance to GLS-targeted therapies, it also points towards potential drug synergies that could be exploited in combination treatments. For example, the drug metformin has been identified to have a profound impact on fuel dependence in cancer cells, as it can decrease glucose oxidation and increase glutamine dependency in prostate cancers both ex vivo and in vivo [19, 27].
In addition to influencing gene expression via epigenetic mechanisms, recent work has implicated glutamine as a signaling agent that can promote cancer progression independently of its metabolic role. As an example, extracellular glutamine was recently shown to activate the transcription factor STAT3 to promote cancer cell proliferation [64]. Intracellular glutamine as well can stimulate cell signaling pathways, indirectly activating the mechanistic target of rapamycin complex 1 (mTORC1) [65]. Furthermore, efflux of intracellular glutamine via SLC7A5 can drive leucine uptake, another amino acid that is required for mTORC1 activation [66]. Recent work has also connected glutamine metabolism to NOTCH signaling in acute lymphoblastic leukemia (ALL). Inhibiting glutaminolysis and autophagy synergistically enhances the antileukemic effects of anti-NOTCH1 therapies, providing another example of the importance of glutamine metabolism in vivo [16].
Targeting glutamine metabolism for cancer therapy
The diverse roles played by glutamine in proliferating cells, supplying carbon and reduced nitrogen for biosynthetic reactions and redox homeostasis, present opportunities for targeting glutamine metabolism for cancer therapy [67]. A number of approaches are conceivable, including depletion of glutamine in blood serum, blockade of cellular glutamine uptake, and inhibition of enzymes involved in glutamine synthesis or catabolism [62]. L-asparaginases, which are routinely used to treat ALL patients, catalyze the deamidation of both asparagine and glutamine, leading to depletion of both of these amino acids in serum [68]. Since most ALL cells are auxotrophic for asparagine, L-asparaginase effectively starves them of this nutrient. The glutamine hydrolysis activity of L-asparaginases is also thought to contribute to their cytotoxicity, in part because glutamine is required for de novo synthesis of asparagine by asparagine synthetase, one potential resistance mechanism to asparagine starvation [68].
Early clinical trials of glutamine antimetabolites revealed unacceptable systemic toxicity, indicating that more selective approaches, for example by defining the tumors that might respond to lower doses, are required to disrupt glutamine metabolism in cancer patients [69]. Two classes of GLS inhibitors have been identified, based on the lead compound molecules ‘968’ and BPTES [11, 12]. The only potential drug to progress beyond preclinical studies, to our knowledge, is the inhibitor CB-839, which is currently being evaluated in Phase I clinical trials [8]. CB-839 is based on the BPTES scaffold, has an IC50 value against recombinant human GLS of <50 nM [10], and has shown efficacy against cultured cancer cell lines and xenograft models for triple-negative breast cancer and multiple myeloma [9, 10, 59]. Consistent with studies showing that GLS-mediated glutamine catabolism is required for in vivo growth of some tumor types but not others, only certain cancers are sensitive to GLS inhibition, and selection of appropriate target patient groups for CB-839 treatment is essential [8]. Efforts to develop small-molecule inhibitors of the GLS2 isozyme have been limited, presumably because of the conflicting literature reports concerning the importance of this enzyme in cancer (see Text Box 2). However, lead compounds with sub-micromolar IC50 values against recombinant GLS2 were recently identified, and inhibit growth of liver and lung cancer cell lines [70]. It remains to be seen whether GLS2 could be targeted for treatment of tumors such as neuroblastoma, which frequently exhibit elevated GLS2 expression downstream of N-Myc.
Text Box 2. GLS2, the other glutaminase.
Two genes encode glutaminase enzymes in mammals, GLS and GLS2. At least two functional isoforms, GAC and KGA (collectively referred to as GLS), arise from the GLS gene as a result of alternative splicing. Similarly, the GLS2 gene encodes at least two isoforms, LGA and GAB (collectively referred to as GLS2), through a surrogate promoter mechanism. In healthy individuals, GLS2 is expressed primarily in the liver, brain, and pancreas, whereas GLS expression is ubiquitous and highest in kidney, lymphocytes, brain, and enterocytes. The GLS and GLS2 proteins are highly homologous, and most reports indicate that both are localized to mitochondria [6, 93, 94].
In liver, the organ in which GLS2 is most abundant, tumorigenesis is frequently accompanied by downregulation of the GLS2 transcript, although immunohistochemistry staining indicates a less consistent pattern at the protein level [15]. Similarly, GLS2 is downregulated in glioblastoma relative to surrounding brain tissue (as is GLS), and forced overexpression of GLS2 in liver cancer or glioblastoma cell lines suppresses proliferation and/or formation of xenograft tumors [95, 96]. In contrast, GLS2 is elevated in NMYC-amplified neuroblastoma, and in this context is important for in vivo tumor progression, such that high expression correlates with poor prognosis [97]. GLS2 is also upregulated in lung tumors, colon tumors, and radiation-resistant cervical cancers, where it is important for in vivo tumorigenesis [98, 99].
The discovery that GLS2 is frequently downregulated in liver cancers, together with early work indicating a pro-oncogenic role for GLS, initially led to the proposal that the two isozymes play opposing roles during tumorigenesis. However, more recent findings indicate that in certain contexts, GLS2 can be important for tumor progression. One possible explanation for the discrepancies is that these homologous enzymes do not have fundamentally different roles, but are simply subject to distinct modes of regulation. Indeed, whereas expression of the GLS gene is enhanced by oncogenic transcription factors associated with cell-cycle progression [44, 46], transcription of GLS2 is driven both by the tumor suppressor p53 in response to oxidative stress [96], and also by the proto-oncoprotein N-myc [97]. Disruption of p53 function likely explains some instances of downregulated GLS2 expression in tumors. However, there are some intrinsic differences between the GLS and GLS2 enzymes that could lead to selective pressure for the former over the latter. In the presence of phosphate, GLS has a lower KM for glutamine than does GLS2, resulting in a much higher catalytic efficiency. Furthermore, it was recently reported that the GLS2 C-terminus, binds to and inhibits activation of the small GTPase Rac1, resulting in suppressed migration, invasion, and metastasis of liver cancer cell lines [100].
A number of other nodes of cellular glutamine/glutamate metabolism have been proposed as attractive targets for cancer therapy, including transaminases [71], glutamine and glutamate transporters [51], asparagine synthetase [72], and glutamate dehydrogenase [73]. However, to date, studies remain preclinical and there is a lack of potent and selective inhibitors of these proteins, although each of these targets has scientific promise [67].
Concluding Remarks
Our understanding of the roles played by glutamine in cancer is evolving rapidly, and recent work has provided new insights and also has raised a number of questions. It is now clear that there is a not a single ‘metabolic map’ or ‘metabolic switch’ describing cancer cell metabolism [74], and the fate of glutamine varies with a range of parameters, including the tissue of origin of a cancer, the genetic aberrations which drive it, the tumor microenvironment, and possibly diet and host metabolism. The collective effect of these variables is striking, such that the metabolic phenotypes of cancer cells range from those that are highly dependent on catabolism of exogenous glutamine, to those that accumulate glutamine via de novo synthesis and are self-sufficient for glutamine requirements. Adding an additional level of complexity, recent studies have demonstrated that metabolic phenotype can change when cancer cells are transitioned between different culture systems, or between ex vivo cell culture and in vivo animal model environments. Standard cell culture media contain excesses of certain nutrients relative to their physiological concentrations, while completely lacking other nutrients such as acetate or aspartate which can play important roles in tumor metabolism [75]. However, these variables are controllable, and we anticipate that future studies will further characterize how metabolic phenotype responds to changing nutrient availability. We also anticipate that advances in metabolic labeling and modeling methods [37, 76–80], as well as the development of subcellular compartment-specific metabolite isolation methods [81], will reveal new insights into the compartmentalization of glutamine metabolism in cancer cells.
Although the glutamine dependence of some NSCLC cells is lost when they are grown in vivo, the canonical anaplerotic role of glutamine prevails in other contexts in vivo [9, 10, 12, 14–16, 30], and clinical trials of the GLS inhibitor CB-839 have yielded some promising results [8] suggesting that these recent observations in NSCLC models is certainly not general to all cancers. It is also important to note that the dispensability of glutamine in certain circumstances does not imply that anaplerosis in these cells is not required; as discussed above, cancer cells can find alternative ways to maintain an anaplerotic flux. Continued progress in targeting glutamine metabolism for cancer therapy will likely require identification of synergistic drug combinations along with careful selection of appropriate target patient groups, which could be aided by new techniques for imaging tumor glutamine metabolism in vivo [82, 85]. Collectively, recent findings on the complexities of cancer cell glutamine metabolism in vivo and ex vivo, far from ‘completing’ the field, have generated many new questions (see Outstanding Questions), and set the scene for future studies to provide novel and biologically relevant insights.
Trends.
The role of glutamine in cancer metabolism is more complex than previously appreciated.
Glutaminase inhibition effectiveness in vivo is highly dependent on tumor cell origin and tumor microenvironment.
Animal and anchorage independent cell culture studies can greatly complement monolayer cell culture studies and may reveal unique metabolic patterns.
The synthesis or uptake, and the utilization, of glutamine in cancer cells is highly flexible and dependent on cell origin, oncogenic drivers and the tumor microenvironment.
Some tumor types rely on catabolism of exogenous glutamine, and might be effectively targeted by therapeutic regimes involving glutaminase inhibition.
Different metabolic pathways, including glutamine catabolism, can achieve TCA cycle anaplerosis.
Outstanding Questions.
What causes the glutamine metabolic heterogeneity observed between cells grown in cell culture and in animal models?
Do the changes in metabolic phenotype that occur when NSCLC cells are transitioned between in vivo and ex vivo environments apply to other cancer types?
To what extent do epigenetic phenomena lead to resistance to drugs targeting glutamine metabolism?
Does diet or host metabolism including liver physiology play a role in the requirements of glutamine metabolism in tumors?
Acknowledgments
The authors thank Ralph DeBerardinis and members of the Cerione and Locasale labs for their helpful comments. The authors apologize to any authors whose work could not be included owing to space limitations. Support from the National Institutes of Health, R01CA193256 (JWL), R00CA168997 (JWL), is gratefully acknowledged. AAC is supported by a graduate fellowship from the King Abdullah International Medical Research Center under the Ministry of National Guard Health Affairs.
Glossary
- Anaplerosis
The process of replenishing metabolic pathway intermediates. For example, carbon that is lost from the TCA cycle to supply biosynthetic reactions can be replenished by glutamine-derived α-KG, glucose-derived oxaloacetate, etc.
- Auxotroph
an organism that is unable to synthesize a particular compound required for its growth.
- Catabolism
Describes metabolic pathways that breakdown macromolecules into smaller units and release energy. As opposed to Anabolism, which describes energy-consuming biosynthetic metabolic pathways that construct macromolecules to build biomass of the cell.
- Epigenetics
The study of heritable changes in gene expression that does not involve changes to the underlying DNA sequence. Often encompassing chemical modifications to the DNA and Histones.
- Extracellular Vesicles
Bilayered membrane-enclosed packages that are shed by various cell types including cancer cells and can contain important cargo that facilitates paracrine signaling. These vesicles can be divided into two broad classes according to size and the unique underlying mechanisms of their biogenesis. A large vesicle class with a diameter of 0.2–1 um called microvesicles, which are plasma membrane-derived and result from its budding and fission. And a much smaller class with a diameter of 0.04 to 0.1 um known as exosomes, that are derived from multi-vesicular bodies that reroute and fuse to the plasma membrane for exocytosis [101–103].
- ex vivo
taking place outside of a living organism with minimal deviation from natural conditions.
- Glutaminolysis
The metabolic breakdown of glutamine.
- Immunometabolism
The changes in intracellular metabolic pathways in immune cells that alter their function.
- in vivo
taking place in a living organism
- Macropinocytosis
A regulated actin-dependent form of endocytosis, which enables the cell to engulf extracellular macromolecules such as proteins.
- Warburg Effect
Also known as aerobic glycolysis, is the phenomenon of increased glucose uptake coupled to lactate secretion, regardless of O2 availability in cancer cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23(1):27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351–9. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagarajan A, Malvi P, Wajapeyee N. Oncogene-directed alterations in cancer cell metabolism. Trends Cancer. 2016;2(7):365–377. doi: 10.1016/j.trecan.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016;41(3):211–8. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016 doi: 10.1038/nrc.2016.131. [DOI] [PubMed] [Google Scholar]
- 7.Jiang L, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532(7598):255–8. doi: 10.1038/nature17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garber K. Cancer anabolic metabolism inhibitors move into clinic. Nat Biotechnol. 2016;34(8):794–5. doi: 10.1038/nbt0816-794. [DOI] [PubMed] [Google Scholar]
- 9.Bromley-Dulfano S, et al. Antitumor Activity Of The Glutaminase Inhibitor CB-839 In Hematological Malignances. Blood. 2013;122(21):4226–4226. [Google Scholar]
- 10.Gross MI, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13(4):890–901. doi: 10.1158/1535-7163.MCT-13-0870. [DOI] [PubMed] [Google Scholar]
- 11.Robinson MM, et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) Biochem J. 2007;406(3):407–14. doi: 10.1042/BJ20070039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang JB, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18(3):207–19. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davidson SM, et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016;23(3):517–28. doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shroff EH, et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci U S A. 2015;112(21):6539–44. doi: 10.1073/pnas.1507228112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiang Y, et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest. 2015;125(6):2293–306. doi: 10.1172/JCI75836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herranz D, et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med. 2015;21(10):1182–9. doi: 10.1038/nm.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kung HN, Marks JR, Chi JT. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011;7(8):e1002229. doi: 10.1371/journal.pgen.1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Son J, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–5. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fendt SM, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013;73(14):4429–38. doi: 10.1158/0008-5472.CAN-13-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakrabarti G, et al. Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ss-lapachone. Cancer Metab. 2015;3:12. doi: 10.1186/s40170-015-0137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reid MA, et al. IKKbeta promotes metabolic adaptation to glutamine deprivation via phosphorylation and inhibition of PFKFB3. Genes Dev. 2016;30(16):1837–51. doi: 10.1101/gad.287235.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eagle H. Nutrition needs of mammalian cells in tissue culture. Science. 1955;122(3168):501–14. doi: 10.1126/science.122.3168.501. [DOI] [PubMed] [Google Scholar]
- 23.Jain M, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336(6084):1040–4. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosios AM, et al. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev Cell. 2016;36(5):540–9. doi: 10.1016/j.devcel.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Timmerman LA, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24(4):450–65. doi: 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan M, et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol. 2016;18(10):1090–1101. doi: 10.1038/ncb3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, et al. Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab. 2016;24(5):728–739. doi: 10.1016/j.cmet.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sellers K, et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest. 2015;125(2):687–98. doi: 10.1172/JCI72873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng T, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108(21):8674–9. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matre P, et al. Inhibiting glutaminase in acute myeloid leukemia: metabolic dependency of selected AML subtypes. Oncotarget. 2016 doi: 10.18632/oncotarget.12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stumvoll M, et al. Role of glutamine in human carbohydrate metabolism in kidney and other tissues. Kidney Int. 1999;55(3):778–92. doi: 10.1046/j.1523-1755.1999.055003778.x. [DOI] [PubMed] [Google Scholar]
- 32.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123(9):3678–84. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tardito S, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol. 2015;17(12):1556–68. doi: 10.1038/ncb3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuneva MO, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15(2):157–70. doi: 10.1016/j.cmet.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marin-Valencia I, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15(6):827–37. doi: 10.1016/j.cmet.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hensley CT, et al. Metabolic Heterogeneity in Human Lung Tumors. Cell. 2016;164(4):681–94. doi: 10.1016/j.cell.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christen S, et al. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 2016;17(3):837–848. doi: 10.1016/j.celrep.2016.09.042. [DOI] [PubMed] [Google Scholar]
- 38.Dupuy F, et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015;22(4):577–89. doi: 10.1016/j.cmet.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 39.Chang CH, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162(6):1229–41. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ho PC, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162(6):1217–28. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakaya M, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40(5):692–705. doi: 10.1016/j.immuni.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blagih J, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 2015;42(1):41–54. doi: 10.1016/j.immuni.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 43.O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–65. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao P, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762–5. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bott AJ, et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015;22(6):1068–77. doi: 10.1016/j.cmet.2015.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lukey MJ, et al. The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat Commun. 2016;7:11321. doi: 10.1038/ncomms11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicolay BN, et al. Loss of RBF1 changes glutamine catabolism. Genes Dev. 2013;27(2):182–96. doi: 10.1101/gad.206227.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reynolds MR, et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. 2014;33(5):556–66. doi: 10.1038/onc.2012.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGuirk S, et al. PGC-1alpha supports glutamine metabolism in breast cancer. Cancer Metab. 2013;1(1):22. doi: 10.1186/2049-3002-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nguyen TV, et al. Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol Cell. 2016;61(6):809–20. doi: 10.1016/j.molcel.2016.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhutia YD, et al. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015;75(9):1782–8. doi: 10.1158/0008-5472.CAN-14-3745. [DOI] [PubMed] [Google Scholar]
- 52.Kamphorst JJ, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75(3):544–53. doi: 10.1158/0008-5472.CAN-14-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Commisso C, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497(7451):633–7. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palm W, et al. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell. 2015;162(2):259–70. doi: 10.1016/j.cell.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davidson SM, et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat Med. 2016 doi: 10.1038/nm.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao H, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife. 2016;5:e10250. doi: 10.7554/eLife.10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang L, et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016;24(5):685–700. doi: 10.1016/j.cmet.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Santana SM, et al. Cancerous epithelial cell lines shed extracellular vesicles with a bimodal size distribution that is sensitive to glutamine inhibition. Phys Biol. 2014;11(6):065001. doi: 10.1088/1478-3975/11/6/065001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coloff JL, et al. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell Metab. 2016;23(5):867–80. doi: 10.1016/j.cmet.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 60.Smith B, et al. Addiction to Coupling of the Warburg Effect with Glutamine Catabolism in Cancer Cells. Cell Rep. 2016;17(3):821–836. doi: 10.1016/j.celrep.2016.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hao Y, et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat Commun. 2016;7:11971. doi: 10.1038/ncomms11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lukey MJ, Wilson KF, Cerione RA. Therapeutic strategies impacting cancer cell glutamine metabolism. Future Med Chem. 2013;5(14):1685–700. doi: 10.4155/fmc.13.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kinnaird A, et al. Metabolic control of epigenetics in cancer. Nat Rev Cancer. 2016 doi: 10.1038/nrc.2016.82. [DOI] [PubMed] [Google Scholar]
- 64.Cacace A, et al. Glutamine activates STAT3 to control cancer cell proliferation independently of glutamine metabolism. Oncogene. 2016 doi: 10.1038/onc.2016.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jewell JL, et al. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science. 2015;347(6218):194–8. doi: 10.1126/science.1259472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nicklin P, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136(3):521–34. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martinez-Outschoorn UE, et al. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2016 doi: 10.1038/nrclinonc.2017.1. [DOI] [PubMed] [Google Scholar]
- 68.Covini D, et al. Expanding targets for a metabolic therapy of cancer: L-asparaginase. Recent Pat Anticancer Drug Discov. 2012;7(1):4–13. doi: 10.2174/157489212798358001. [DOI] [PubMed] [Google Scholar]
- 69.Ahluwalia GS, et al. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther. 1990;46(2):243–71. doi: 10.1016/0163-7258(90)90094-i. [DOI] [PubMed] [Google Scholar]
- 70.Lee YZ, et al. Discovery of selective inhibitors of Glutaminase-2, which inhibit mTORC1, activate autophagy and inhibit proliferation in cancer cells. Oncotarget. 2014;5(15):6087–101. doi: 10.18632/oncotarget.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Korangath P, et al. Targeting Glutamine Metabolism in Breast Cancer with Aminooxyacetate. Clin Cancer Res. 2015;21(14):3263–73. doi: 10.1158/1078-0432.CCR-14-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gutierrez JA, et al. An inhibitor of human asparagine synthetase suppresses proliferation of an L-asparaginase-resistant leukemia cell line. Chem Biol. 2006;13(12):1339–47. doi: 10.1016/j.chembiol.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin L, et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell. 2015;27(2):257–70. doi: 10.1016/j.ccell.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strickaert A, et al. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene. 2016 doi: 10.1038/onc.2016.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mayers JR, Vander Heiden MG. Famine versus feast: understanding the metabolism of tumors in vivo. Trends Biochem Sci. 2015;40(3):130–40. doi: 10.1016/j.tibs.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park JO, et al. Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat Chem Biol. 2016;12(7):482–9. doi: 10.1038/nchembio.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hackett SR, et al. Systems-level analysis of mechanisms regulating yeast metabolic flux. Science. 2016;354(6311) doi: 10.1126/science.aaf2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dai Z, Locasale JW. Understanding metabolism with flux analysis: From theory to application. Metab Eng. 2016 doi: 10.1016/j.ymben.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dai Z, et al. A Flux Balance of Glucose Metabolism Clarifies the Requirements of the Warburg Effect. Biophys J. 2016;111(5):1088–100. doi: 10.1016/j.bpj.2016.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Buescher JM, et al. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol. 2015;34:189–201. doi: 10.1016/j.copbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen WW, et al. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell. 2016;166(5):1324–1337 e11. doi: 10.1016/j.cell.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schulte ML, et al. Non-Invasive Glutamine PET Reflects Pharmacological Inhibition of BRAFV600E In Vivo. Mol Imaging Biol. 2016 doi: 10.1007/s11307-016-1008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Y, Shen J. Simultaneous quantification of glutamate and glutamine by J-modulated spectroscopy at 3 Tesla. Magn Reson Med. 2016;76(3):725–32. doi: 10.1002/mrm.25922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Venneti S, et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med. 2015;7(274):274ra17. doi: 10.1126/scitranslmed.aaa1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hassanein M, et al. Preclinical Evaluation of 4-[18F]Fluoroglutamine PET to Assess ASCT2 Expression in Lung Cancer. Mol Imaging Biol. 2016;18(1):18–23. doi: 10.1007/s11307-015-0862-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gao X, et al. Metabolic interactions with cancer epigenetics. Mol Aspects Med. 2016 doi: 10.1016/j.mam.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27(4):599–608. doi: 10.1093/annonc/mdw013. [DOI] [PubMed] [Google Scholar]
- 88.Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. 2012;13(5):312–21. doi: 10.1038/nrm3334. [DOI] [PubMed] [Google Scholar]
- 89.Moloughney JG, et al. mTORC2 Responds to Glutamine Catabolite Levels to Modulate the Hexosamine Biosynthesis Enzyme GFAT1. Mol Cell. 2016;63(5):811–26. doi: 10.1016/j.molcel.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ferrer CM, V, Sodi L, Reginato MJ. O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J Mol Biol. 2016;428(16):3282–94. doi: 10.1016/j.jmb.2016.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Garcia-Gimenez JL, Pallardo FV. Maintenance of glutathione levels and its importance in epigenetic regulation. Front Pharmacol. 2014;5:88. doi: 10.3389/fphar.2014.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Garcia-Gimenez JL, et al. Histone h3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxid Redox Signal. 2013;19(12):1305–20. doi: 10.1089/ars.2012.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Katt WP, Cerione RA. Glutaminase regulation in cancer cells: a druggable chain of events. Drug Discov Today. 2014;19(4):450–7. doi: 10.1016/j.drudis.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44(D1):D1251–7. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Szeliga M, Albrecht J. Opposing roles of glutaminase isoforms in determining glioblastoma cell phenotype. Neurochem Int. 2015;88:6–9. doi: 10.1016/j.neuint.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 96.Suzuki S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A. 2010;107(16):7461–6. doi: 10.1073/pnas.1002459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xiao D, et al. Myc promotesglutaminolysis in human neuroblastoma through direct activation of glutaminase 2. Oncotarget. 2015;6(38):40655–66. doi: 10.18632/oncotarget.5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Giacobbe A, et al. p63 regulates glutaminase 2 expression. Cell Cycle. 2013;12(9):1395–405. doi: 10.4161/cc.24478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xiang L, et al. Knock-down of glutaminase 2 expression decreases glutathione, NADH, and sensitizes cervical cancer to ionizing radiation. Biochim Biophys Acta. 2013;1833(12):2996–3005. doi: 10.1016/j.bbamcr.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 100.Zhang C, et al. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. Elife. 2016;5:e10727. doi: 10.7554/eLife.10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Antonyak MA, Cerione RA. Microvesicles as mediators of intercellular communication in cancer. Methods Mol Biol. 2014;1165:147–73. doi: 10.1007/978-1-4939-0856-1_11. [DOI] [PubMed] [Google Scholar]
- 102.Desrochers LM, Antonyak MA, Cerione RA. Extracellular Vesicles: Satellites of Information Transfer in Cancer and Stem Cell Biology. Dev Cell. 2016;37(4):301–9. doi: 10.1016/j.devcel.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lo Cicero A, Stahl PD, Raposo G. Extracellular vesicles shuffling intercellular messages: for good or for bad. Curr Opin Cell Biol. 2015;35:69–77. doi: 10.1016/j.ceb.2015.04.013. [DOI] [PubMed] [Google Scholar]