

ABSTRACT
Insulin receptor substrate 4 (IRS4) belongs to a family of cytoplasmic docking proteins mediating signals from cell surface receptors to downstream effectors. While IRS1 and IRS2 mediate signals from an active receptor, we found that IRS4 hyperactivates the phosphatidylinositol phosphate kinase (PI3K)-pathway independent of upstream signals and is irresponsive to feedback regulation causing cancer and resistance to human epidermal growth factor receptor 2 (HER2) targeted therapy.
KEYWORDS: Breast cancer, HER2, IRS4, lapatinib, PI3-Kinase, therapy resistance, trastuzumab
Hyperactivation of the PI3K/AKT/mTOR signaling pathway (phosphatidylinositol phosphate kinase, PI3K-pathway) is common in almost all human cancer types, including breast cancer. Dysregulation of this pathway most frequently occurs through activating mutations in the PIK3CA gene, encoding the catalytic subunit of PI3K. This results in an increase in membrane phosphatidylinositol 3,4,5-trisphosphate (PIP3), which recruits protein kinase B (PKB, commonly known as Akt) to the plasma membrane for phosphorylation, causing activation of numerous downstream effector molecules that promote tumorigenesis. In addition, loss or decreased expression of the tumor suppressor phosphatase and tensin homolog (PTEN), the major antagonist of PI3K, or gain-of-function mutations or amplifications in upstream receptor tyrosine kinases (RTKs), including the receptors for insulin and Insulin-like growth factor 1 (IGF1), activate the PI3K-pathway in many cancers. Both these receptors signal via the insulin receptor substrate (IRS) proteins: scaffolding proteins that transmit signals to intracellular signaling cascades, including the PI3K-pathway. IRSs lack intrinsic kinase activity, but upon binding, the activated RTK phosphorylates several tyrosine residues in the C-terminal region of these proteins, which subsequently act as docking sites for downstream signaling molecules.1 In humans, the IRS-family consists of three closely related members (IRS1, IRS2, and IRS4), of which IRS1 and IRS2 are by far the best studied members. The rather universally expressed IRS1 and IRS2 genes are known to play essential and non-redundant roles in postnatal growth and glucose homeostasis, respectively, and their overexpression has been implicated in cancer, although in a highly context-dependent fashion.1,2 In contrast, IRS4 is less well studied and appears silent in normal adult tissues.
In recent years, we have performed several mouse mammary tumor virus (MMTV)-mediated insertional mutagenesis screens to search for genes that induce mammary tumors in various mouse models.3-5 A frequently tagged gene in these screens was Irs4, but not the closely related family members Irs1 and Irs2. We recently reported that IRS4 has a growth factor independent activity, in contrast to IRS1 and IRS2.5 Specifically, we demonstrated that IRS4 has a high basal signal transduction activity and a sustained activity upon upstream stimulation due to a lacking Src homology phosphatase 2 (Shp2)-binding site. In IRS1 and IRS2, this phosphatase is recruited by specific phosphotyrosines in their carboxyl-termini, leading to tyrosine dephosphorylation of the IRSs, consequently preventing docking and further activation of downstream effectors. Hence, IRS4 is unresponsive to this strong feedback regulation and hyperactivates the PI3K-pathway without significant upstream RTK activation (see Fig. 1A for a schematic overview). Instead of being subjected to post-transcriptional regulation, IRS4 is strictly transcriptionally regulated, explaining why its expression is limited to a few embryonic tissues and even more confined postnatally.1,5 However, in line with our in vitro and in vivo experiments that established IRS4 as an oncogenic driver, we found that IRS4 is expressed in a relatively small but significant subset of human breast cancers (6–15%) and we showed that survival of patients with tumors expressing IRS4 was significantly reduced.
Figure 1.
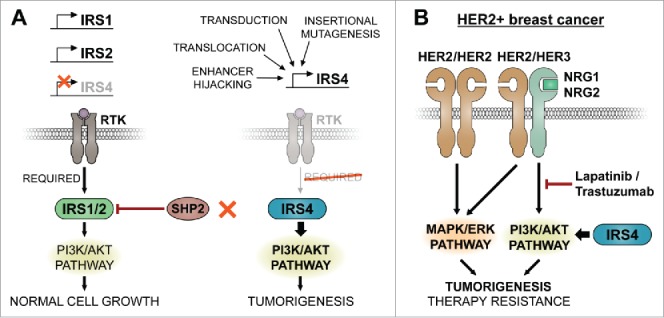
IRS4 signaling in malignant cells and therapy resistance. (A) Simplified IRS (insulin receptor substrate) 2-induced phosphatidylinositol phosphate kinase (PI3K) signaling cascade in normal cells expressing IRS1 and IRS2 (left) or cells expressing IRS4 (right). In most normal cells either IRS1 or IRS2, or both IRSs are expressed, whereas IRS4 is rarely expressed. IRS1 and IRS2 activity is kept in check by negative feedback via SHP2 (Src homology phosphatase 2) mediated tyrosine dephosphorylation. In cancer cells, various mutagenic events may activate IRS4, which is irresponsive to SHP2 mediated feedback and hyperactivates the PI3K pathway leading to tumor growth. (B) Tumorigenesis requires continues stimulation of the PI3K and mitogen-activated protein kinase (MAPK)-pathways. The HER2/HER3 (human epidermal growth factor receptor 2/3) heterodimer may provide both these signals in a subset of tumors, where HER2 provides the MAPK signal and HER3 the PI3K signal. IRS4 synergistically enhances HER2-induced tumorigenesis by hyperactivating the PI3K pathway. Trastuzumab (humanized monoclonal antibodies against HER2) and lapatinib (a tyrosine kinase inhibitor inhibiting HER2 kinase activity) prevent the oncogenic signals of HER2 and HER3, but this is circumvented by IRS4-induced hyperactivation of the PI3K pathway, leading to therapy resistance. NRG1 and NRG2: ligands for HER3. RTK: receptor tyrosine kinase. Thickness of the arrows indicates strength of signaling. Red “X” indicates no interaction.
We have not elucidated the mechanism behind IRS4 upregulation in these tumors, but in pediatric T-cell acute lymphoblastic leukemia (T-ALL), strong IRS4 upregulation has been reported due to chromosomal translocation, bringing the gene under the transcriptional control of T-cell receptor β regulatory elements.6 A somewhat related mechanism was reported at the same time of our study, revealing IRS4 as a candidate pan-cancer gene that is activated due to “enhancer hijacking” in 10 different tumor types, most prominently lung squamous carcinomas and cervical squamous carcinomas.7 Here, cis-regulatory elements such as enhancers were found to be rearranged and juxtaposed to IRS4. This resembles the activation of Irs4 by MMTV-proviral integrations in our insertional mutagenesis screens,3-5 where the proviral transcriptional enhancers interact with the Irs4-promoter, upregulating the gene. It may be worthwhile assessing the activating mechanisms of IRS4 in human breast cancer as well, particularly in cases of acquired therapy resistance (see below).
Regardless the mechanism of upregulation, we show that the clinical implications of IRS4-expression are substantial. It is well recognized that a hyperactivated PI3K-pathway can induce resistance to various therapies (reviewed in Ref.8). We found that IRS4 was mainly expressed in human epidermal growth factor receptor 2 (HER2, encoded by the ERBB2 gene) positive and triple negative, but scarcely in luminal A or B breast cancers.5 HER2-targeting therapy using monoclonal antibodies trastuzumab or pertuzumab, and the tyrosine kinase inhibitor lapatinib greatly improves the prognosis of HER2+ breast cancer patients. However, both primary and secondary resistance are common and are often associated with PI3K-pathway hyperactivation.9 Indeed, we showed that expression of IRS4 in various cell lines with ERBB2 overexpression greatly reduced the sensitivity to HER2-directed therapeutic agents.5 Moreover, we observed that IRS4 synergistically accelerates tumorigenesis in vitro and in vivo when co-expressed with HER2, most likely due to the combined potent activation of the PI3K-pathway by IRS4 and the RAS/RAF/MEK/ERK (mitogen-activated protein kinase, MAPK) pathway by HER2 (see Fig. 1B for a schematic overview). IRS4-expression could also rapidly be attained in naive HER2+ breast cancer cell lines by culturing the cells for several passages in medium with increasing concentrations of trastuzumab or lapatinib, indicating selection for IRS4-expressing cells under the pressure of the drugs.5 Hence, our data suggest that IRS4 can cause both primary resistance to trastuzumab or lapatinib, as well as acquired resistance during treatment, thus likely plays a role in relapse in breast cancer patients. Importantly, we also showed that treating HER2+ breast cancer cells expressing IRS4 with a HER2-targeting drug in combination with a PI3K-specific inhibitor abrogated IRS4-mediated resistance, even in suboptimal doses.5 This finding may inspire the field to keep investigating the combination of HER2-targeted drugs with PI3K, AKT and/or mTOR inhibitors, despite the limited success of initial clinical trials. Indeed, the BOLERO-3 trial, in which patients with trastuzumab-resistant HER2+ breast cancer were treated with a combination of trastuzumab and the mTOR inhibitor everolimus in an effort to restore sensitivity to the HER2-targeting drug showed clear benefit to such combined treatment in patients with tumors exhibiting high PI3K-pathway activity.10
In conclusion, IRS4 is an oncogene that plays a role in various types of human cancer by driving PI3K-pathway activity constitutively. We propose IRS4 as a novel clinical biomarker for PI3K-pathway hyperactivation and, perhaps most importantly, for HER2-targeted therapy resistance.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Giovannone B, Scaldaferri ML, Federici M, Porzio O, Lauro D, Fusco A, Sbraccia P, Borboni P, Lauro R, Sesti G, et al.. Insulin receptor substrate (IRS) transduction system: distinct and overlapping signaling potential. Diabetes Metab Res Rev 2000; 16:434-41; PMID:11114102; http://dx.doi.org/ [DOI] [PubMed] [Google Scholar]
- 2.Mardilovich K, Pankratz SL, Shaw LM. Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun Signal 2009; 7:14; PMID:19534786; http://dx.doi.org/ 10.1186/1478-811X-7-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klijn C, Koudijs MJ, Kool J, ten Hoeve J, Boer M, de Moes J, Akhtar W, van Miltenburg M, Vendel-Zwaagstra A, Reinders MJ, et al.. Analysis of tumor heterogeneity and cancer gene networks using deep sequencing of MMTV-induced mouse mammary tumors. PLoS One 2013; 8:e62113; PMID:23690930; http://dx.doi.org/ 10.1371/journal.pone.0062113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Theodorou V, Kimm MA, Boer M, Wessels L, Theelen W, Jonkers J, Hilkens J. MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat Genet 2007; 39:759-69; PMID:17468756; http://dx.doi.org/ 10.1038/ng2034 [DOI] [PubMed] [Google Scholar]
- 5.Ikink GJ, Boer M, Bakker ERM, Hilkens J. IRS4 induces mammary tumorigenesis and confers resistance to HER2-targeted therapy through constitutive PI3K/AKT-pathway hyperactivation. Nat Commun 2016; 7:13567; PMID:27876799; http://dx.doi.org/ 10.1038/ncomms13567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karrman K, Kjeldsen E, Lassen C, Isaksson M, Davidsson J, Andersson A, Hasle H, Fioretos T, Johansson B. The t(X;7)(q22;q34) in paediatric T-cell acute lymphoblastic leukaemia results in overexpression of the insulin receptor substrate 4 gene through illegitimate recombination with the T-cell receptor beta locus. Br J Haematol 2009; 144:546-51; PMID:19055661; http://dx.doi.org/ 10.1111/j.1365-2141.2008.07453.x [DOI] [PubMed] [Google Scholar]
- 7.Weischenfeldt J, Dubash T, Drainas AP, Mardin BR, Chen Y, Stutz AM, Waszak SM, Bosco G, Halvorsen AR, Raeder B, et al.. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet 2016; 49:65-74 advance online publication; PMID: 27869826; http://dx.doi.org/ 10.1038/ng.3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown KK, Toker A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000 Prime Rep 2015; 7:13; PMID:25750731; http://dx.doi.org/24742739 10.12703/P7-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilks ST. Potential of overcoming resistance to HER2-targeted therapies through the PI3K/Akt/mTOR pathway. The Breast 2015; 24:548-55;PMID:26187798; http://dx.doi.org/24742739 10.1016/j.breast.2015.06.002 [DOI] [PubMed] [Google Scholar]
- 10.Andre F, O'Regan R, Ozguroglu M, Toi M, Xu B, Jerusalem G, Masuda N, Wilks S, Arena F, Isaacs C, et al.. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol 2014; 15:580-91; PMID:24742739; http://dx.doi.org/ 10.1016/S1470-2045(14)70138-X [DOI] [PubMed] [Google Scholar]