

ABSTRACT
In a recent publication in Science Signaling, we showed that a Mes molecular subtype of epithelial ovarian cancer (EOC) harboring epithelial-mesenchymal transition (EMT) features has a unique signaling network downstream of the GAS6/AXL pathway. Our finding leads to a potential strategy for treating the Mes subtype of EOC by targeting AXL.
Keywords: AXL, EMT, epithelial ovarian cancer
Epithelial ovarian cancer (EOC) is a clinically and molecularly complex and heterogeneous disease.1 It remains the most lethal gynecologic malignancy and more effective systemic therapeutic approaches beyond traditional surgery and chemotherapy are clearly required to improve patient outcomes. Recent advances in our understanding of the underlying molecular aberrations in EOC and in the development of new drugs for EOC are now providing unique opportunities for precision medicine in the management of this disease. With approval of the anti-vascular endothelial growth factor (VEGF) antibody bevacizumab for use in front-line and recurrent EOC treatment and the first poly ADP ribose polymerase (PARP) inhibitor, Olaparib, for use in the treatment of recurrent BRCA1/2-mutant EOC by the US Foods and Drugs Administration (FDA) and European Medicines Agency (EMA), it is almost certain that we are only seeing the tip of the iceberg in a new era of personalized therapy for patients with EOC, with numerous therapeutic possibilities yet to be explored.
Our group has previously identified EOC gene expression molecular subtypes (GEMS) that could be used to predict the outcomes of EOC patients.2 Four of the sub-types were identified as Epithelial-A (Epi-A), Epithelial-B (Epi-B), Mesenchymal (Mes), and Stem-A. These tumors have varying abilities to undergo epithelial-mesenchymal transition (EMT), in which epithelial cells transform into mesenchymal cells associated with aggressive, invasive, and metastatic cancer. The Epi-A and Epi-B subtypes have low EMT status whereas the Mes and Stem-A subtypes have high EMT status.3 In our recent study,4 the Mes subset of EOC was shown to be dependent on the unique pattern of signaling mediated by the receptor tyrosine kinase (RTK) AXL. AXL was known to be overexpressed in advanced EOC, particularly in tumors that have metastasized. However, it was unclear whether AXL functioned differently between the above-mentioned EOC GEMS. In our new study, we further identified the sustained temporal activation of AXL and its subsequent crosstalk with other RTKs as a unique feature of the Mes subtype. We discovered that the Mes subtype has lower levels of Dual Specificity Phosphatase 4 (DUSP4) that, together with AXL-RTK crosstalk, enables tumors with the Mes subtype to propagate and sustain AXL signals through recurrent activation of phosphorylated extracellular signal-regulated kinase (pERK). The sustained AXL-pERK signaling axis contributes to aggressive behavior in terms of enhanced motility and invasion. This mechanism of signaling amplification is absent in other subtypes such as Epi-A, in which elevated DUSP4 levels and lack of AXL-RTK crosstalk result in a linear and transient signaling schema. This implies that the Mes subtype of EOC cells has been rewired to enhance AXL-pERK activation. In addition, we showed that the Mes subtype is highly enriched in an AXL-signature. Therefore, it is highly plausible that the EOC Mes subtype would more sensitive to AXL-targeted therapy. Our data are consistent with findings from research groups working on other cancers.5-7
What remains elusive, however, is how the AXL signaling network contributes to platinum sensitivity in EOC. In terms of the correlation between GEMS and platinum sensitivity, the data from one small series of 23 paired primary “platinum-sensitive” and recurrent “platinum resistant” tumor samples are particularly intriguing.8 Labeling this small cohort of paired samples with our GEMS profiling scheme (Fig. 1) revealed 4 Epi-A (17.4%), 8 Epi-B (34.8%), 6 Mes (26.1%), 3 Stem-A (13.0%), and 2 Stem-B (8.7%) tumors in the platinum-sensitive primary group. During relapse, 3 of the 4 Epi-A (75%), 6 of the 8 Epi-B (75%), and 2 of the 3 Stem-A (66.7%) tumors switched to the Mes subtype upon gaining platinum resistance. Most of the Mes tumors remained Mes, with only 1 out of 6 switching to Epi-B (16.7%). In general, 10 out of the 23 pairs (43.5%) maintained the same GEMS during transition from platinum sensitive to platinum resistant. The Mes subtype was the most dominant subtype if there was a GEMS switch during the acquisition of platinum resistance; 11 of the 13 pairs (84.6%) that showed GEMS switch became Mes upon acquiring platinum resistance. Also, the Mes subtype appeared to be stable during disease progression, with very rare subtype drift. Therefore, it can be concluded that the Mes subtype seems to be the default GEMS upon acquiring platinum resistance. Since amplified AXL signaling is highly correlated with the Mes subtype,4 we can assume that the distribution of AXL-signature positive population will follow the pattern of Mes. Indeed, in the above mentioned 23 paired tumors expression of the AXL-signature was significantly enriched in the platinum-resistant relapse samples. However, 7 out of the 23 (30.4%) platinum-resistant relapse tumors were non-Mes, and platinum-resistant EOC remains a heterogeneous population of patients with different resistant mechanisms. The estimated population of Mes subtype within the platinum-resistant relapse EOC population is approximately 70%.
Figure 1.
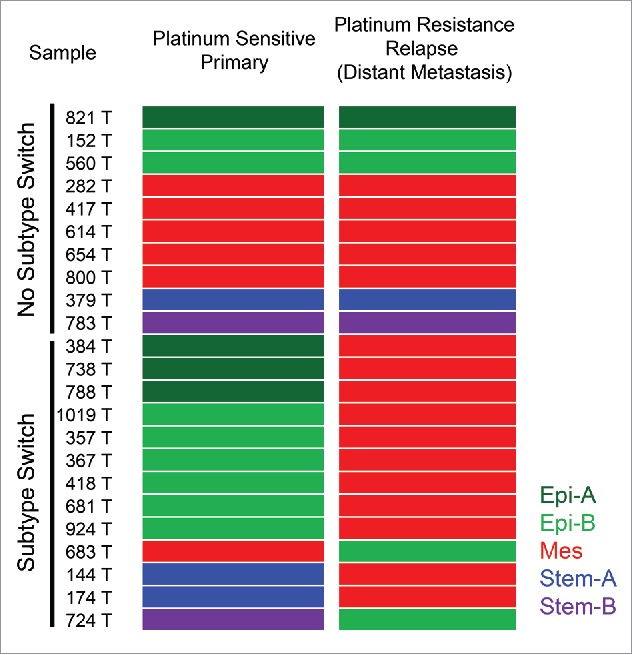
Molecular subtype switch in platinum-resistant relapsed epithelial ovarian cancer (EOC). Samples of the cohort of E-MTAB-611, which consisted of 23 paired platinum-sensitive primary ovarian tumors (left colored panel) and platinum-resistant relapsed tumors at distant metastatic sites (right colored panel), were classified as with (Subtype Switch) or without (No Subtype Switch) subgroup switching with annotation of the 5 gene expression molecular subtypes (GEMS) as follows: Epi-A (dark green); Epi-B (light green); Mes (red); Stem-A (blue); Stem-B (violet).
There are no adequate preclinical data to support the hypothesis that anti-AXL therapy would restore platinum sensitivity. In comparisons of the isogenic-paired cell lines PEO1 (platinum sensitive) and PEO4 (platinum resistant) derived from the same EOC patient, AXL expression was in fact lost in PEO4.4 These two cell lines were both labeled as Epi-A subtype based on the GEMS profiling, suggesting that platinum resistance can follow mechanisms that are independent of GEMS subtype switch and AXL signaling. Of note, the PEO1 line is a BRCA2 mutant whereas BRCA2 function is restored in the PEO4 line as the result of a secondary BRCA2 mutation.9 Therefore, the platinum resistance mechanism demonstrated by this cell line pair would most likely to be related to partial restoration of BRCA2 function. From the preclinical data described in Antony et al., 2016, the GI50 value of the selective AXL inhibitor R428 against the AXL-null PEO4 line was extremely high at 195.9 μM in vitro. Transfection of PEO4 with functional AXL resulted in a significant reduction of the GI50 to R428 of 30.48 μM in vitro. However, this GI50 is still much higher than the average in vitro GI50 of Mes lines of 2.91 μM. Therefore, based on the assumption that anti-AXL therapy could reverse platinum resistance, it is not clear what would be the optimal dose of R428 to restore platinum sensitivity in a heterogeneous group of patients with different subtypes.
Nevertheless, these data clearly demonstrated the necessity to stratify EOC patients based on either Mes subtype or the status of AXL signaling in both the platinum-sensitive and -resistant settings. Anti-AXL therapy would most likely exert cytotoxic effects as a single agent toward tumors dependent on the EMT and AXL signaling pathway. Downstream signaling molecules such as pERK might serve as reliable pharmacodynamic biomarkers for anti-AXL therapy. Therefore, it would be sensible to start with a clinical study to test the hypothesis by administering anti-AXL treatment as a single agent in a carefully preselected patient population. Considering platinum-resistant EOC patients for anti-AXL therapy without GEMS stratification would probably not be an optimal strategy.
Looking forward, positioning anti-AXL therapy as the first-line treatment in combination with platinum-based chemotherapy in selected patients with Mes subtype EOC might be a promising strategy. Given our limited understanding of how AXL signaling might interfere with platinum sensitivity, combination use of anti-AXL with conventional chemotherapy warrants further bench research. It is worth exploring the interaction between AXL signaling and the homologous recombination (HR) repair pathway within the Mes subtype because a good percentage of Mes tumors harbor the “BRCAness” phenotype manifested by either BRCA mutation or HR deficiency. Would a certain subset of patient further benefit from combination of anti-AXL and PARP inhibitor treatment? One recent study10 demonstrated that in non-small cell lung cancers (NSCLCs), triple-negative breast cancers (TNBCs), and head and neck squamous cell carcinomas (HNSCCs), downregulation or inhibition of AXL not only directly reversed the EMT phenotype, but also led to decreased expression of DNA repair genes and diminished HR efficiency and RAD51 foci formation. As a result, AXL inhibition caused a state of HR deficiency in the cells, making them sensitive to PARP inhibition. This could be a future combination strategy to extend the therapeutic indications for PARP inhibitors beyond the BRCAness phenotype.
In summary, the EOC field has entered an exciting period when our understanding of the disease biology and available therapeutic options are converging and will hopefully accelerate the development of novel therapeutic strategies that will improve outcomes for patients with EOC. AXL signaling appears to be an emerging therapeutic target in this arena, and with the advent of AXL-targeting agents such as selective AXL inhibitors or antibodies future clinical studies to further elucidate the therapeutic relevance of this pathway in EOC are eagerly anticipated.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work is supported by National Research Foundation (NRF) Singapore and the Singapore Ministry of Education under its Research Centers of Excellence initiative, National Medical Research Council (NMRC) under its Center Grant scheme to National University Cancer Institute (NCIS).
Author contributions
R.Y.H, J.A, and D.S.T wrote the manuscript. T.Z.T. performed the GEMS profiling of the Marchini data set in Fig. 1.
References
- 1.Bowtell DD, et al.. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer 2015; 15:668-79; PMID:26493647; http://dx.doi.org/ 10.1038/nrc4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan TZ, et al.. Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol Med 2013; 5:983-98; PMID:23666744; http://dx.doi.org/25214461 10.1002/emmm.201201823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan TZ, et al.. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med 2014; 6:1279-93; PMID:25214461; http://dx.doi.org/ 10.15252/emmm.201404208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antony J, et al.. The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype-specific therapeutic target for ovarian cancer. Sci Signal 2016; 9:ra97; PMID:27703030; http://dx.doi.org/ 10.1126/scisignal.aaf8175 [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, et al.. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012; 44:852-60; PMID:22751098; http://dx.doi.org/ 10.1038/ng.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gjerdrum C, et al.. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A 2010; 107:1124-9; PMID:20080645; http://dx.doi.org/ 10.1073/pnas.0909333107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byers LA, et al.. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013; 19:279-90; PMID:23091115; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchini S, et al.. Resistance to platinum-based chemotherapy is associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Eur J Cancer 2013; 49:520-30; PMID:22897840; http://dx.doi.org/ 10.1016/j.ejca.2012.06.026 [DOI] [PubMed] [Google Scholar]
- 9.Sakai W, et al.. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res 2009; 69:6381-6; PMID:19654294; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balaji K, et al.. AXL Inhibition Suppresses the DNA Damage Response and Sensitizes Cells to PARP Inhibition in Multiple Cancers. Mol Cancer Res 2016; PMID:27671334; http://dx.doi.org/ 10.1158/1541-7786.MCR-16-0157 [DOI] [PMC free article] [PubMed] [Google Scholar]