

Abstract
Background/Purpose
Transforming growth factor β activated kinase 1 (TAK1) is a key MAPKKK family protein in interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), or toll-like receptor signaling. We examined the posttranslational modification of TAK1 and its therapeutic regulation in rheumatoid arthritis (RA).
Methods
The effect of TAK1, IRAK-1, or TRAF6 inhibition was evaluated in IL-1β-induced human RA synovial fibroblasts (RA-FLS). Western blotting, immunoprecipitation, and 20S proteasome assay were used to study the ubiquitination process in RA-FLS. The efficacy of epigallocatechin-3-gallate (EGCG), a potent anti-inflammatory molecule, in regulating these processes in RA-FLS was evaluated. Molecular docking was performed to examine the interaction of EGCG with human TAK1, IRAK-1, and TRAF6. These findings were validated using a rat adjuvant-induced arthritis (AIA) model.
Results
Inhibition of TAK1, not IRAK-1 or TRAF6, completely abrogates IL-1β-induced IL-6 and IL-8 synthesis in RA-FLS. EGCG inhibits TAK1 phosphorylation at Thr184/187 and occupies the C174 position, an ATP-binding site, to inhibit its kinase activity. EGCG pretreatment also inhibits K63-autoubiquitination of TRAF6, a post-translational modification essential for TAK1 autophosphorylation, by forming stable hydrogen-bond at the K124 position on TRAF6. Furthermore, EGCG enhances associated deubiquitinase expression to rescue proteins from proteasomal degradation. Western blot analyses on the joint homogenates from rat AIA show a significant increase in K48-linked polyubiquitination, TAK1 phosphorylation, and TRAF6 expression when compared to the naïve group. Administration of EGCG (50 mg/kg/day) for 10 days ameliorates AIA by reducing TAK1 phosphorylation and K48 polyubiquitination.
Conclusions
This study provides rationale for targeting TAK1 for RA treatment by EGCG.
Keywords: Rheumatoid arthritis, synovial fibroblasts, ubiquitination, TAK1, EGCG, posttranslational modification
Introduction
In rheumatoid arthritis (RA), the increased expression of interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and IL-6 in the synovial microenvironment contributes to joint pain, inflammation and tissue destruction (1). While the discovery of highly effective biological therapies in the last decade has transformed the therapeutic landscape for RA treatment, the ever-increasing numbers of non- or partial responders concomitant with the associated morbidity and mortality pose a significant socioeconomic and clinical challenge. The success of pharmacological approaches to clinically develop tofacitinib, a pan-Janus kinase (JAK) inhibitor, has opened a new chapter of small-molecule therapeutics in the treatment of RA (2). While the success of this inhibitor related to clinical efficacy and safety remains untested, this has accelerated research for the discovery of novel small-molecule kinase-directed therapeutics in RA (3, 4). Thus, validation of a target protein that is critical in cytokine signaling networks may yield higher efficacy and a better clinical response (4).
Recent studies on the intracellular signaling pathways have identified transforming growth factor β-activated kinase 1 (TAK1) as an interesting therapeutic target for inflammatory diseases and cancer (5). TAK1 is a mitogen activated protein kinase kinase kinase (MAPKKK) that mediates activation of downstream MAPKs (JNK and p38) and nuclear factor-κB (NF-κB) pathways in response to IL-1β, TNF-α, or Toll-like receptor (TLR) stimulation (6, 7). Among these cytokines, IL-1β plays an important pathological role in RA (8, 9). Surprisingly, a recombinant form of human IL-1 receptor antagonist, anakinra, lacked clinical efficacy in the treatment of RA and elicited serious infections such as pneumonia and cellulitis (10, 11). While the biologic agents that target TNF-α (etanercept) and IL-6 (tocilizumab) are clinically successful in the management of RA, therapeutic approaches aimed at developing small-molecule inhibitors with more efficient blocking of IL-1β signaling pathways are desired for RA and other IL-1-driven diseases (12).
IL-1β binding to IL-1R1 initiates an autophosphorlyation of IL-1 receptor activating kinase-4 (IRAK-4), which further recruits IRAK-1 to Myd88 and subsequently autophosphorylates to trigger its activation and consequent degradation (6). TRAF6, an E3 ubiquitin ligase, forms a complex with IRAK-1 that dissociates from the receptor and translocates to the cytoplasm to recruit and activate TAK1 by K63 mono (Lys 34) and polyubiquitination (13, 14). TAK1 activation at its kinase domain threonine (Thr) 184/187 results in MAPKs phosphorylation and IκB kinase (IKK) activation leading to IκBα degradation and NF-κB activation (15).
While TAK1 is a key signaling protein in IL-1β-induced gene expression, its highly regulated posttranslational modifications such as autophosphorylation, ubiquitination, or deubiquitination by IRAK-1 or TRAF6 defines its role in disease pathogenesis (16). Despite established roles of MAPKs and NF-κB pathways in IL-1β signaling in RA, significant gaps remain in our understanding of the role of adaptor proteins proximal to IL-1R1 (IRAK-1/TRAF6/TAK1) to strike therapeutic advantages for effectively inhibiting IL-1β signaling in RA. Studies from our lab have shown the efficacy of epigallocatechin-3-gallate (EGCG), a potent anti-inflammatory molecule, in regulating IL-1β-induced IL-6 and chemokine production and matrix metalloproteinase-2 (MMP-2) activation in human RA synovial fibroblasts (RA-FLS) by inhibiting the activation of JNK, p38, and NF-κB (17, 18). These findings suggest that EGCG may be interfering with the proteins proximal to IL-1R to suppress multiple inflammatory signaling pathways. Thus, the present study was carried out with a two-pronged approach, aimed at defining the role and underlying mechanism of TAK1 activation in RA and testing the selectivity of EGCG in regulating these events in RA-FLS and in a rat adjuvant-induced arthritis (AIA) model of human RA. Our results showed that EGCG selectively inhibits TAK1 activation by blocking its phosphorylation at the Thr184/187 ATP-binding site and hinders the association of TRAF6 and TAK1 through downregulation of TRAF6-associated K63 autoubiquitination. These findings suggest that TAK1 is a potential therapeutic target in RA and EGCG may be developed as a TAK1 inhibitor for the treatment of RA and other inflammatory diseases.
Materials and Methods
Antibodies and Reagents
Recombinant human IL-1β and TNF-α were purchased from R&D Systems (Minneapolis, MN). EGCG (>95% pure) was purchased from Sigma (Cat# 4143). The 20S proteasome activity assay was purchased from Cayman Chemical (Ann Arbor, MI). Goat anti-rabbit and goat anti-mouse horseradish peroxidase-linked secondary antibodies were purchased from Cell Signaling Technology (Beverly, MA). Inhibitors for the signaling protein IRAK-1 (N-(2-Morpholinylethyl)-2-(3-nitrobenzoylamido)-benzimidazole) and TAK1 (5Z-7-Oxozeaenol) were from EMD Millipore (Billerica, MA), and TRAF6 control and inhibitor peptides were from Imgenex/Novus (Littleton, CO). Human IL-6 and IL-8 ELISA assay duo kits were purchased from R&D Systems (Minneapolis, MN). Rabbit anti IRAK-1 (Sc-7883), mouse monoclonal TRAF6 (sc-8409), and mouse monoclonal β-actin (sc-47778) were purchased from Santa Cruz Biotech, (Santa Cruz, CA). Anti-FK-2 (Enzo Life Sciences), Anti-phospho TAK1 Ser184/187 (#4508), anti-phospho IRAK4 Thr345/Ser346 (#11927), anti-IRAK-4 (#4363), anti-MyD88 (#4238), anti-K63 polyubiquitin (#5621), and anti-K48 polyubiquitin (#8081) antibody were purchased from Cell Signaling Technology, anti-phospho TAK1 Ser439 (ab109404), anti-TAK1 (ab109526), and anti-TRAF6 (ab33915) were from Abcam (Cambridge, MA).
Culture of human RA-FLS
FLS were isolated from RA synovium obtained according to the Institutional Review Board (IRB) approved protocol in compliance with the Helsinki Declaration from patients who had undergone total joint replacement surgery or synovectomy and processed as described previously (17).
Treatment of RA-FLS
To evaluate the time-dependent activation of IL-1β-induced signaling pathways and the protective effect of EGCG treatment, RA-FLS (2 × 105/well) were plated in 6-well plates with or without EGCG (2.5–20 μM) for overnight (12–14 hours), followed by IL-1β (10 ng/ml) stimulation for 30 minutes (for signaling studies) or 24 hours to evaluate the production of IL-6 and IL-8 in the conditioned media.
To study the effect of IL-1β stimulation on IRAK-1 degradation, RA-FLS were stimulated with IL-1β (10 ng/ml) for 0–120 mins. To further study the effect of MTX or dexamethasone on IL-1β-induced IRAK-1 degradation, RA-FLS were pretreated with MTX (10 μM) or Dexa (20 μM) for an hour followed by IL-1β stimulation of 30 minutes.
IRAK-1 in vitro kinase activity
IRAK-1 kinase activity was determined using a fluorescence based in vitro kinase assay (ADAPTA™ Kinase Assay, Life Technologies). Briefly, the 2X IRAK-1/Histone H3 (1–20) peptide mixture was prepared in 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, 1 mM EGTA. The final 10 μL kinase reaction was carried out for 60 minutes consisting of 100 μM ATP, EGCG (10 nM – 20 μM), 3.17–30.5 ng IRAK-1, and 100 μM Histone H3 (1–20) peptide in 32.5 mM HEPES pH 7.5, 0.005% BRIJ-35, 5 mM MgCl2, 0.5 mM EGTA. After an hour of incubation, 5 μL of Detection Mix was added, followed by the emission read at 665 nM.
20S proteasome activity assay
20S proteasome activity was determined in RA-FLS using an assay kit (Cayman Chemical) as described earlier (19).
Western blotting analysis
Western blot analysis was performed as described earlier (17, 18). Specific details are provided in Supplementary data.
Immunoprecipitation Assay
RA-FLS were grown in 150 mm dishes up to 80% confluence, overnight starved with or without 20 μM EGCG, followed by IL-1β stimulation for 30 minutes. Cells were washed 2 times in ice cold 1X PBS, lysed in 500 μl of RIPA buffer as described earlier, then utilized for immunoprecipitation assays as described in Supplementary data.
Rat adjuvant-induced arthritis (AIA)
Female Lewis rats, ~100 g (Harlan Laboratories, Indianapolis, IN), were injected subcutaneously at the base of the tail with 300 μL (5 mg/ml) of lyophilized Mycobacterium butyricum (Difco Laboratories, Detroit, MI) in sterile mineral oil. The day of adjuvant injection was considered day 0. Ankle circumferences were measured on day 17 by the blinded observer as described previously (17). Healthy (naïve) rats group served as a control for AIA group. In the treatment group, EGCG (50 mg/kg, i.p.) was administered as described in our earlier study (17). EGCG (50 mg/kg) in rats corresponds to 480 mg of human equivalent dose based on the body-surface area ratio (20). The ankle circumferences of both the hind ankles from each animal were averaged and ‘n’ is represented as the number of animals used in each of the experimental groups. All animal studies were approved by the university’s IACUC committee.
Molecular Modeling Studies
Ligand preparation
EGCG ((2R,3R)-5,7-dihydroxy-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-1-benzopyran-3-yl 3,4,5-trihydroxybenzoate) was first optimized as a ligand at B3LYP/6-311++G** level of calculation using jaguar2014.3 and then prepared using the LigPrep module for docking in the Glide module of Schrodinger suite 2014.3. Further details regarding the in silico modeling and docking studies is provided in Supplementary data.
Human Phospho-kinase antibody array
To evaluate the effect of EGCG on other RA-FLS kinases involved in important pathophysiological processes, we utilized human antibody array kit (Cat#ARY003B; R&D systems, MN). Further experimental details of the assay are provided in Supplementary data.
Statistical analysis
Statistical analysis was performed using a Kruskal-Wallis nonparametric test followed by a Mann-Whitney U test to evaluate the statistical significance of group differences in measured parameters from IL-6 and IL-8 protein expression or Western blotting studies in RA-FLS. Student’s t-test was performed to calculate statistical differences between the means of the different protein variables obtained from in vivo findings. P values (2-tailed) less than 0.05 were considered significant.
Results
TAK1 regulates IL-1β induced IL-6 and IL8 production
IL-1β is a master cytokine for local and systemic inflammation. RA-FLS stimulation with IL-1β (10 ng/ml) resulted in more than a 180- and 250-fold increase in IL-6 and IL-8 production, respectively (Fig. 1A & 1B; p<0.001). Pretreatment with EGCG (2.5–20 μM) resulted in IL-6 and IL-8 inhibition at 10 and 20 μM concentrations (p<0.05). To test if the chronic exposure of EGCG at nanomolar concentrations may produce similar inhibitory effect to blunt IL-1β-induced IL-6 and IL-8 production, RA-FLS were treated with EGCG (1–1000 nM, daily dose; in 5% FBS-RPMI 1640) for 7 days, followed by serum starvation, and stimulation with IL-1β (10 ng/ml) for 8 and 24 hours (supplementary Fig. S1). The result of the analysis showed that EGCG treatment even at nanomolar concentrations was effective in inhibiting IL-1β-induced IL-6 (20–35%) and IL-8 (15–20%) in a dose-dependent manner (Fig. S1; p<0.01 for IL-6).
Fig. 1. TAK1 regulates IL-1β-induced IL-6 and IL-8 production in RA-FLS. (A.
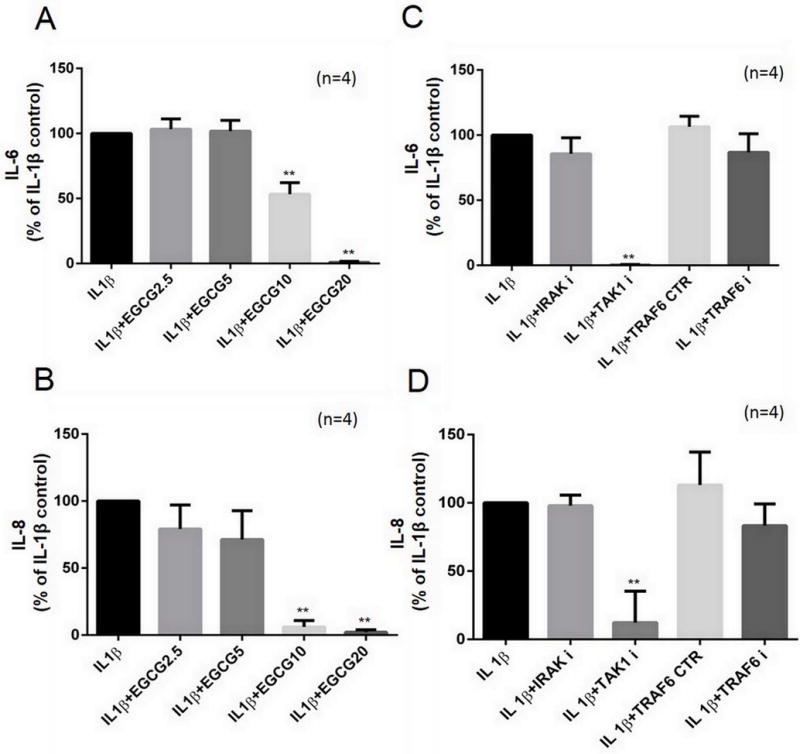
and B) RA-FLS were pretreated with EGCG (2.5–20 μM) overnight, followed by IL-1β (10 ng/ml) stimulation for 24 hours. IL-6 and IL-8 production was determined in the conditioned media using commercially available ELISA kits. (C and D) RA-FLS were preincubated with signaling inhibitors for IRAK-1 (N-(2-Morpholinylethyl)-2-(3-nitrobenzoylamido)-benzimidazole; 50 μM), TAK1 (5Z-7-Oxozeaenol; 5 μM), TRAF6 (inhibitor peptide; 50 μM), or control peptide for TRAF6 (50 μM) for 2 hours, followed by stimulation with IL-1β (10 ng/ml) for 24 hours to determine IL-6 and IL-8 production in the conditioned media. The values are represented as mean ±SEM of n=4 experiments using different donors. **p<0.01 for IL-1β vs IL-1β+EGCG; **p<0.01 for IL-1β vs IL-1β+inhibitors;
To test the role of IRAK-1, TAK1, or TRAF6 in IL-1β-induced IL-6 and IL-8 production, RA-FLS were pretreated with inhibitors of IRAK-1 [N-(2-Morpholinylethyl)-2-(3-nitrobenzoylamido)-benzimidazole, 50 μM], TAK1 [(5Z)-7-oxozeaenol, 5 μM], or TRAF6 inhibitor peptide or control peptide, 50 μM], followed by IL-1β stimulation. Resulting analysis on the conditioned media showed that only TAK1 inhibition completely abrogated IL-1β-induced IL-6 and IL-8 production (Figs. 1C & 1D; p<0.05). These results also underscore therapeutic value of TAK1 regulation in RA and the effectiveness of EGCG in suppressing IL-1β-induced IL-6 and IL-8 production.
EGCG inhibits IRAK-1 kinase activity but not its proteasomal degradation
IL-1β-induced activation of NF-κB and MAPK pathways is tightly regulated by molecules such as IRAK-1/TRAF6/TAK1 (6, 21). Since not much is known in terms of therapeutic value of these signal transducers in RA, we performed a time-dependent study with IL-1β stimulation in human RA-FLS. The result of the study showed that IL-1β-induced a rapid degradation of IRAK-1, followed by the phosphorylation of TAK1 at active site (Thr184/187) reaching a peak at 30 minutes in response to IL-1β stimulation (Fig. 2A). However, TRAF6 expression remained unchanged suggesting that its autoubiquitination, not the expression, is critical in IL-1β-induced signaling mechanisms. Further evaluation of EGCG (10–20 μM) pretreatment on IRAK-1 protein expression showed that EGCG was unable to rescue IL-1β-induced spontaneous IRAK-1 degradation in RA-FLS (Fig. 2B). Interestingly, pretreatment with methotrexate or dexamethasone also had no effect on IL-1β-induced IRAK-1 degradation at 30 minutes. To further investigate the mechanism of IRAK-1 proteasomal degradation in RA-FLS, we immunoprecipitated global ubiquitinated proteins from cell lysates using an FK-2 monoclonal antibody and probed for IRAK-1 expression. The results showed that IRAK-1 is heavily ubiquitinated in response to IL-1β stimulation and EGCG had no influence on this process (Fig. 2C). Studies suggest that IRAK-1 phosphorylation and subsequent degradation, but not IRAK-1 kinase activity, is an important event in the activation of downstream signaling cascades (22–24). This prompted us to confirm the effect of EGCG on IRAK-1 activity using an ADAPTA-kinase assay in vitro, which showed a ~66% inhibition of IRAK-1 activity by EGCG at a 1 μM concentration (Fig. 2D).
Fig. 2. EGCG inhibits IRAK-1 kinase activity, but not its proteasomal degradation. (A).
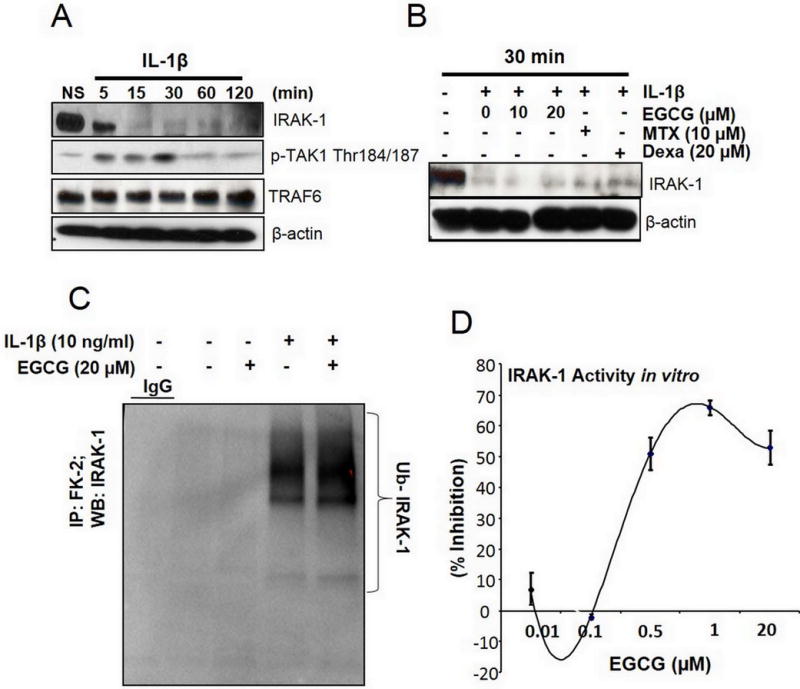
Time-dependent activation of the proximal signaling proteins (IRAK-1, TRAF6, and pTAK1 Thr184/187) in RA-FLS was studied using Western immunoblotting upon IL-1β stimulation for 5–120 minutes. (B) RA-FLS were pretreated with EGCG (10–20 μM, overnight), methotrexate (MTX; 10 μM, 2 hours), or dexamethasone (Dexa; 20 μM, 2 hours), followed by IL-1β stimulation for 30 minutes. IRAK-1 protein degradation was determined using Western immunoblotting. (C) RA-FLS treated with IL-1β and/or EGCG (20 μM) were immunoprecipitated with global ubiquitin (FK-2) or IgG (M2 Flag) and probed to study ubiquitinated IRAK-1. (D) Inhibition of the IRAK-1 in vitro kinase activity by EGCG was tested using an ADAPTA kinase assay as per the manufacturer’s instructions. Western immunoblots shown are the representatives from the 3 or 4 experiments repeated on different RA-FLS donors.
EGCG selectively inhibits phosphorylation of TAK1 at Thr184/187 ATP-binding site
TAK1 is a central mediator of TNF-α, IL-1β, or TLRs signal transduction pathways (25). Studies also suggest that TAK1 phosphorylation at Thr184/187 is a critical determinant of its kinase activity (26, 27). We evaluated the effect of EGCG in regulating IL-1β-induced pTAK1 (Thr184/187) in RA-FLS (Fig. 3A). Pretreatment of EGCG (2.5–20 μM) inhibited pTAK1 (Thr184/187) expression in RA-FLS dose-dependently (Fig. 3A; p<0.01 for 20 μM). To obtain further molecular insights, we studied the effect of EGCG on other IL-1β signaling proteins proximal to IL-1R such as MyD88, IRAK-4, and phospho-IRAK-4. Our results show that MyD88 and IRAK-4 expression remain unchanged with different treatments, while IL-1β induced phosphorylation of IRAK-4 at Thr345/Ser346 was modestly inhibited by EGCG in RA-FLS (Fig. 3B). However, this partial reduction in the phosphorylation of IRAK-4 by EGCG was unable to rescue IRAK-1 degradation in these samples (Fig. 3B).
Fig. 3. EGCG selectively inhibits phosphorylation of TAK1 at the Thr184/187 site to inhibit its kinase activity. (A).
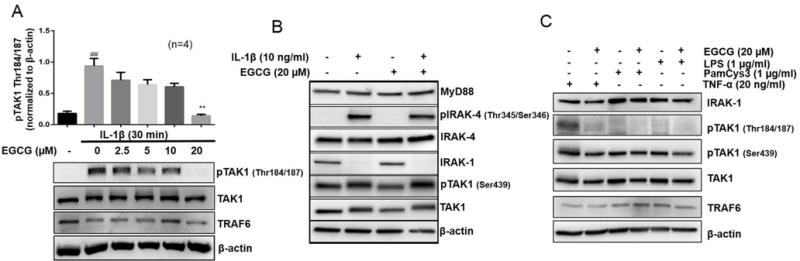
RA-FLS were pretreated with EGCG (2.5–20 μM), followed by IL-1β stimulation for 30 minutes. Cell lysates were analyzed for pTAK1 (Thr184/187), TAK1, TRAF6, and β-actin. (B) RA-FLS treated with IL-1β and/or EGCG (20 μM) were analyzed for MyD88, p-IRAK-4 (Thr345/Ser346), IRAK-4, IRAK-1, pTAK1 (Ser439), total TAK1, or β-actin to validate the selectivity of EGCG for pTAK1 (Thr 184/187). (C) RA-FLS were pretreated with EGCG (20 μM), TLR2 agonist (PamCys3, 1 μg/ml), or TLR4 agonist (lipopolysaccharide, LPS; 1 μg/ml), followed by stimulation with TNF-α (20 ng/ml) for 30 minutes. Cell lysates were analyzed for the expression of IRAK-1, TRAF6, pTAK1 (Thr184/187), pTAK1 (Ser439), total TAK1, and β-actin. The results in each subfigure represents the experiments repeated on four RA-FLS from different donors. ## p<0.01 for NS vs IL-1β; **p<0.01 for IL-1β vs IL-1β+ EGCG.
To validate EGCG’s selectivity for the Thr184/187 active site of TAK1, we determined the effect of EGCG on the serine 439 (Ser439) phosphorylation site of TAK1 protein (Fig. 3B). Interestingly, EGCG had no effect on IL-1β-induced activation of pTAK1 (Ser439) in RA-FLS, suggesting its selectivity in regulating TAK1 activation by inhibiting the phosphorylation at Thr184/187 site within the kinase activation loop of TAK1 (28). To further confirm whether this TAK1 inhibitory activity of EGCG was selective for IL-1β, we performed similar experiments with TNF-α, TLR2 agonist (PamCys3), or TLR4 agonist (LPS) alone or in combination with EGCG (Fig. 3C). The results of the study showed that only TNF-α was a potent inducer of pTAK1 (Thr184/187) and EGCG was effective in blocking TNF-α-induced pTAK1 (Thr184/187) in RA-FLS (Fig. 3C). Overall, these findings suggest a novel mechanism of cytokine activated TAK1 kinase regulation by EGCG in RA-FLS.
To test the effect of EGCG on other known kinases involved in several pathophysiological processes, we analyzed EGCG and/or IL-1β treated RA-FLS lysates for kinome assay using the human Phospho-kinase antibody array (R&D systems, MN). This array simultaneously detects the relative site-phosphorylation of 43 kinases and 2 related proteins (Supplementary Figs. S2A and S2B). The result of the kinase array showed that IL-1β-induced majority of kinases in RA-FLS within 30 minutes of stimulation. However, EGCG pretreatment had modest to no effect on most of the kinases (Figs. S2A and S2B). However, since these findings from the antibody array are qualitative and open to interpretation, further quantitative analysis on the kinases of interest may provide clearer and specific insights. In realms of the present study, these results suggest that EGCG preferentially inhibits IL-1β-induced phosphorylation and activation of TAK1 (Thr184/187) to suppress downstream signaling pathways such as p38, JNK, and NF-κB in RA-FLS, as observed in our previous studies (17, 18).
EGCG inhibits 20S proteasome activity and enhances deubiquitinase expression in RA-FLS
The ubiquitin-proteasome system plays an important role in posttranslational modification of proteins that are either destined to be degraded or stabilized for further signaling (16). A K48-linked Lys chains ‘prime’ protein to be recognized by 26S proteasome for degradation, whereas K63-linked Lys chains are tagged to the proteins that are required for stability and sustained signaling (29). However, the ubiquitination process has never been studied in depth in realms of RA pathogenesis. To explore this, we immunoprecipitated total polyubiquitinated proteins using a FK-2 antibody in IL-1β and/or EGCG treated RA-FLS and analyzed for K63- and K48-linked ubiquitination (Fig. 4). The results showed that EGCG had no effect on K63 or K48 ubiquitination by itself, but markedly enhanced K63, not K48, ubiquitination in the presence of IL-1β (Fig. 4A). A similar trend in the cell lysates from the remaining input suggests that EGCG may be enhancing the global K63 ubiquitination process to stabilize certain proteins required to block IL-1β signaling in RA-FLS (Fig. 4B). However, further analysis of 20S proteasome activity in a similarly treated lysates showed that IL-1β-induced proteasome activity in RA-FLS was increased by ~40%, which was significantly reduced in EGCG’s presence by ~50% (Fig. 4C; p<0.05). This suggests that despite being ineffective in downregulating K48-linked ubiquitination, EGCG may block further processing of these proteins in the 26S proteasome tunnel. One postulated mechanism could be the possible role of deubiquitinase (DUB) enzymes that hydrolyze polyubiquitin chains on the tagged proteins, thus, rescuing them from degradation (30). Analysis of the associated and non-associated DUBs, such as UCHL2, USP14, UCH37, and UBC9 and DUB associated with core 20S proteasome (PSMD13) in RA-FLS showed that in the presence of IL-1β, EGCG increased the expression of UCH37 and USP14, DUBs known to preferentially hydrolyze K48-linked polyubiquitin chains, with a marginal effect on unassociated DUB such as UCHL2 (Fig. 4D). However, the expression of PSMD13, a 26S proteasome non-ATP regulatory core subunit, remains unchanged with EGCG treatment suggesting that it selectively inhibits 20S proteasome activity without influencing the degradation process.
Fig. 4. EGCG inhibits K63 autoubiquitination of TRAF6 and upregulates the expression of deubiquitinases in RA-FLS. (A).
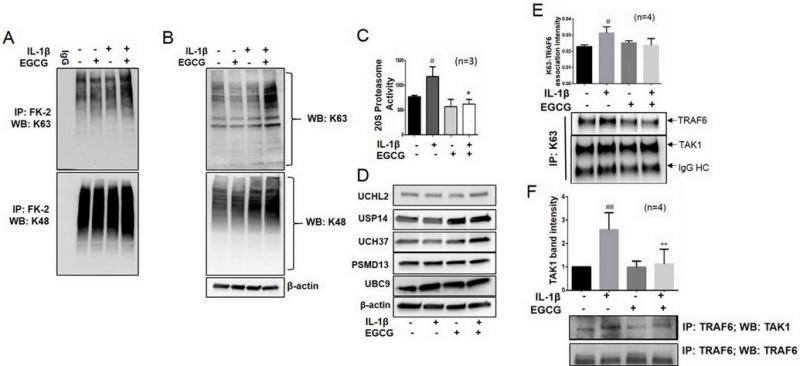
RA-FLS treated with IL-1β and/or EGCG (20 μM) were immunoprecipitated with global ubiquitin (FK-2) or IgG and analyzed for K63 (upper panel) or K48 (lower panel) linked ubiquitination pattern. (B) Input from the same treatment was utilized for detection of global K63- (upper) or K48- (lower) linked polyubiquitination in RA-FLS. (C) RA-FLS (0.5 × 104/well in a 96-well plate) were treated with IL-1β and/or EGCG (20 μM) for 30 minutes and analyzed for 20S proteasome activity using a commercially available kit (Cayman Chemical). Values are the mean ± SEM of experiments performed in three different RA-FLS donors. (D) RA-FLS treated with IL-1β and/or EGCG (20 μM) for 30 minutes were lysed and probed for the expression of proteasome associated and non-associated deubiquitinases. (E) RA-FLS (5 × 106/150 mm dish) treated with IL-1β and/or EGCG (20 μM) were immunoprecipitated with a K63 antibody and probed for TAK1 and TRAF6 ubiquitination. Values represented in the graph are mean ± SEM of experiment performed in four different RA-FLS donors. (F) Cell lysates from RA-FLS treated similarly to Fig. 4E were immunoprecipitated with a TRAF6 antibody and probed with TAK1 to analyze the effect of EGCG in interfering with TRAF6 and TAK1 association in RA-FLS. Values represent mean ± SEM of experiment performed in four different RA-FLS donors. ## p<0.01, # p<0.05 for NS vs IL-1β; **p<0.01, *p<0.05 for IL-1β vs IL-1β+ EGCG.
EGCG interferes TRAF6-TAK1 association by inhibiting K63 autoubiquitination of TRAF6
Autoubiquitination of TRAF6 at K124 is directly linked to NF-κB activation and its nuclear translocation (31). To examine the effect of EGCG on K63 autoubiquitination of TAK1 and TRAF6 in response to IL-1β, we immunoprecipitated K63-linked proteins from IL-1β and/or EGCG treated cell lysates and probed for TAK1 and TRAF6 proteins (Fig. 4E). Although we did not observe marked change in K63-mediated ubiquitination of TAK1, but saw ~30% increase in K63-mediated ubiquitination of TRAF6 with IL-1β treatment, which inhibited in the presence of EGCG by ~30% (Fig. 4E). To further understand the impact of the inhibition of TRAF6 autoubiquitination, we immunoprecipitated cell lysates with TRAF6 and probed for its association with TAK1. Interestingly, we found that IL-1β treated samples exhibited ~2.5-fold higher TRAF6/TAK1 association compared to the untreated (NS) group (Fig. 4F; p<0.01). However, EGCG alone or in presence of IL-1β inhibited TRAF6 association of TAK1 in RA-FLS, in part, by preventing its K63-mediated autoubiquitination (Fig. 4F).
EGCG occupies ATP-binding sites of IRAK-1 and TAK1 to inhibit kinase activity
Molecular modeling of the IRAK-1 protein based on the known 3D structure of IRAK-4 was utilized for conducting in silico molecular docking studies (Fig. 5A). We found that the hydroxyl group of a trihydroxyphenyl ring of EGCG perfectly positions (~1.6 Å) to make a hydrogen (H)-bond with the D358 and N345, and π-π interaction at F223 to stabilize EGCG in the binding pocket, resulting in its inhibitory activity. D358 is a conserved aspartate residue and an ATP-binding site for IRAK-1 as obtained from the PROSITE-ProRule: PRU00159 (Uniprot.org). The hydroxyl group of trihydroxybenzoate ring shows strong H-bond with the E214 and weak H-bond with K239. The benzoate nucleus fits well in a pocket formed by various lipophilic or aromatic residues. It is in interaction with hydrophobic residues L347 and V226 and perfectly positioned to form π-π interaction with Y288. Thus, binding of EGCG at these sites result in a reversible inhibition of IRAK-1 activity as further confirmed earlier with an ADAPTA in vitro kinase assay.
Fig. 5. Unique insights from the in silico molecular docking studies of EGCG binding on IRAK-1, TAK1-TAB1 complex, and TRAF6. (A).
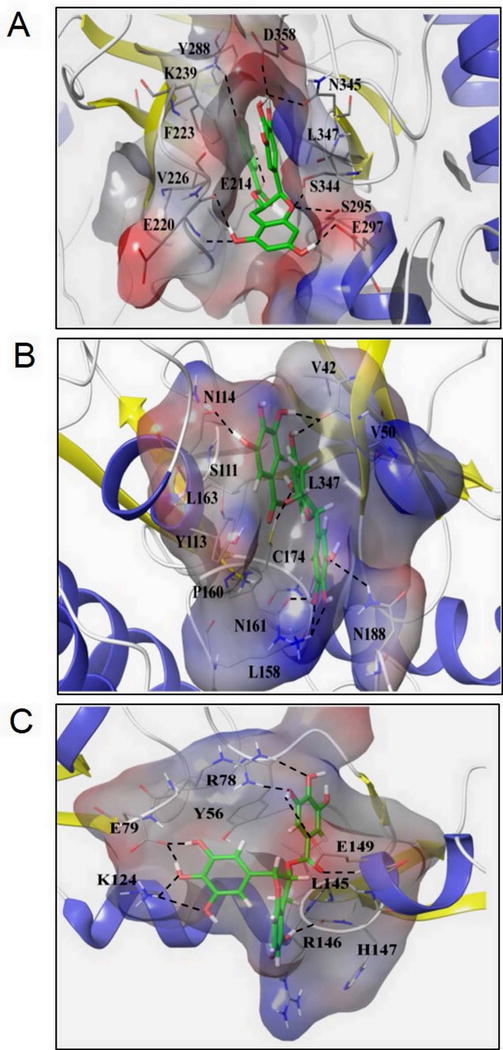
Molecular docking of EGCG on a constructed IRAK-1 structure showed the hydroxyl group of the trihydroxyphenyl ring of EGCG perfectly positions (1.6 Å) to make an H-bond with D358, a conserved aspartate residue and ATP-binding site, and N345, which inhibits IRAK-1 kinase activity. (B) EGCG forms an H-bond at C174, an ATP-binding site, on the TAK1-TAB1 complex and stabilizes its position in the binding pockets by forming hydrophobic links to distal V42, V50, and L163 residues that happen to be TAK1 nucleotide binding regions and a π-π interaction at Y113, thus serving as a potent and reversible TAK1 inhibitor. (C) Side chains of E79 and K124 residues on TRAF6 play significant roles in fastening the position of trihydroxyphenyl ring of EGCG through the H-bond network, thereby inhibiting its K63 autoubiquitination.
Molecular docking studies of EGCG with a TAK1-TAB1 complex revealed an interesting finding (Fig. 5B). The hydroxyl group of a benzopyran ring in EGCG completely occupies the position in the cavity that facilitates strong H-bond formation with the C174 residue (Fig. 5B). Importantly, (5Z)-7-Oxozeaenol, a known TAK1 inhibitor, acts on C174 to epitomize irreversible covalent binding that results in a permanent abrogation of TAK1 kinase activity (32). In addition to binding with C174, EGCG stabilizes its position in the binding pockets by forming hydrophobic links to distal V42, V50, and L163 residues that happen to be TAK1 nucleotide binding regions and π-π interaction at Y113 with the trihydroxybenzoate ring of EGCG. The trihydroxyphenyl ring fits well in the outer pocket formed by various lipophilic residues and forms an H-bond with the K158, N161, and N188 residues by its hydroxyl groups and stabilizes its position by hydrophobic interaction with the P160 site, thus acting as a potent reversible TAK1 inhibitor.
In terms of TRAF6, the almost linear structure of this molecule provides a limited opportunity due to its shallow binding pockets for hydrophobic interactions with EGCG (Fig. 5C). The hydroxyl group of the trihydroxybenzoate ring of ECGC forms the H-bond network with the side chain of R78 and E149, π-π interaction with Y56, and hydrophobic interaction with the L145 to provide stability. The backbone of R146 and H147 are also involved in the formation of H-bond with the hydroxyl group of benzopyran ring. However, mechanistically important, the side chains of E79 and K124 residues play a significant role in fastening the position of a trihydroxyphenyl ring through H-bond network. Since K124 is an important site for K63 autoubiquitination of TRAF6, these findings provide mechanistic insights into the observed inhibitory effect of EGCG on TRAF6 K63-linked autoubiquitination in RA-FLS.
EGCG modulates in vivo ubiquitination process to ameliorate rat AIA
A rat AIA model was used to study in vivo ubiquitination associated with RA and the modulatory effect of EGCG. We have earlier shown that EGCG (100 mg/kg) daily for 10 days inhibits IL-6 production in serum and joint homogenates of AIA rats (17). In the present study, we observed that the administration of EGCG (50 mg/kg/day i.p.) during the disease onset was more effective in ameliorating AIA as evident from a significant reduction in the ankle circumferences (Fig. 6A; p<0.01; n=6). Western blot analysis of the joint homogenates showed a modest decrease in K63 ubiquitinated proteins in the AIA group compared to the naïve group, which was reversed with EGCG pretreatment (Fig. 6B). However, a marked increase in K48-linked ubiquitinated proteins was observed in AIA joint homogenates, which was effectively inhibited with EGCG administration back to almost naïve group levels (Fig. 6C; p<0.05). These findings clearly indicate that EGCG differentially modulates K63- and K48-linked ubiquitination that results in the amelioration of rat AIA.
Fig. 6. EGCG modulates the in vivo ubiquitination process to inhibit rat AIA. (A).
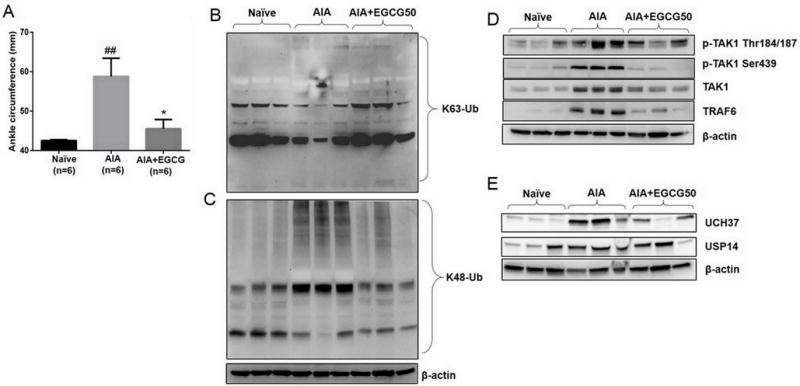
EGCG (50 mg/kg/day, i.p.) administration for 10 days (starting at day 7) ameliorated rat AIA as evident from reduced ankle circumferences. The ankle circumferences of both the hind ankles from each animal were averaged and ‘n’ is represented as the number of animals used in each of the experimental groups. (B and C) Joint homogenates (30 μg per sample) from naïve, AIA, and EGCG treated rats were analyzed for the expression of global K63- and K48-linked ubiquitination. (D) The same homogenates were analyzed for the expression of pTAK1 (Thr184/187), pTAK1 (Ser439), total TAK1, TRAF6, and β-actin. (E) The effect of EGCG administration was evaluated on the modulation of in vivo deubiquitinase enzymes associated with the removal ubiquitin chains. ## p<0.01 for Naïve vs AIA; *p<0.05 for AIA vs AIA+EGCG.
Further correlating these findings with the expression of signaling intermediates, we found that the levels of pTAK1 (Thr184/187), pTAK1 (Ser439), total TAK1, TRAF6, and UCH37 and USP14 were markedly increased in AIA joints as compared to the naïve joints (Figs. 6D & 6E; p<0.05 or p<0.01 for all the studied proteins except K63; statistical analysis in Fig. S3A–G). Interestingly, joint homogenates from EGCG-treated rats showed a marked decreased in the expression of these proteins, which was comparable to the levels observed in the naïve group (Figs. 6 and S3A–G; p<0.05 for pTAK1 (Ser439), total TAK1, and TRAF6). These findings suggest that EGCG may blunt cytokine-signaling pathways in the arthritic ankles by regulating TAK1 and other associated proteins by interfering post-translational modifications to inhibit RA pathogenesis.
Discussion
Our study provides previously unidentified mechanism of TAK1 regulation in human RA-FLS and rat AIA by modulating the ubiquitin proteasome system to inhibit IL-1β-induced signaling pathways. Importantly, our study provides mechanistic insights into the posttranslational ubiquitination processes that govern RA-FLS TAK1 activation and the mechanism of EGCG’s interaction with IRAK-1/TRAF6/TAK1 that results in the inhibition of TAK1 activity. Given the safety profile of EGCG in humans (33), our study provides a platform to design TAK1 inhibitors that can effectively disrupt the integrated cytokine signaling to limit their role in RA pathogenesis.
IL-1β is a master regulator of inflammation and tissue destruction in several autoimmune diseases such as RA, gout, and type 2 diabetes (8). From therapeutic stand-point, only IL-1R antagonists were pursued for clinical testing to block the effect of IL-1 in the treatment of RA. Despite the failure of anakinra and given the pathological significance of IL-1 in RA, further therapeutic approaches are warranted with the aim of testing agents that are more potent inhibitors of IL-1 signaling pathways. In this regard, IRAK-4 kinase inhibitors have shown promise in pre-clinical testing (12). However, recent studies suggest the differential role of IRAK-4 in regulating cytokine production in different cell types (34). Of particular interest, the inhibition of IRAK-4 activation in human dermal fibroblasts was not effective in regulating IL-1R- or TLR agonists-induced IL-6 production, whereas its inhibition in human monocytes was effective in reducing IL-6 levels (34). These findings suggest that the kinase activities of IRAK-1/-4 are redundant in regulating inflammatory cytokine production in human cells, including RA-FLS (35). Given the role of IRAK-1/-4 in controlling immune alterations and innate responses to pathogens, several multi-omics and computational intensive approaches warn of impaired innate immune responses with IRAK-1/-4 inhibitors (36). In addition, transcriptome analysis using IL-1 receptor antagonist and 13 gene partners, including MyD88, IRAK-1/-4, and TRAF6, suggest that identifying protein target proximal to IL-1 receptor may elicit serious adverse effects as observed with anakinra treatment (37). This opens therapeutic avenues for validating other signaling proteins involved in IL-1β mediated inflammation and tissue destruction.
An ideal position of TAK1 in the inflammatory signaling cascades triggered by IL-1β, also TNF-α, makes it an attractive therapeutic target (28, 38). Although TAK1 inhibition using siRNA approach has shown some promise for RA (39), there is a complete lack of understanding of its posttranslational regulation in RA pathogenesis. Binding of IL-1β to its receptor IL-1R leads to the recruitment of adaptor proteins and kinases such as MyD88, IRAK-1 and IRAK-4. IRAK-4 phosphorylates IRAK-1 and facilitates TRAF6 recruitment to the complex and IRAK-1 degradation (40). Being an ubiquitin E3 ligase, TRAF6 undergoes K63-linked autoubiquitination that follows the recruitment of TAK1-TAB1 or the TAB2/3 complex and the induction of TAK1 kinase activity. TAK1 eventually phosphorylates MKK4/7-JNK and MKK3/6-p38 MAPK, and IKK, resulting in nuclear translocation of the transcription factors activation protein-1 (AP-1) and NF-κB (5). Just like methotrexate and dexamethasone, EGCG pretreatment was unable to block IRAK-1 degradation, however, it markedly inhibited in vitro IRAK-1 kinase activity. Molecular docking studies using IRAK-1 homology structural modeling provides evidence that EGCG can tightly occupy ATP-binding sites resulting in complete blockade of its kinase activity. However, several studies point to the fact that IRAK-1 phosphorylation and subsequent degradation, not its kinase activity, is an important event for the activation of downstream signaling cascades (22–24).
Ubiquitination is an important posttranslational modification that regulates various cellular processes including cell survival, apoptosis, and signaling (16, 41). Ubiquitination is carried out by the ubiquitin-protease system, which involves E1, E2, and E3 ubiquitin ligases that identify the proteins for ubiquitin ligation, proteases that are involved in degrading proteins, and the proteasome that safeguards highly specific nature of degradation system and prevents non-specific protein degradation. The process is tightly regulated by two key chain-linking lysine residues, K48 and K63 (5, 16). K48-linked polyubiquitination of the protein primes it for proteasomal degradation, whereas, K63-linked ubiquitination of the protein stabilizes it for further cell signaling and function. Another class of cysteine proteases, deubiquitinase, can hydrolyze and remove these ubiquitin chains to reverse the fate of the protein (30). In our studies, we found that IL-1β-induced K63 autoubiquitination of TRAF6 was markedly inhibited by EGCG. Further molecular insights using molecular docking studies validate that EGCG forms H-bond networks with TRAF6 at the E79 and K124 positions, and π-π interactions with Y56 that create an electron cloud and exceptional stability to EGCG for holding on to critical TRAF6 sites. Site-specific K63 autoubiquitination of TRAF6 is a critical determinant of IKK activity (31, 42). Although we have shown EGCG to inhibit NF-κB activation in a variety of cell types (17, 18, 43–45), this study provides evidence of TAK1 as the actual molecular target for its anti-rheumatic activity.
Only autoubiquitinated TRAF6 is capable of associating with and autophosphorylating TAK1 (46, 47). Thus, blocking K63 autoubiquitination of TRAF6 showed a profound effect on this process and a marked decrease in association of TRAF6 with TAK1 and the subsequent inhibition of p-TAK1 (Thr 184/187) in RA-FLS. While TRAF6 has been an attractive therapeutic target for inflammatory diseases, it is required for optimal physiological signaling mechanisms, making it a riskier therapeutic target. This study provides a unique mechanism of therapeutically regulating TRAF6 via reversible inhibition of its function through stable H-bond formation by EGCG without jeopardizing its physiological functions. Rationalizing drug design approaches in light of these findings may yield more potent and safer drugs for the treatment of RA and other inflammatory diseases. However, further studies are warranted to understand the effect of EGCG on E1 or E2 ubiquitin ligases, which associate with TRAF6 to activate MAPK and IKK through TAK1.
The progress in the development of RA therapies suggests that the existing drugs for RA are expensive, immunosuppressive, and sometimes unsuitable for long-term use (48, 49). Additionally, the existing RA therapies are B-cell-, T-cell-, and cytokine-directed and no existing medication for RA targets FLS despite an established body of molecular and clinical evidence suggesting their role in RA pathogenesis (50–52). This may be attributed to our limited understanding of the molecular mechanisms that govern their invasive and destructive potentials. In this study, we provide the rational for targeting RA-FLS TAK1 for amelioration of RA and a unique mechanism through which EGCG inhibits the interaction between signaling molecules important in cytokine signaling, ultimately inhibiting inflammation and tissue destruction in RA.
Supplementary Material
Acknowledgments
This study was supported by the NIH grant AR063104 (S.A.), The Arthritis Foundation Innovative Research Grant (S.A.), and the Start-up funds from Washington State University. Authors thank National Disease Research Interchange (NDRI), and Cooperative Human Tissue Network (CHTN) for providing RA synovial tissues. Authors thank our longtime collaborator, Dr. David A. Fox (Division of Rheumatology, University of Michigan Medical School), for a critical review of this manuscript and helpful suggestions. Authors also thank Ms. Maria Beamer for her technical support in animal studies.
Footnotes
CONFLICT OF INTEREST The authors declare no conflict of interest.
References
- 1.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9(1):24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. 2014;57(12):5023–38. doi: 10.1021/jm401490p. [DOI] [PubMed] [Google Scholar]
- 3.Fox DA. Kinase inhibition-a new approach to the treatment of rheumatoid arthritis. The N Eng J Med. 2012;367(6):565–7. doi: 10.1056/NEJMe1206315. [DOI] [PubMed] [Google Scholar]
- 4.Meier FM, McInnes IB. Small-molecule therapeutics in rheumatoid arthritis: scientific rationale, efficacy and safety. Best Prac Clin Res Rheumatol. 2014;28(4):605–24. doi: 10.1016/j.berh.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 5.Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol Sci. 2012;33(10):522–30. doi: 10.1016/j.tips.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 6.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412(6844):346–51. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270(5244):2008–11. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 8.Dinarello CA, van der Meer JW. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol. 2013;25(6):469–84. doi: 10.1016/j.smim.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191(2):313–20. doi: 10.1084/jem.191.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salliot C, Dougados M, Gossec L. Risk of serious infections during rituximab, abatacept and anakinra treatments for rheumatoid arthritis: meta-analyses of randomised placebo-controlled trials. Ann Rheum Dis. 2009;68(1):25–32. doi: 10.1136/ard.2007.083188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buch MH, Bingham SJ, Seto Y, McGonagle D, Bejarano V, White J, et al. Lack of response to anakinra in rheumatoid arthritis following failure of tumor necrosis factor alpha blockade. Arthritis Rheum. 2004;50(3):725–8. doi: 10.1002/art.20115. [DOI] [PubMed] [Google Scholar]
- 12.Bahia MS, Kaur M, Silakari P, Silakari O. Interleukin-1 receptor associated kinase inhibitors: potential therapeutic agents for inflammatory- and immune-related disorders. Cell Signal. 2015;27(6):1039–55. doi: 10.1016/j.cellsig.2015.02.025. [DOI] [PubMed] [Google Scholar]
- 13.Cui W, Xiao N, Xiao H, Zhou H, Yu M, Gu J, et al. beta-TrCP-mediated IRAK1 degradation releases TAK1-TRAF6 from the membrane to the cytosol for TAK1-dependent NF-kappaB activation. Mol Cell Biol. 2012;32(19):3990–4000. doi: 10.1128/MCB.00722-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou H, Yu M, Fukuda K, Im J, Yao P, Cui W, et al. IRAK-M mediates Toll-like receptor/IL-1R-induced NFkappaB activation and cytokine production. EMBO J. 2013;32(4):583–96. doi: 10.1038/emboj.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10(10):1199–207. doi: 10.1038/ncb1780. [DOI] [PubMed] [Google Scholar]
- 16.Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev. 2012;246(1):95–106. doi: 10.1111/j.1600-065X.2012.01108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmed S, Marotte H, Kwan K, Ruth JH, Campbell PL, Rabquer BJ, et al. Epigallocatechin-3-gallate inhibits IL-6 synthesis and suppresses transsignaling by enhancing soluble gp130 production. Proc Natl Acad Sci USA. 2008;105(38):14692–7. doi: 10.1073/pnas.0802675105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmed S, Pakozdi A, Koch AE. Regulation of interleukin-1beta-induced chemokine production and matrix metalloproteinase 2 activation by epigallocatechin-3-gallate in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2006;54(8):2393–401. doi: 10.1002/art.22023. [DOI] [PubMed] [Google Scholar]
- 19.Ahmed S, Silverman MD, Marotte H, Kwan K, Matuszczak N, Koch AE. Down-regulation of myeloid cell leukemia 1 by epigallocatechin-3-gallate sensitizes rheumatoid arthritis synovial fibroblasts to tumor necrosis factor alpha-induced apoptosis. Arthritis Rheum. 2009;60(5):1282–93. doi: 10.1002/art.24488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freireich EJ, Gehan EA, Rall DP, Schmidt LH, Skipper HE. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer Chemother Rep. 1966;50(4):219–44. [PubMed] [Google Scholar]
- 21.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398(6724):252–6. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 22.Knop J, Martin MU. Effects of IL-1 receptor-associated kinase (IRAK) expression on IL-1 signaling are independent of its kinase activity. FEBS Lett. 1999;448(1):81–5. doi: 10.1016/s0014-5793(99)00322-1. [DOI] [PubMed] [Google Scholar]
- 23.Li X, Commane M, Burns C, Vithalani K, Cao Z, Stark GR. Mutant cells that do not respond to interleukin-1 (IL-1) reveal a novel role for IL-1 receptor-associated kinase. Mol Cell Biol. 1999;19(7):4643–52. doi: 10.1128/mcb.19.7.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maschera B, Ray K, Burns K, Volpe F. Overexpression of an enzymically inactive interleukin-1-receptor-associated kinase activates nuclear factor-kappaB. Biochem J. 1999;339(Pt 2):227–31. [PMC free article] [PubMed] [Google Scholar]
- 25.Ajibade AA, Wang HY, Wang RF. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013;34(7):307–16. doi: 10.1016/j.it.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Singhirunnusorn P, Suzuki S, Kawasaki N, Saiki I, Sakurai H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-beta-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J Biol Chem. 2005;280(8):7359–68. doi: 10.1074/jbc.M407537200. [DOI] [PubMed] [Google Scholar]
- 27.Yu Y, Ge N, Xie M, Sun W, Burlingame S, Pass AK, et al. Phosphorylation of Thr-178 and Thr-184 in the TAK1 T-loop is required for interleukin (IL)-1-mediated optimal NFkappaB and AP-1 activation as well as IL-6 gene expression. J Biol Chem. 2008;283(36):24497–505. doi: 10.1074/jbc.M802825200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kilty I, Jones LH. TAK1 selective inhibition: state of the art and future opportunities. Future Med Chem. 2015;7(1):23–33. doi: 10.4155/fmc.14.138. [DOI] [PubMed] [Google Scholar]
- 29.Adhikari A, Xu M, Chen ZJ. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene. 2007;26(22):3214–26. doi: 10.1038/sj.onc.1210413. [DOI] [PubMed] [Google Scholar]
- 30.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550–63. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 31.Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I kappa B kinase activation. J Biol Chem. 2007;282(6):4102–12. doi: 10.1074/jbc.M609503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu J, Powell F, Larsen NA, Lai Z, Byth KF, Read J, et al. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)-7-Oxozeaenol. ACS Chem Biol. 2013;8(3):643–50. doi: 10.1021/cb3005897. [DOI] [PubMed] [Google Scholar]
- 33.Chow HH, Cai Y, Hakim IA, Crowell JA, Shahi F, Brooks CA, et al. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin Cancer Res. 2003;9(9):3312–9. [PubMed] [Google Scholar]
- 34.Cushing L, Stochaj W, Siegel M, Czerwinski R, Dower K, Wright Q, et al. Interleukin 1/Toll-like receptor-induced autophosphorylation activates interleukin 1 receptor-associated kinase 4 and controls cytokine induction in a cell type-specific manner. J Biol Chem. 2014;289(15):10865–75. doi: 10.1074/jbc.M113.544809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song KW, Talamas FX, Suttmann RT, Olson PS, Barnett JW, Lee SW, et al. The kinase activities of interleukin-1 receptor associated kinase (IRAK)-1 and 4 are redundant in the control of inflammatory cytokine expression in human cells. Mol Immunol. 2009;46(7):1458–66. doi: 10.1016/j.molimm.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 36.Tieri P, Zhou X, Zhu L, Nardini C. Multi-omic landscape of rheumatoid arthritis: re-evaluation of drug adverse effects. Front Cell Dev Biol. 2014;2:59. doi: 10.3389/fcell.2014.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao Y, Jiao Y, Wang L, Huang Y, Postlethwaite A, Stuart J, et al. Anakinra as an interleukin 1 receptor antagonist, complicated genetics and molecular impacts-from the point of view of mouse genomics. Int Immunopharmacol. 2012;13(1):28–36. doi: 10.1016/j.intimp.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hammaker DR, Boyle DL, Inoue T, Firestein GS. Regulation of the JNK pathway by TGF-beta activated kinase 1 in rheumatoid arthritis synoviocytes. Arthritis Res Ther. 2007;9(3):R57. doi: 10.1186/ar2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Courties G, Seiffart V, Presumey J, Escriou V, Scherman D, Zwerina J, et al. In vivo RNAi-mediated silencing of TAK1 decreases inflammatory Th1 and Th17 cells through targeting of myeloid cells. Blood. 2010;116(18):3505–16. doi: 10.1182/blood-2010-02-269605. [DOI] [PubMed] [Google Scholar]
- 40.Dai L, Aye Thu C, Liu XY, Xi J, Cheung PC. TAK1, more than just innate immunity. IUBMB life. 2012;64(10):825–34. doi: 10.1002/iub.1078. [DOI] [PubMed] [Google Scholar]
- 41.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7(8):758–65. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nathan JA, Kim HT, Ting L, Gygi SP, Goldberg AL. Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J. 2013;32(4):552–65. doi: 10.1038/emboj.2012.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmed S, Wang N, Lalonde M, Goldberg VM, Haqqi TM. Green tea polyphenol epigallocatechin-3-gallate (EGCG) differentially inhibits interleukin-1 beta-induced expression of matrix metalloproteinase-1 and -13 in human chondrocytes. J Pharmacol Exp Ther. 2004;308(2):767–73. doi: 10.1124/jpet.103.059220. [DOI] [PubMed] [Google Scholar]
- 44.Hafeez BB, Ahmed S, Wang N, Gupta S, Zhang A, Haqqi TM. Green tea polyphenols-induced apoptosis in human osteosarcoma SAOS-2 cells involves a caspase-dependent mechanism with downregulation of nuclear factor-kappaB. Toxicol Appl Pharmacol. 2006;216(1):11–9. doi: 10.1016/j.taap.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 45.Singh R, Ahmed S, Islam N, Goldberg VM, Haqqi TM. Epigallocatechin-3-gallate inhibits interleukin-1beta-induced expression of nitric oxide synthase and production of nitric oxide in human chondrocytes: suppression of nuclear factor kappaB activation by degradation of the inhibitor of nuclear factor kappaB. Arthritis Rheum. 2002;46(8):2079–86. doi: 10.1002/art.10443. [DOI] [PubMed] [Google Scholar]
- 46.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103(2):351–61. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 47.Wu H, Arron JR. TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. BioEssays. 2003;25(11):1096–105. doi: 10.1002/bies.10352. [DOI] [PubMed] [Google Scholar]
- 48.Furst DE. The risk of infections with biologic therapies for rheumatoid arthritis. Semin Arthritis Rheum. 2010;39(5):327–46. doi: 10.1016/j.semarthrit.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 49.Khanna D, McMahon M, Furst DE. Safety of tumour necrosis factor-alpha antagonists. Drug Saf. 2004;27(5):307–24. doi: 10.2165/00002018-200427050-00003. [DOI] [PubMed] [Google Scholar]
- 50.Chang SK, Noss EH, Chen M, Gu Z, Townsend K, Grenha R, et al. Cadherin-11 regulates fibroblast inflammation. Proc Natl Acad Sci USA. 2011;108(20):8402–7. doi: 10.1073/pnas.1019437108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315(5814):1006–10. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- 52.Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233(1):233–55. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.