

Summary
To determine the effect of glucocorticoids (GCs) on endothelial dysfunction (ED) and on traditional cardiovascular (CV) risk factors in the adjuvant‐induced arthritis (AIA) rat model. At the first signs of AIA, a high dose (HD) [10 mg/kg/day, intraperitoneally (i.p.), GC‐HD] or low dose (LD) (1 mg/kg/day, i.p., GC‐LD) of prednisolone was administered for 3 weeks. Endothelial function was studied in aortic rings relaxed with acetylcholine (Ach) with or without inhibitors of nitric oxide synthase (NOS), cyclooxygenase 2 (COX‐2), arginase, endothelium derived hyperpolarizing factor (EDHF) and superoxide anions ( °) production. Aortic expression of endothelial NOS (eNOS), Ser1177‐phospho‐eNOS, COX‐2, arginase‐2, p22phox and p47phox was evaluated by Western blotting analysis. Arthritis scores, blood pressure, heart rate and blood levels of cytokines, triglycerides, cholesterol and glucose were measured. GC‐HD but not GC‐LD reduced arthritis score significantly and improved Ach‐induced relaxation (P < 0·05). The positive effect of GC‐HD resulted from increased NOS activity and EDHF production and decreased COX‐2/arginase activities and ° production. These functional effects relied upon increased phospho‐eNOS expression and decreased COX‐2, arginase‐2 and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase expression. Despite the lack of effect of GC‐LD on ED, it increased NOS and EDHF and down‐regulated ° pathways but did not change arginase and COX‐2 pathways. GC‐HD increased triglycerides levels and blood pressure significantly (P < 0·05). Both doses of GCs decreased to the same extent as plasma interleukin (IL)‐1β and tumour necrosis factor (TNF)‐α levels (P < 0·05). Our data demonstrated that subchronic treatment with prednisolone improved endothelial function in AIA via pleiotropic effects on endothelial pathways. These effects occurred independently of the deleterious cardiometabolic effects and the impact of prednisolone on systemic inflammation.
Keywords: adjuvant‐induced arthritis, endothelial dysfunction, glucocorticoids, mechanisms
Introduction
Rheumatoid arthritis (RA) is one of the most common chronic autoimmune diseases characterized by articular and extra‐articular manifestations, including cardiovascular (CV) diseases, the latter accounting for 30–50% of all deaths 1. To date, the exact reasons for this excess CV risk are not known, but RA‐dependent factors, traditional CV risk factors and iatrogenic factors are likely to play a role 2. These factors are able to modify the phenotype of endothelial cells, thereby leading to endothelial dysfunction (ED), i.e. a functional and reversible alteration of endothelial cells leading to a shift of the actions of the endothelium towards reduced vasodilation, proinflammatory state, proliferative and prothrombotic properties. The presence of ED is recognized as the Primum movens of atherogenesis, and as such is a seminal target for reducing CV risk in RA 3.
Glucocorticoids (GCs) were the first drugs to treat a woman with RA successfully in 1948 4. Despite a significant evolution in the treatment of RA, the majority of patients still use GCs alone or in combination with disease‐modifying anti‐rheumatic drugs (DMARDs). There is a general awareness of the potential harmful CV effects of GCs 4 but, surprisingly, the CV effects of GCs in case of inflammatory diseases such as RA remain subject to debate 5, 6. GCs might increase CV risk (CVR) by increasing CV risk factors (dyslipidaemia, hypertension, insulin resistance). However, GCs might also decrease CV risk by decreasing the systemic and/or vascular inflammation. In addition, GCs may modulate vascular function directly, as GC receptors are expressed by both endothelial and vascular smooth muscle cells (VSMC) 7. Recent reviews addressing the effects of GCs on CV risk in RA yielded conflicting results. GCs were found to increase CV and/or mortality risk 8, 9, 10, to improve CV/mortality prognosis 11 or to have no or an uncertain effect 12, 13. Regarding the endothelial function specifically, only five studies in RA investigated the effects of GCs on ED, and reported that GCs had no deleterious effect 14, 15, 16, 17 or even beneficial effects on vascular function 18. Unfortunately, these few studies suffer from important shortcomings due to a high level of polymedication with other anti‐rheumatic drugs, the lack of control group, the small sample size, the heterogeneity in dosage or duration of GC treatment. In this context, as randomization to GCs versus placebo in a long‐term clinical trial would be difficult to conduct and perhaps even not feasible in RA, the use of animal models of arthritis is a benchmark choice to address this question. In the present study, we investigated the effect of a treatment with prednisolone on endothelial function in AIA rats. Endothelial function was studied on isolated aortic rings on day 33 after arthritis induction (i.e. at a time at which ED has been observed previously in this model 19), and the mechanisms involved were dissected. The effect of GC on disease activity and systemic inflammation was assessed by measurement of arthritis score and plasma interleukin (IL)−1β and tumour necrosis factor (TNF)‐α levels, respectively. We also measured blood pressure, heart rate, glycaemia and blood lipid levels to assess whether the treatment modified CV risk factors.
Material and methods
Animals
Six‐week‐old male Lewis rats (n = 96) were purchased from Janvier (Le Genest‐Saint‐Isle, France). Animals were kept under a 12 h–12 h light–dark cycle and allowed free access to food and water. The experimental procedures were approved by the local committee for ethics in animal experimentation no. 2012/018‐CD of Franche‐Comté University (Besançon, France), and complied with the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines.
Induction and clinical evaluation of the arthritis model
Adjuvant arthritis was induced by a single intradermal injection at the base of the tail of 120 µl of 1 mg of heat‐killed Mycobacterium butyricum (Difco, Detroit, MI, USA) suspended in 0·1 ml of mineral oil (Freund's incomplete adjuvant; Difco). Rats were weighed daily and monitored for clinical signs of arthritis. The scoring system was employed as follows 20: arthritis of one finger scores 0·1, weak and moderate arthritis of one big joint (ankle or wrist) scores 0·5 and intense arthritis of one big joint scores 1. Tarsus and ankle were considered as the same joint. Sum of joints scores of four limbs leads to an arthritis score of maximum 6 to each rat.
Drug treatment
At the beginning of arthritis (i.e. at days 11–12 post‐immunization) AIA rats were randomized in three groups. One group received intraperitoneally (i.p.) prednisolone phosphate sodium salt at 13·4 mg/kg/day (equivalent to 10 mg/kg/day prednisolone) [GC high dose (GC‐HD), n = 32] or at 0·134 mg/kg/day (equivalent to 0·1 mg/kg/day prednisolone) [GC low dose (GC‐LD), n = 32] for 3 weeks (subchronic treatment). Another group received saline at 1 ml/kg/day (i.p.) for 3 weeks (vehicle, n = 32). The HD of prednisolone corresponds to a dose that results in a significant reduction in arthritis severity 21. The LD of prednisolone was chosen on the basis of preliminary experiments aiming to find a subtherapeutic dose on clinical arthritis in this model. The LD was used as a pharmacological tool to determine whether a vascular effect of GC might occur independently of the reduction of arthritis severity.
Tissue collection, blood pressure and heart rate measurements
Twenty‐one days after treatment initiation, rats were anaesthetized with pentobarbital (60 mg/kg, i.p.). Blood pressure and heart rate were measured after cannulation of the left carotid artery and connection of the catheter to a pressure recorder system (Easy Graf, Gould, USA) under rectal temperature control. Blood was then withdrawn from the abdominal artery and centrifuged to obtain serum and plasma stored at −80°C until analysis. Thoracic aortas were removed and were used either immediately for the group of rats used for vascular reactivity studies or frozen in liquid nitrogen and stored at −80°C until Western blot analysis.
Vascular reactivity
At the end of the treatment period (day 33 post‐immunization, i.e. the time at which ED is present in the AIA model 19, 22, 23, 24) thoracic aorta was excised, cleaned of connective tissue and cut into rings ∼2 mm in length. Rings were suspended in Krebs solution (mol/l: NaCl 118, KCl 4·65, CaCl2 2·5, KH2PO4 1·18, NaHCO3 24·9, MgSO4 1·18, glucose 12, pH 7·4), maintained at 37°C and aerated continuously with 95% O2, 5% CO2 for isometric tension recording in organ chambers. After a 90‐min equilibration period under a resting tension of 2 g, the presence of functional endothelium was verified by the ability of acetylcholine (Ach, 10−6 mol/l) to induce more than 70% relaxation in rings preconstricted with phenylephrine (PE, 10−6 mol/l). In some rings, endothelium was removed mechanically. The completeness of endothelial denudation was confirmed by absence of relaxation in response to Ach (10−6 mol/l). To determine whether GCs improved endothelial function, rings with intact endothelium were constricted with PE (10−6 mol/l) and endothelium‐dependent relaxation to Ach (10− 11−10− 4 mol/l) was compared between GCs and vehicle group. To investigate the mechanisms involved, rings were incubated previously for 1 h with the non‐selective nitric oxide synthase (NOS) inhibitor NW‐nitro‐L‐arginine methyl ester (L‐NAME, 10−4 mol/l), arginase inhibitor Nw‐hydroxy‐nor‐L‐arginine (nor‐NOHA, 10−4 mol/l), superoxide dismutase mimetic (SOD) Tempol (10−4 mol/l), selective cyclooxygenase‐2 (COX‐2) inhibitor (NS‐398, 10−5 mol/l) and Ca2+‐dependent K+ channel inhibitors apamin (10−7 mol/l) and charybdotoxin (10−7 mol/l), respectively. Endothelium‐denuded rings were used to determine the vasoconstrictive response to noradrenalin (NE, 10−11−10−4 mol/l) and the vasorelaxant response to the NO‐donor sodium nitroprussiate (SNP, 10−11−10−4 mol/l) after preconstriction with PE 10−6 mol/l.
Western blot analysis
To investigate whether the effects of GCs on endothelial function relied upon changes in protein expression, the protein content of eNOS and phospho‐Ser1177‐eNOS (P‐eNOS, an activated form of eNOS at serine 1177 that produces a sustained release of endothelial NO), arginase‐2, COX‐2, p22phox and p47phox [membrane‐bound and cytosolic components, respectively, of nicotinamide adenine dinucleotide phosphate (NADPH oxidase), a major vascular enzyme responsible for ° production], was assessed in homogenates of thoracic aortas from vehicle and GC groups by Western blotting analysis, as described previously in detail 25 (see Supporting information).
Blood measurements
Total cholesterol and triglycerides were measured in serum (Vista; Siemens, Malvern, PA, USA) and glucose was measured in blood using a glycometer (GlucoMen; Menarini Diagnostics, Florence, Italy). Plasma levels of proinflammatory cytokines [interleukin (IL)‐1β] and tumour necrosis factor (TNF)‐α were measured using Milliplex magnetic bead panel kits (eBioscience, Vienna, Austria), analysed using a Luminex MAGPIX system (Luminex Corporation; Houston, TX, USA) and Milliplex Analyst software (Millipore, St Charles, MO, USA). The limits of detection provided by the manufacturer for IL‐1β and TNF‐α were 13 and 3·78 pg/ml, respectively.
Data and statistical analysis
Values are presented as means ± standard error of the mean (s.e.m.). Data were analysed using GraphPad Prism software version 5.0. Contractile responses to NE were expressed as the percentage of the maximum response to KCl 100 mmol/l. Relaxant responses to SNP and Ach were expressed as the percentage of relaxation of the contractile response to PE 10−6 mol/l. Concentration–response curves to Ach, SNP and NE in vehicle and GC were compared by two‐way analysis of variance (anova) for repeated measures. In each group (vehicle, GC‐HD, GC‐LD), concentration–response curves to Ach with or without a specific inhibitor were compared by two‐way anova for repeated measures. The two factors of anova were concentration (of Ach, SNP or PE) and treatment (vehicle, GC‐LD or GC‐HD) or pharmacological inhibitor (LNAME, etc.). When necessary, to understand more clearly the effect of inhibitors, the results were expressed as the area under the curve (AUC) calculated from the individual concentration–response curves. Comparison between two values was assessed by unpaired Student's t‐test or Mann–Whitney U‐test when data were not normally distributed. The analysis of the relationship between two parameters was determined by linear regression analysis and Spearman's correlation coefficient was calculated between these variables. P < 0·05 was considered statistically significant.
Results
Effects of GC on clinical and biological parameters
Compared to vehicle, as expected, GC‐LD did not reduce arthritic score (Fig. 1a). Similarly, SBP, diastolic blood pressure (DBP), heart rate, glycaemia and triglyceride levels were unchanged compared to vehicle, whereas total cholesterol levels were reduced (Table 1). Conversely, GC‐HD decreased arthritis scores dramatically in AIA (Fig. 1b) and induced higher SBP and DBP (P < 0·05), higher triglyceride levels (P < 0·001), lower cholesterol levels (P < 0·01) and slightly increased glycaemia, although the difference was not significant (Table 1). Regarding plasma cytokine levels, GC‐HD and GC‐LD reduced significantly IL‐1β (−69 and −75% versus vehicle, respectively) and TNF‐α levels (−70 and −75% versus vehicle, respectively) to the same extent (Table 1).
Figure 1.
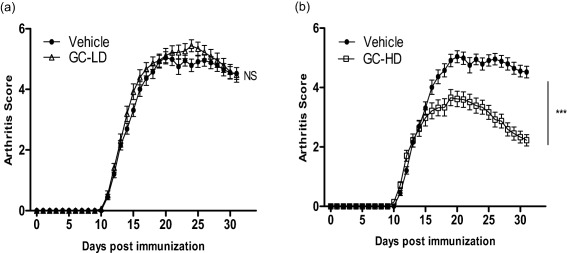
Evolution of arthritis in vehicle and glucocorticoid (GC)‐treated rats. Arthritis scores were plotted over time after adjuvant‐induced arthritis (AIA) induction in rats treated with (a) a low dose (LD) of prednisolone (0·1 mg/kg/day, GC‐LD), (b) a high dose (HD) of prednisolone (10 mg/kg/day, GC‐HD) and compared to saline‐treated rats (vehicle). Values are the mean ± standard error of the mean (s.e.m.) (n = 32 rats/group). ***P < 0·001.
Table 1.
Effect of glucocorticoids on physiological and biological parameters
| Vehicle | GC‐LD | GC‐HD | |
|---|---|---|---|
| SBP (mm Hg) | 101 ± 4 | 102 ± 4 | 115 ± 4* |
| DBP (mm Hg) | 65 ± 4 | 64 ± 4 | 79 ± 3* |
| Heart rate (bpm) | 225 ± 13 | 229 ± 12 | 218 ± 8 |
| Total cholesterol (g/l) | 0·89 ± 0·03 | 0·80 ± 0·01* | 0·77 ± 0·03** |
| Triglycerides (g/l) | 0·56 ± 0·05 | 0·57 ± 0·05 | 0·95 ± 0·09*** |
| Blood glucose (g/l) | 1·04 ± 0·03 | 1·10 ± 0·05 | 1·10 ± 0·04 |
| TNF‐α (pg/ml) | 28·9 ± 7·0 | 7·2 ± 2·4* | 8·7 ± 3·2* |
| IL‐1β (pg/ml) | 67·2 ± 14·7 | 16·3 ± 5·2* | 21·3 ± 6·8* |
*P < 0·05; **P < 0·01; ***P < 0·001. SBP = systolic blood pressure; DBP = diastolic blood pressure; MAP = mean arterial blood pressure; TNF = tumour necrosis factor; IL = interleukin; GC‐HD = glucocorticoids high dose; GC‐LD = GC low dose. All parameters were measured in adjuvant‐induced arthritis (AIA) rats at day 33 post‐immunization after 21 days of treatment (intraperitoneally) with saline (vehicle) or prednisolone at a low dose (0·1 mg/kg/day, GC‐LD) or a high dose (10 mg/kg/day, GC‐HD). Values are expressed as means ± standard error of the mean (n = 10–20 rats per group).
The low dose of GC did not improve endothelial function in AIA but induced some endothelial phenotypical changes
To determine whether GC might modify endothelial function independently of their effect on arthritis severity, the concentration–response curves to Ach were compared between GC‐LD and vehicle‐treated AIA rats. As shown in Fig. 2a, GC‐LD did not change Ach‐induced vasorelaxation. To ascertain that this effect was not due to impaired response of VSMCs to vasoconstrictive stimulus or to the relaxant effect of NO, the effect of GC‐LD on NE‐induced vasoconstriction and on SNP‐induced vasodilation was determined on endothelium‐denuded aortic rings. Results demonstrated that the response to the NO‐donor SNP (Fig. 2b) or to NE (Fig. 2c) was not different between GC‐LD and vehicle rats.
Figure 2.
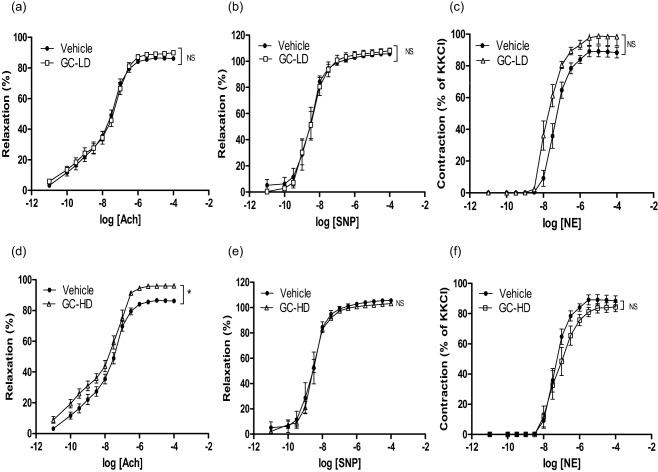
Vascular reactivity to vasodilators and vasoconstrictive agents in adjuvant‐induced arthritis (AIA) treated with prednisolone. Experiments were performed on thoracic aortic rings harvested at the end of the treatment period (21 days after the onset of arthritis) of AIA rats treated with saline (vehicle) or with prednisolone at low dose (LD) (0·1 mg/kg/day, GC‐LD) or high dose (HD) (10 mg/kg/day, GC‐HD). Concentration‐response curve of Ach in endothelium‐intact aortic rings preconstricted with phenylephrine (PE) 10−6 mol/l (a,d), sodium nitroprussiate (SNP) on endothelium‐denuded aortic rings preconstricted with PE 10−6 mol/l (b,e) and noradrenalin (NE) on endothelium‐denuded aortic rings (c,f). Values are the mean ± standard error of the mean (s.e.m.) (n = 6–15 aortic rings from 6–15 rats per group). **P < 0·01, by two‐way analysis of variance (anova) for repeated measures.
Despite the absence of effect on endothelial function, certain endothelial pathways involved in Ach‐relaxant effects were modified by this subtherapeutic dose of GC. Incubation of rings with L‐NAME significantly blunted Ach‐associated relaxation in both the vehicle (Fig. 3a) and GC‐LD groups (Fig. 3b). However, the effect of L‐NAME was greater in GC‐LD compared to vehicle (% reduction of AUC: 78 ± 5 versus 69 ± 7, P = 0·036), thus suggesting that NOS activity was enhanced by GC‐LD. This finding was confirmed by the higher expression of P‐eNOS (the active form of eNOS) (Fig. 3d) and total eNOS expression (Fig. 3f) in the GC‐LD group. In vehicle AIA, as a reflection of increased ° production, Tempol improved Ach‐induced vasorelaxation (Fig. 4a). The treatment of AIA with GC‐LD abolished the impact of Tempol on Ach‐induced relaxation (Fig. 4b). This effect was due, at least partly, to a decrease in p22phox expression (Fig. 4d), p47phox expression being unchanged by the treatment (Fig. 4f). A deficit in endothelium‐derived hyperpolarizing factor (EDHF) production is present in vehicle AIA, as apamin and charybdotoxin did not reduce but, on the contrary, improved Ach‐induced vasodilation (Fig. 5a). GC‐LD restored the EDHF contribution significantly, as attested by the reduction of Ach‐induced relaxation (Fig. 5b). By contrast, GC‐LD had no effect on arginase activity (Fig. 5e) and COX‐2 activity (Fig. 5h). In summary, the low dose of GC increased NOS activity and EDHF production and decreased ° production, but these effects were not sufficient to translate into an improvement of endothelial function.
Figure 3.
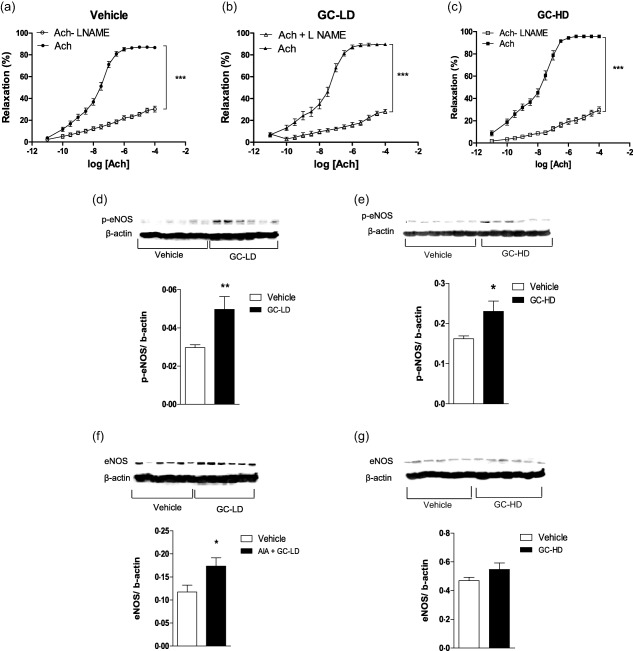
Effects of low dose (LD) and high dose (HD) of glucocorticoid (GC) on nitric oxide synthase (NOS) pathway. Experiments were performed on thoracic aortic rings harvested at the end of the treatment period (21 days after the onset of arthritis) of adjuvant‐induced arthritis (AIA) rats treated with saline (vehicle), or a low dose of prednisolone (0·1 mg/kg/day, GC‐LD) or a high dose of prednisolone (10 mg/kg/day, GC‐HD). Cumulative concentration‐response curves of acetylcholine (Ach) were obtained after incubation or not with NW‐nitro‐L‐arginine methyl ester (L‐NAME) at 10−4 mol/l (a–c). Expression of endothelial nitric oxide synthase (eNOS) (f,g) and its phosphorylated form at serine 1177, P‐eNOS (d,e), were evaluated in aortas by Western blotting. Values from the concentration–response curves are the mean ± standard error of the mean (s.e.m.) (n = 7–15 rings from 7–15 rats per group), compared by two‐way analysis of variance (anova) for repeated measures. Values from immunoblots are the mean ± s.e.m. (n = 6 rats per group), compared by the Mann–Whitney U‐test. *P < 0.05; ***P < 0·001.
Figure 4.
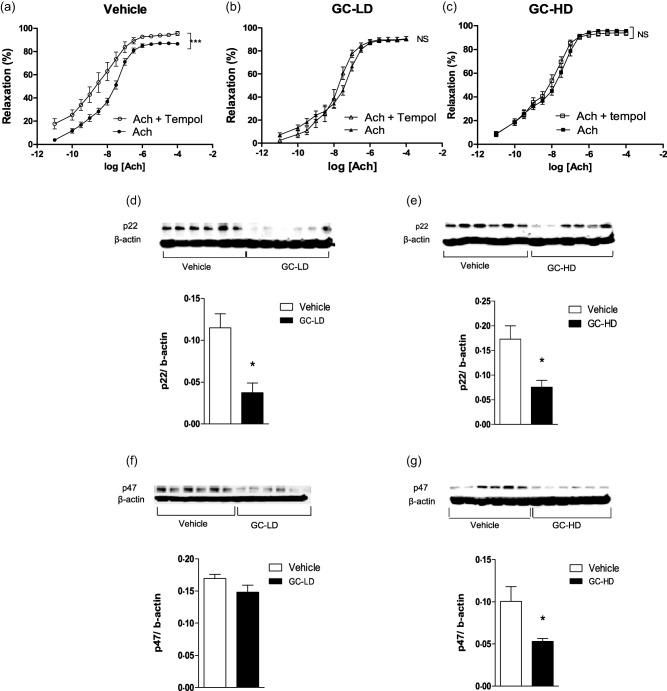
Effects of low dose (LD) and high dose (HD) of glucocorticoid (GC) on superoxide anions production. Experiments were performed on thoracic aortic rings harvested at the end of the treatment period (21 days after the onset of arthritis) of adjuvant‐induced arthritis (AIA) rats treated with saline (vehicle) or with a low dose of prednisolone (0·1 mg/kg/day, GC‐LD) or with a high dose of prednisolone (10 mg/kg/day, GC‐HD). Cumulative concentration–response curves of acetylcholine (Ach) were obtained after incubation or not with Tempol at 10−4 mol/l (a–c). Expression of the subunits of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase p22phox (d,e) and p47phox (f,g) were evaluated in aortas by Western blotting. Values from the concentration–response curves are the mean ± standard error of the mean (s.e.m.) (n = 8–15 rings from 8–15 rats per group), compared by two‐way analysis of variance (anova) for repeated measures. Values from immunoblots are the mean ± s.e.m. (n = 6 rats per group), compared by the Mann–Whitney U ‐test. *P < 0·05; ***P < 0·001.
Figure 5.
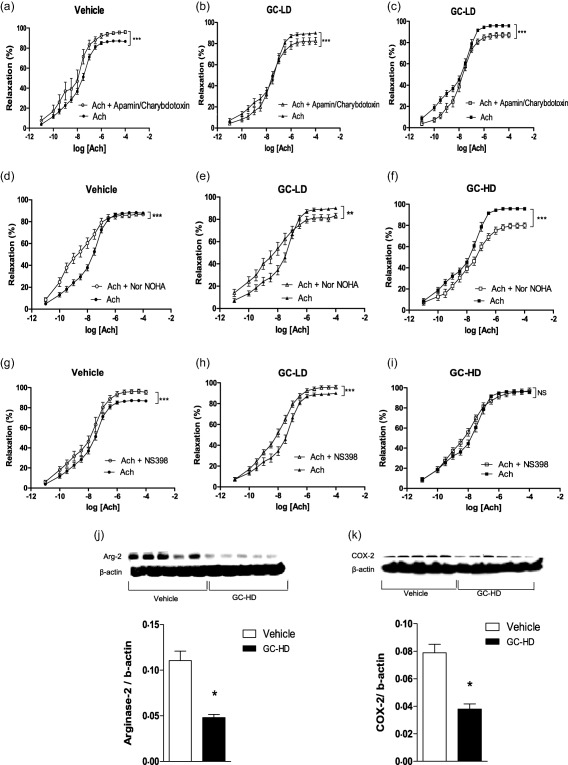
Effects of low dose (LD) and high dose (HD) of glucocorticoid (GC) on endothelium derived hyperpolarizing factor (EDHF), arginase, cyclooxygenase 2 (COX‐2) pathways. Experiments were performed on thoracic aortic rings harvested at the end of the treatment period (21 days after the onset of arthritis) of adjuvant‐induced arthritis (AIA) rats treated with saline (vehicle) or with a low dose of prednisolone (0·1 mg/kg/day, GC‐LD) or with a high dose of prednisolone (10 mg/kg/day, GC‐HD). Cumulative concentration–response curves of acetylcholine (Ach) were obtained after incubation or not with Apamin and charybdotoxin at 10−7 mol/l (a–c), Nw‐hydroxy‐nor‐L‐arginine (NOHA) at 10−4 mol/l (d–f) or NS‐398 at 10−5 mol/l (g–i). Expression of arginase 2 (j) and COX‐2 (k) were evaluated in aortas by Western blotting. Values from the concentration–response curves are the mean ± standard error of the mean (s.e.m.) (n = 8–15 rings from 8–15 rats per group), compared by two‐way analysis of variance (anova) for repeated measures. *P < 0·05; ***P < 0·001. Values from immunoblots are the mean ± s.e.m. (n = 6 rats per group), compared by the Mann–Whitney U‐test. *P < 0·05; ***P < 0·001.
The high dose of GC enhanced endothelial function in AIA via pleiotropic endothelial mechanisms
Contrary to GC‐LD, GC‐HD improved endothelial function significantly, as attested by the improvement of Ach‐induced relaxation compared to vehicle AIA (Fig. 2d). No difference regarding NE‐induced vasoconstriction and SNP‐induced relaxation was observed between GC‐HD and vehicle (Fig 2e, f). The beneficial effect of GC‐HD relies on increased NOS activity (the L‐NAME‐induced reduction of AUC of Ach was greater in GC‐HD, −82 ± 2%, than in the vehicle group, −69 ± 7%, P = 0·035, Fig. 3c), as confirmed by the increase in P‐eNOS expression (Fig. 3e). Conversely, total eNOS expression was unchanged (Fig. 3g). GC‐HD led to a decrease in ° production, as attested by the lack of effect of Tempol on Ach‐induced relaxation (Fig. 4c) secondary to decreased expression of both p22phox (Fig. 4e) and p47phox (Fig. 4g) expression. GC‐HD restored the EDHF contribution in AIA (Fig. 5c). Whereas incubation of aortic rings with the arginase inhibitor nor‐NOHA improved relaxation in vehicle (Fig. 5d), consistent with the increased deleterious activity of this enzyme in AIA 18, nor‐NOHA decreased the response to Ach in GC‐HD group (Fig. 5f). These data indicated that GC‐HD decreased arginase activity, an effect associated with a blunted expression of arginase 2 (Fig. 5j). Moreover, GC‐HD decreased COX‐2 activity (the effect of NS‐398 on Ach‐induced relaxation is abolished in the GC‐HD group, Fig. 5i), as well as COX‐2 expression (Fig. 5k). Overall, the results indicate that the HD of GCs induced changes in the major endothelial pathways activated by Ach leading to the improvement of endothelial function in AIA.
Of note, no correlation was found between arthritis score and Ach‐induced relaxation (AUC) (r = –0·316, P = 0·7) (all AIA rats from the three groups, data not shown).
Discussion
Although ED is acknowledged as the early vascular event leading to atherogenesis and, in turn, high risk of morbidity in RA 3, only a few studies were designed to identify therapeutic options able to reduce this vascular impairment. With regard to the effect of GC therapy, certain studies reported a positive effect or the lack of effect on endothelial function in RA patients 14, 15, 16, 17, 18. These discrepant results may be explained by the great heterogeneity in RA disease stage, dosages and treatment duration of GCs, or therapies associated with GCs among these studies. Using an animal model of RA that offers the unique opportunity to standardize these different points, the present study revealed that ED and disease activity are both reduced by the clinically efficient GC‐HD, but refractory to the inefficient GC‐LD, suggesting a contribution of decreased arthritis severity to the observed effects on endothelial function. In our conditions, the HD of prednisolone was associated with a negative effect on cardiometabolic parameters, such as blood pressure and lipid levels. Thus, even though the transposition of the dose used in an animal model to doses used in clinical conditions is somewhat speculative, it can be assumed that the HD used in this study in AIA may be related to an intermediate or HD of prednisolone in RA patients (i.e. > 7·5 mg per day) 26. Unexpectedly, both doses changed endothelial phenotype in AIA rats. Therefore, even if the reduction of arthritis probably contributes to the effect of GC, this result, associated with the lack of correlation between arthritis score and endothelial function, suggests that a direct effect of GC on endothelial cells is also involved. However, while the two GC doses share the same positive effect on NOS activity (increase), O2–° (decrease) and EDHF (increase) production, only the HD decreased the COX‐2 and arginase pathways. These data corroborate the seminal role of the balance between COX‐2 and NOS/arginase pathways in endothelial dysfunction identified previously in AIA 24, and demonstrate that the positive effect of GCs on endothelial function requires a combination of concomitant effects on NOS, arginase, COX‐2 and ° pathways, as observed previously with other drugs such as arginase inhibitors or etanercept 18, 24. The pleiotrophic effects of GC on endothelial pathways suggest that, at least partly, GC acts on a common endothelial effector. One candidate might be the p38 mitogen‐activated protein kinase (MAPK) signalling pathway, activated by inflammation 27, which activates eNOS 28, arginase 29 and COX‐2 30, the activity of which was shown to be reduced by GCs, at least in macrophages 31. Further studies are warranted to confirm this hypothesis.
In an attempt to link the effect of GC on ED to their effect on systemic inflammation, plasma IL‐1β and TNF‐α, two dominant mediators of immune‐mediated joint disease in RA patients and experimental models 32, were measured. Preclinical studies suggested previously that circulating proinflammatory cytokines such as IL‐1β and TNF‐α could be reliable biomarkers of ED in RA 22. Moreover, both cytokines were reported to induce ED acutely 33, 34. In contrast, the present study reported a similar decrease in plasma levels of these cytokines by GCs irrespective of its impact on endothelial function in AIA rats. This apparently conflicting result does not question the involvement of inflammatory processes in ED elicited by AIA, but rather supports the idea that the effect of drugs on ED in RA patients cannot be predicted from circulating proinflammatory cytokine levels. Rather, it supports the idea that circulating cytokines are not a reliable marker of vascular inflammation, as suggested previously in other tissues 35. In addition, these data provide arguments that the effect of GCs on endothelial pathways relies – at least partly – on a direct vascular effect. Our results are consistent with the previously reported lack of association between cumulative inflammation and endothelial function in RA patients 36, 37, 38. They are also in line with a study in RA, showing that DMARDs‐treated RA patients who used GCs had a better endothelial function than the non‐GC users, but that endothelial function (measured by flow‐mediated vasodilation) was not correlated with systemic inflammation in this cohort (C‐reactive protein levels and erythrocyte sedimentation rate) 18. Such disconnection between the effect of a treatment on ED and on biological inflammation resonates with previous findings with other drugs than GCs. A recent meta‐analysis reported that despite a decrease in biological inflammation, anti‐TNF‐α therapy induced no changes in ED 39. Similarly, pioglitazone decreased markers of inflammation in RA patients, while it did not change endothelial function 40, 41. From a clinical perspective, these results suggest that biological signs of inflammation are not useful to assess the impact of GCs on endothelial function.
GC therapy was suggested to increase CV risk as a result of GC‐induced dyslipidaemia, hypertension and insulin resistance 13. In the present study, the subchronic administration of GC in AIA rats improved endothelial function despite an elevation in blood pressure and triglyceride levels. This finding suggests that the effects of GCs on traditional CV risk factors are not predictive of their effects on endothelial function in RA, in agreement with evidence that classical CV risk factors are minimally explaining the CV risk in RA 42. Our results resonate with a study reporting that RA patients receiving DMARD alone or DMARD + prednisolone for 5 years exhibited the same endothelial function, despite a more frequent hypertension and dyslipidaemia in the GC group 14. However, although not investigated in the present study, we cannot exclude that the undesirable cardiometabolic actions of prednisolone would outweigh its positive vascular effects after longer treatment duration. Of note, as blood pressure levels are determined by vascular tone in resistance vessels, a differential effect of GC on ED among distinct vascular beds cannot be excluded.
In conclusion, the present study demonstrated that a subchronic treatment with prednisolone used at a clinically efficient dose improved endothelial function in the AIA model independently of its negative cardiometabolic effects. Contrary to the traditional thinking that GCs increased CV risk, our data suggest that GCs might prove beneficial for vascular function in early RA. From a clinical perspective, as ED and increased CV risk occurs early after RA onset 43, studying the effects of GCs on ED when used as a bridge therapy for other DMARDs in early RA would be of particular interest.
Funding sources
This work was supported by funding from the French Ministry for Higher Education and Research.
Disclosures
None.
Acknowledgements
The authors thank Dr C. Moussard for the analysis of cholesterol and triglyceride levels, M. Nappey for her technical assistance for tissue collection and blood pressure measurements, R. Bordy for his help for plasma cytokines measurements and Alice Monnier for her help for Western blot analysis.
References
- 1. Avina‐Zubieta JA, Choi HK, Sadatsafav M, Etminan M, Esdaile JM, Lacaille D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta‐analysis of observational studies. Arthritis Rheum 2008; 59:1690–7. [DOI] [PubMed] [Google Scholar]
- 2. Giles JT. Cardiovascular disease in rheumatoid arthritis: current perspectives on assessing and mitigating risk in clinical practice. Best Pract Res Clin Rheumatol 2015; 29:597–613. [DOI] [PubMed] [Google Scholar]
- 3. Steyers CM, 3rd , Miller FJ. Jr. Endothelial dysfunction in chronic inflammatory diseases. Int J Mol Sci 2014; 15:11324–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pieringer H, Pichler M. Cardiovascular morbidity and mortality in patients with rheumatoid arthritis: vascular alterations and possible clinical implications. Q J Med 2011; 104:13–26. [DOI] [PubMed] [Google Scholar]
- 5. Boers M. Drugs and cardiovascular risk in inflammatory arthritis: another case of glucocorticoid‐bashing? Ann Rheum Dis 2015; 74:e33. [DOI] [PubMed] [Google Scholar]
- 6. Roubille C, Richer V, Starnino T et al Response to: ‘Drugs and cardiovascular risk in inflammatory arthritis: another case of glucocorticoid‐bashing?’ by Dr Boers. Ann Rheum Dis 2015; 74:e34. [DOI] [PubMed] [Google Scholar]
- 7. Goodwin JE. Glucocorticoids and the cardiovascular system. Adv Exp Med Biol 2015; 872:299–314. [DOI] [PubMed] [Google Scholar]
- 8. del Rincón I, Battafarano DF, Restrepo JF, Erikson JM, Escalante A. Glucocorticoid dose thresholds associated with all‐cause and cardiovascular mortality in rheumatoid arthritis. Arthritis Rheumatol 2014; 66:264–72. [DOI] [PubMed] [Google Scholar]
- 9. Listing J, Kekow J, Manger B et al Mortality in rheumatoid arthritis: the impact of disease activity, treatment with glucocorticoids, TNFα inhibitors and rituximab. Ann Rheum Dis 2015; 74:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wei L, MacDonald TM, Walker BR. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann Intern Med 2004; 141:764–70. [DOI] [PubMed] [Google Scholar]
- 11. Naranjo A, Sokka T, Descalzo MA et al Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST‐RA study. Arthritis Res Ther 2008; 10:R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ajeganova S, Svensson B, Hafström I, BARFOT Study Group . Low‐dose prednisolone treatment of early rheumatoid arthritis and late cardiovascular outcome and survival: 10‐year follow‐up of a 2‐year randomised trial. BMJ Open 2014; 4:e004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ruyssen‐Witrand A, Fautrel B, Saraux A, Le Loët X, Pham T. Cardiovascular risk induced by low‐dose corticosteroids in rheumatoid arthritis: a systematic literature review. Joint Bone Spine 2011; 78:23–30. [DOI] [PubMed] [Google Scholar]
- 14. Hafström I, Rohani M, Deneberg S, Wörnert M, Jogestrand T, Frostegård J. Effects of low‐dose prednisolone on endothelial function, atherosclerosis, and traditional risk factors for atherosclerosis in patients with rheumatoid arthritis‐a randomized study. J Rheumatol 2007; 34:1810–6. [PubMed] [Google Scholar]
- 15. Ikonomidis I, Lekakis JP, Nikolaou M et al Inhibition of interleukin‐1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation 2008; 117:2662–9. [DOI] [PubMed] [Google Scholar]
- 16. Radhakutty A, Mangelsdorf BL, Drake SM et al Effect of acute and chronic glucocorticoid therapy on insulin sensitivity and postprandial vascular function. Clin Endocrinol 2016; 84:501–8. [DOI] [PubMed] [Google Scholar]
- 17. Foster W, Carruthers D, Lip GY, Blann AD. Inflammation and microvascular and macrovascular endothelial dysfunction in rheumatoid arthritis: effect of treatment. J Rheumatol 2010; 37:711–6. [DOI] [PubMed] [Google Scholar]
- 18. Veselinovic MV, Zivkovic VI, Toncev S et al Carotid artery intima‐media thickness and brachial artery flow‐mediated vasodilatation in patients with rheumatoid arthritis. Vasa 2012; 41:343–51. [DOI] [PubMed] [Google Scholar]
- 19. Prati C, Berthelot A, Kantelip B, Wendling D, Demougeot C. Treatment with the arginase inhibitor Nw‐hydroxy‐nor‐L‐arginine restores endothelial function in rat adjuvant‐induced arthritis. Arthritis Res Ther 2012; 1:R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sakaguchi N, Takahashi T, Hata H et al Altered thymic T‐cell selection due to a mutation of the ZAP‐70 gene causes autoimmune arthritis in mice. Nature 2003; 426:454–60. [DOI] [PubMed] [Google Scholar]
- 21. Abdin AA, Abd El‐Halim MS, Hedeya SE, El‐Saadany AA. Effect of atorvastatin with or without prednisolone on Freund's adjuvant induced‐arthritis in rats. Eur J Pharmacol 2012; 676:34–40. [DOI] [PubMed] [Google Scholar]
- 22. Totoson P, Maguin‐Gaté K, Nappey M, Wendling D, Demougeot C. Endothelial dysfunction in rheumatoid arthritis: mechanistic insights and correlation with circulating markers of systemic inflammation. PLOS ONE 2016; 11:e0146744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haruna Y, Morita Y, Komai N et al Endothelial dysfunction in rat adjuvant‐induced arthritis: vascular superoxide production by NAD (P) H oxidase and uncoupled endothelial nitric oxide synthase. Arthritis Rheum 2006; 54:1847–55. [DOI] [PubMed] [Google Scholar]
- 24. Totoson P, Maguin‐Gaté K, Nappey M et al Microvascular abnormalities in adjuvant‐induced arthritis: relationship with macrovascular endothelial dysfunction and markers of endothelial activation. Arthritis Rheumatol 2015; 67:1203–13. [DOI] [PubMed] [Google Scholar]
- 25. Totoson P, Maguin‐Gaté K, Prigent‐Tessier A et al Etanercept improves endothelial function via pleiotropic effects in rat adjuvant‐induced arthritis. Rheumatology 2016; 55:1308–17. [DOI] [PubMed] [Google Scholar]
- 26. Joana Fonseca F, Abdelkhalik Mohamed AA, Emery P. Glucocorticoids and rheumatoid arthritis Rheum. Dis Clin North Am 2016; 42:33–46. [DOI] [PubMed] [Google Scholar]
- 27. Kragholm K, Newby LK, Melloni C. Emerging treatment options to improve cardiovascular outcomes in patients with acute coronary syndrome: focus on losmapimod. Drug Des Devel Ther 2015; 9:4279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anter E, Thomas SR, Schulz E, Shapira OM, Vita JA, Keaney JF. Activation of endothelial nitric‐oxide synthase by the p38 MAPK in response to black tea polyphenols. J Biol Chem 2004; 279:46637–43. [DOI] [PubMed] [Google Scholar]
- 29. Toque HA, Romero MJ, Tostes RC et al p38 Mitogen‐activated protein kinase (MAPK) increases arginase activity and contributes to endothelial dysfunction in corpora cavernosa from angiotensin‐II‐treated mice. J Sex Med 2010; 7:3857–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caughey GE, Cleland LG, Penglis PS, Gamble JR, James MJ. Roles of cyclooxygenase (COX)‐1 and COX‐2 in prostanoid production by human endothelial cells: selective up‐regulation of prostacyclin synthesis by COX‐2. J Immunol 2001; 167:2831–8. [DOI] [PubMed] [Google Scholar]
- 31. Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage glucocorticoid receptors regulate Toll‐like receptor 4‐mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 2007; 109:4313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stolina M, Bolon B, Middleton S et al The evolving systemic and local biomarker milieu at different stages of disease progression in rat adjuvant‐induced arthritis. J Clin Immunol 2009; 29:158–74. [DOI] [PubMed] [Google Scholar]
- 33. Jiménez‐Altayó F, Briones AM, Giraldo J, Planas AM, Salaices M, Vila E. Increased superoxide anion production by interleukin‐1beta impairs nitric oxide‐mediated relaxation in resistance arteries. J Pharmacol Exp Ther 2006; 316:42–52. [DOI] [PubMed] [Google Scholar]
- 34. Wimalasundera R, Fexby S, Regan L, Thom SA, Hughes AD. Effect of tumour necrosis factor‐alpha and interleukin 1beta on endothelium‐dependent relaxation in rat mesenteric resistance arteries in vitro. Br J Pharmacol 2003; 138:1285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ernberg M, Christidis N, Ghafouri B et al Effects of 15 weeks of resistance exercise on pro‐inflammatory cytokine levels in the vastus lateralis muscle of patients with fibromyalgia. Arthritis Res Ther 2016; 18:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sandoo A, Kitas GD, Carroll D, Veldhuijzen van Zanten JJ. The role of inflammation and cardiovascular disease risk on microvascular and macrovascular endothelial function in patients with rheumatoid arthritis: a cross‐sectional and longitudinal study. Arthritis Res Ther 2012; 14:R117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vandhuick T, Allanore Y, Borderie D et al Early phase clinical and biological markers associated with subclinical atherosclerosis measured at 7 years of evolution in an early inflammatory arthritis cohort. Clin Exp Rheumatol 2016; 34:58–67. [PubMed] [Google Scholar]
- 38. Klimek E, Skalska A, Kwaśny‐Krochin B et al Differential associations of inflammatory and endothelial biomarkers with disease activity in rheumatoid arthritis of short duration. Mediators Inflamm 2014; 2014:681635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mathieu S, Couderc M, Pereira B, Soubrier M. The effects of TNF‐alpha inhibitor therapy on arterial stiffness and endothelial dysfunction in rheumatoid arthritis: a meta‐analysis. Semin Arthritis Rheum 2013; 43:e1–2. [DOI] [PubMed] [Google Scholar]
- 40. Marder W, Khalatbari S, Myles JD et al The peroxisome proliferator activated receptor‐γ pioglitazone improves vascular function and decreases disease activity in patients with rheumatoid arthritis. J Am Heart Assoc 2013; 2:e000441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ormseth MJ, Oeser AM, Cunningham A et al Reversing vascular dysfunction in rheumatoid arthritis: improved augmentation index but not endothelial function with peroxisome proliferator‐activated receptor γ agonist therapy. Arthritis Rheumatol 2014; 66:2331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. del Rincón ID, Williams K, Stern MP, Freeman GL, Escalante A. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum 2001; 44:2737–45. [DOI] [PubMed] [Google Scholar]
- 43. de Groot L, Jager NA, Westra J et al Does reduction of disease activity improve early markers of cardiovascular disease in newly diagnosed rheumatoid arthritis patients? Rheumatology 2015; 54:1257–61. [DOI] [PubMed] [Google Scholar]