

ABSTRACT
Our recent work showed that sunitinib exerts dual effect on cancer cells in different dose ranges. In clinically relevant doses, cancer cells tolerate sunitinb cytotoxicity by upregulating pro-survival MCL-1 and activating mTORC1 signaling. Inhibition of MCL-1 or mTORC1 sensitized cancer cells to sunitinib. Analysis of tissues from patients correlated MCL-1/mTORC1 induction with resistance to sunitinib.
KEYWORDS: Autophagy, cell death, MCL-1, mTOR, mTORC1, Sunitinib
Resistance to chemo- and targeted therapy represents a major clinical hurdle in cancer management. In a big part, drug resistance can be attributed to the activation of pro-survival responses in cancer cells. The initial response of cells to stress such as that triggered by anticancer agents is geared toward adaption and resistance through upregulation of survival responses. However, if the stress is overwhelming or prolonged, cell death is initiated. Therefore, the ultimate outcome of exposure to anticancer agents and whether cells can cope and survive or fail and die depend on a variety of factors including the type, duration, and level of the insult as well as the adaptive capacity of cancer cells.
Among targeted therapies introduced recently, sunitinib has become one of the most commonly used first-line therapy for the treatment of several types of cancer. However, resistance to sunitinib has emerged as the major limitation for its clinical use. A high percentage of patients are intrinsically resistant to sunitinib, and the majority of initially responsive patients eventually become resistant in few months, leading to a very modest overall therapeutic benefit.1,2 Sunitinib is a multi-tyrosine kinase inhibitor that modulates a wide array of targets directly or indirectly. It therefore does not come as a surprise that several studies have proposed diverse mechanisms of cytotoxicity as well as resistance of sunitinib. Many of those studies relied on comparison of gene expression profiles of sunitinib-sensitive versus resistant cancer cells and therefore had the shortcoming of not being suitable to identify molecular mechanisms of resistance that do not involve modulation of gene expression such as activation of signaling pathways or modulation of protein degradation. Moreover, most of those studies used concentrations of sunitinib that are higher than those thought to be pharmacologically relevant in patients.3
In our study published recently,4 we applied a unique approach as compared with other studies, focusing on the adaptive pro-survival responses that cells opt to for maintaining their viability and tolerating the cytotoxic effects triggered by sunitinib. We then further analyzed the relevance of those adaptive responses to intrinsic as well as acquired resistance of cancer cells to sunitinib. We particularly focused on the modulation of mTOR signaling and the level of the antiapoptotic Bcl-2 proteins MCL-1, BCL-2 and BCL-XL as crucial determinants of cell survival and resistance to anticancer agents.5,6
Initially, in each of several cell lines representative of different types of cancer; renal cell carcinoma, pancreatic neuroendocrine tumor, colorectal and osteosarcoma, we distinguished sunitinib concentration ranges that are either tolerated from those that exert cytotoxic effects. The thresholds above which sunitinib concentration started to be cytotoxic varied among different cell lines but were generally above the concentration reachable in patients. Our analysis of cell lysates derived from different cell lines treated with doses of sunitinib on either sides of the cell-line-specific threshold showed that sunitinib exerts dual effects on MCL-1 levels and mTOR complex I (mTORC1) signaling in both concentration ranges. In cytotoxic doses, sunitinib triggered a decline in MCL-1 levels and inhibition of mTORC1 signaling activity. Conversely, in lower concentrations, i.e. below the cytotoxic threshold (that are likely to be more clinically relevant), sunitinib treatment resulted in enhancing MCL-1 levels and induction of mTORC1 activity. Consistent with these in vitro results, sunitinib increased MCL-1 levels and mTORC1 activity in murine tumor xenografts. Importantly, we found that induction of MCL-1 and mTORC1 represents pro-survival cellular responses that account for the observed lack of cytotoxicity of clinically relevant doses of sunitinib as inhibition of MCL-1 or mTORC1 greatly sensitized cancer cells to those doses of sunitinib.
Among the wide range of cellular processes regulated by mTORC1, autophagy is a prominent example.6 MCL-1 has also emerged recently as a negative regulator of autophagy through regulation of autophagy mediator Beclin 1.7,8 Consistent with the dual effect of low and high concentrations of sunitinib on mTORC1 and MCL-1, we observed that low and high concentrations of sunitinib inhibit and induce autophagy, respectively. This finding may contribute to explaining the conflict between studies that investigated the interplay between sunitinib and autophagy and reached different conclusions.9,10
Beyond the specific results related to sunitinib, in more general terms, these results highlight an important experimental and conceptual necessity sometimes overlooked by researchers, which is important in exploring the reproducibility of certain results against a wide range of doses. Our results therefore provide a warrant against hasty generalization of results obtained by a single dose of any given drug.
Deeper mechanistic analysis revealed that sunitinib modulates MCL-1 levels by affecting its proteasomal degradation. Furthermore, the dual effect of sunitinib on MCL-1 stability in different dose ranges of sunitinib was attributed to dual effect on GSK3β and ERK phosphorylation, whereas lower doses of sunitinib inhibit GSK3β and activate ERK, and the opposite occurs in higher dose ranges. GSK3β and ERK in turn phosphorylate MCL-1 in different sites leading, respectively, to increasing and decreasing its proteasomal degradation. Modulation of GSK3β by sunitinib in both ranges also mediated the effect on mTOR signaling.
Finally, our analysis of tumor samples derived prior and post treatment from sunitinib-resistant patients provided the proof of concept that the increase in MCL-1 levels and mTORC1 activity upon treatment contributes to resistance to sunitinib.
The reported absence of evident direct cytotoxic effects for clinically relevant doses of sunitinib on cancer cells led to shifting the focus to its antiangiogenic effects and ultimately positioning it in the group of antiangiogenic drugs.3 Our results thus provide detailed molecular explanation for the reported absence of evident direct cytotoxic effects of clinically relevant doses of sunitinib on cancer cells, which led to shifting the focus to its antiangiogenic effects and ultimately positioning it in the group of antiangiogenic drugs.
Taken together, our results indicate that dual modulation of MCL-1 stability and mTORC1 signaling exerted by different dose ranges of sunitinib is a major determinant of resistance or sensitivity of cancer cells to sunitinib (Fig. 1) and further give a rationale for potential synergistic therapeutic benefit of a combination of sunitinib and MCL-1 or mTOR inhibitors that warrants further clinical testing.
Figure 1.
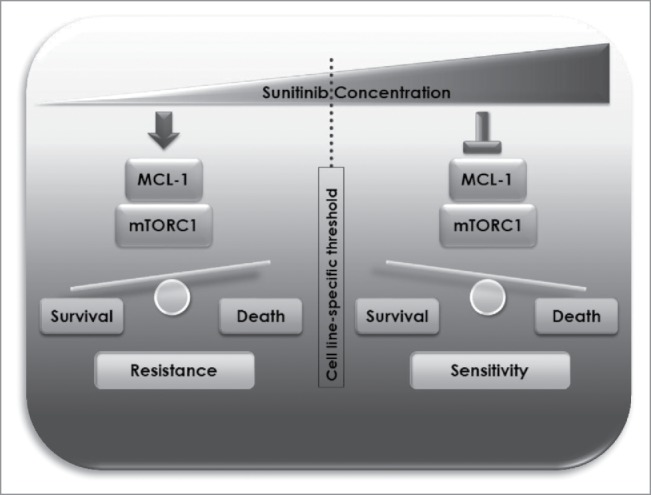
Schematic representation of mechanisms of resistance and sensitivity to sunitinib mediated by dual modulation of MCL-1 and mTORC1 at different dose ranges.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by AIRC (Italian Association for Cancer Research), FUV (Umberto Veronesi Foundation), and INDICAR (Interdisciplinary Cancer Research Postdoctoral Fellowship Program of the University of Vienna) fellowships to ME.
References
- 1.Bottsford-Miller JN, Coleman RL, Sood AK. Resistance and escape from antiangiogenesis therapy: Clinical implications and future strategies. J Clin Oncol 2012; 30(32):4026-34; PMID:23008289; http://dx.doi.org/ 10.1200/JCO.2012.41.9242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morais C. Sunitinib resistance in renal cell carcinoma. J Kidney Cancer VHL 2014; 1(1):1-11; http://dx.doi.org/ 10.15586/jkcvhl.2014.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang D, Ding Y, Li Y, Luo WM, Zhang ZF, Snider J, Vandenbeldt K, Qian CN, Teh BT. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res 2010; 70(3):1053-62; PMID:20103629; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3722 [DOI] [PubMed] [Google Scholar]
- 4.Elgendy, M., Abdel-Aziz, A.K., Renne, S.L., Bornaghi, V., Procopio, G., Colecchia, M., Kanesvaran, R., Toh, C.K., Bossi, D., Pallavicini, I., Perez-Gracia, J.L., Lozano, M.D., Giandomenico, V., Mercurio, C., Lanfrancone, L., Fazio, N., Nole, F., The, B.T., Renne, G., Minucci, S.. Dual modulation of MCL-1 and mTOR determines the response to sunitinib. Journal of Clinical Investigation 2017;127(1):153-168; ; http://dx.doi.org/ 10.1172/JCI84386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cella CA, Minucci S, Spada F, Galdy S, Elgendy M, Ravenda PS, Zampino MG, Murgioni S, Fazio N Dual inhibition of mTOR pathway and VEGF signalling in neuroendocrine neoplasms: From bench to bedside. Cancer Treat Rev 2015(15)130-9 Cancer Treat Rev. 2015 Nov; 41(9):754-60; PMID:26142874; http://dx.doi.org/ 10.1016/j.ctrv.2015.06.008 [DOI] [PubMed] [Google Scholar]
- 6.Laplante M, Sabatini DM. MTOR signaling in growth control and disease. Cell 2012; 149(2):274-293; PMID:22500797; http://dx.doi.org/ 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elgendy M, Ciro M, Saad Eldin A, Belmonte G, Dal Zuffo R, Mirraco C, Mercurio C, Lanfrancone L, Foiani M, Minucci S. Beclin 1 restrains tumorigenesis through Mcl-1 destabilization in an autophagy-independent reciprocal manner. Nat Commun 2014; 5:5637; PMID:25472497; http://dx.doi.org/ 10.1038/ncomms6637 [DOI] [PubMed] [Google Scholar]
- 8.Elgendy M, Minucci S. A novel autophagy-independent, oncosuppressive function of BECN1: Degradation of MCL1. Autophagy 2015; 11:2-4; http://dx.doi.org/ 10.1080/15548627.2015.1029836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao Y, Xue T, Yang X, Zhu H, Ding X, Lou L, Lu W, Yang B, He Q. Autophagy plays an important role in Sunitinib-mediated cell death in H9c2 cardiac muscle cells. Toxicol Appl Pharmacol 2010; 248(1):20-7; PMID:20637791; http://dx.doi.org/ 10.1016/j.taap.2010.07.007 [DOI] [PubMed] [Google Scholar]
- 10.Abdel-aziz AK, Shouman S, El-demerdash E, Elgendy M, Abdel-naim AB Chloroquine as a promising adjuvant chemotherapy together with sunitinib. Chem Biol Interact 2014; Jun 25; 217:28-40; PMID:24751611; http://dx.doi.org/ 10.1016/j.cbi.2014.04.007 [DOI] [PubMed] [Google Scholar]