

Abstract
Familial hypercholesterolemia (FH) is a disease characterized by a triad: elevated low-density lipoprotein (LDL) cholesterol, tendon xanthomas, and premature coronary heart disease. Thus, it can be considered as a model disease for hypercholesterolemia and atherosclerotic cardiovascular disease (ASCVD). For the diagnosis of hetero-FH, the detection of Achilles tendon xanthomas by palpation or on X-ray is an indispensable diagnostic skill in clinical lipidology. To prevent the under-diagnosis and under-treatment of FH, the diagnostic criteria should be more convenient and user-friendly. For a patient with cutaneous or tendon xanthomas, the probability of FH is very high; however, an absence of xanthoma does not rule out FH.
Brown and Goldstein elucidated the pathogenesis of FH by their work on LDL-receptor (LDL-R), for which they were awarded the Nobel Prize in 1985. In the 1950s, FH patients were divided into heterozygous (hetero-) and homozygous (homo-) FH, and diagnosing homo- and hetero-FH based on the phenotypic features of ASCVD or xanthomas frequently became difficult without the DNA analysis of FH genes. It is estimated that heterozygous mutations in the LDL-R or the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene will be found at a combined frequency of 0.005, which corresponds to 1/199 people in the general population in Japan.
Statins and anti-PCSK9 monoclonal antibodies are highly specific and efficient drugs for treating hetero- or homo-FH patients. Most clinical studies have reported an amelioration of ASCVD using long-term statin therapy. Clinical results using anti-PCSK9 monoclonal antibodies will emerge in a few years. In homo-FH patients, mipomersen and lomitapide are expected to yield good results. It is important to sequentially unravel the unrecognized pathogenetic mechanisms of FH to reduce its under-recognition and develop new management strategies for it.
Keywords: Familial hypercholesterolemia (FH), Heterozygous FH, Homozygous FH, Statin, Atherosclerotic cardiovascular disease (ASCVD)
Introduction
Familial hypercholesterolemia (FH) is a common genetic disease, which complicates atherosclerotic cardiovascular disease (ASCVD) because of high levels of low-density lipoprotein (LDL) cholesterol (LDL-C), and can thus be considered as a model disease for hypercholesterolemia and ASCVD1). The first case of FH reported by Fagge in 1873 was associated with cutaneous and tendon xanthomas (Figs. 1, 2, 3, and 4); however, the association of FH with ASCVD or hypercholesterolemia was unknown at that time2). The first homozygous FH (homo-FH) case in Japan was reported by Yamakawa and Kashiwabara in 19223). This 35-year-old man exhibited prominent generalized cutaneous xanthomatosis and severe ASCVD symptoms and signs due to hypercholesterolemia of 526 mg/dL (Fig. 5).
Fig. 1.
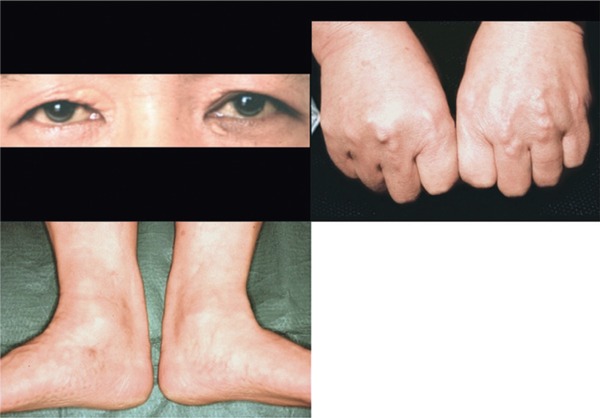
Physical findings in a hetero-FH patient.
S.N. 43 y.o. Male, Achilles tendon thickness (ATT); 17.0 mm, TC 392 mg/dL, TG; 124 mg/dL
Bilateral xanthelasma (+), r-eye; arcus corneae (+), bilateral hands; tendon xanthomas (+), bilateral Achilles tendon xanthomas (+)
Fig. 2.
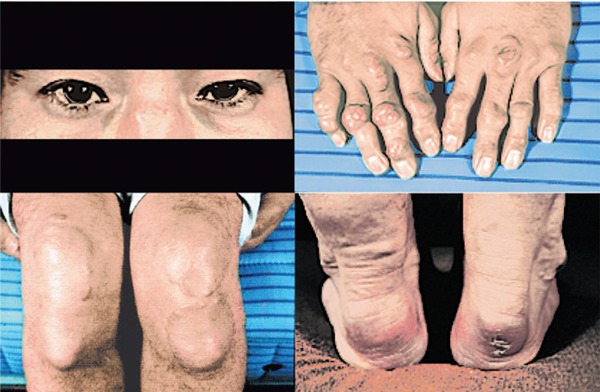
Physical findings in a homo-FH patient.
S.Y. 24 y.o. male, ATT; 42.0 mm, TC 538 mg/dL, TG 80 mg/dL, HDL-C 28 mg/dL, LDL-C 486 mg/dL, LDL-R gene mutations; D280Y/D280Y.
Severe xanthomas on the backs of bilateral hands and nose root. Severe bilateral planar xanthomas around knee joints, bilateral periosteal xanthomas below knee joints, severe ATT; 42.0 mm
Fig. 3.
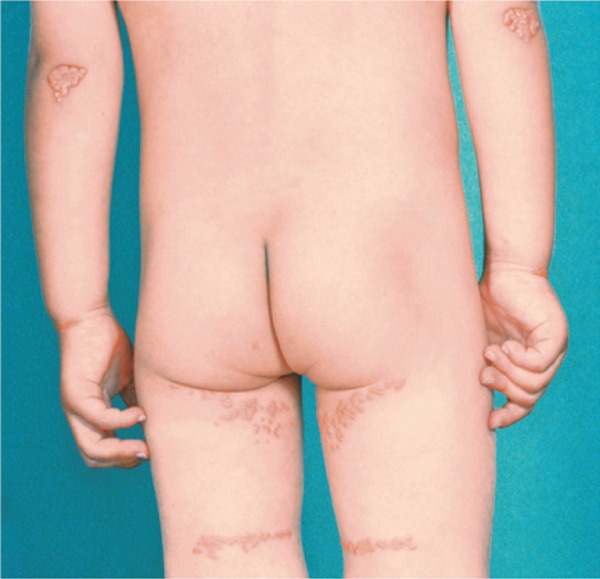
Severe planar and tuberous xanthomatosis in a homo-FH boy.
M.K. 3 y.o. TC 599 mg/dL, TG 61 mg/dL, HDL-C 53 mg/dL, LDL-C 534 mg/dL
Achilles tendon xanthoma (−), His FH genes were double mutations; LDL-R C183S/PCSK9 E32K.
This patient is responsive to statin (pitavastatin) and ezetimibe.
The same double mutations in the FH-related genes (LDL-R and PCSK9) were observed in his younger brother (K.K.), but he has no cutaneous and tendon xanthomas.
Fig. 4.
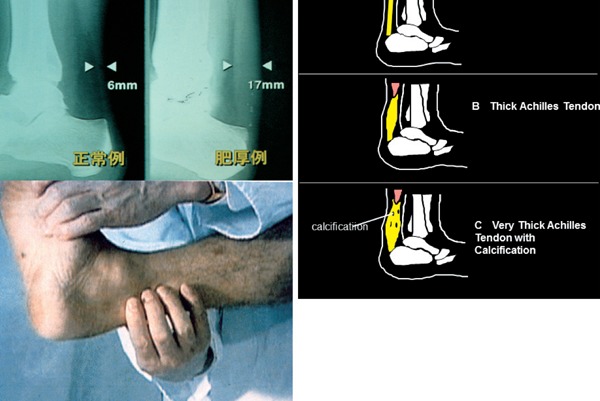
Achilles tendon xanthoma can be easily diagnosed by palpation or on X-ray. (Left; 6.0 mm, Right; 17.0 mm)
Fig. 5.
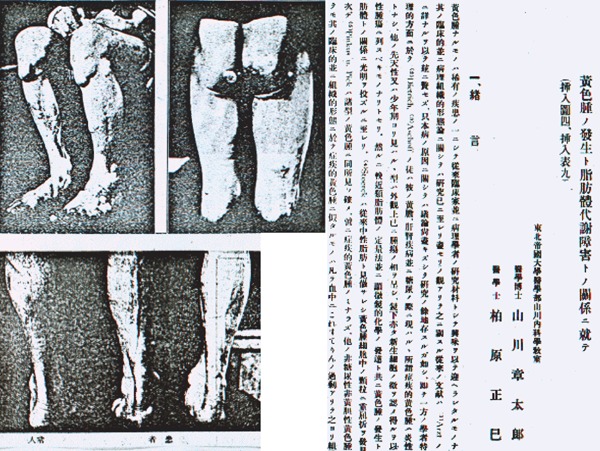
The first case of homo-FH in Japan was reported by Yamakawa and Kashiwabara in 1922. This 35-year-old man showed prominent generalized cutaneous and tendon xanthomatosis and severe ASCVD symptoms and signs due to hypercholesterolemia of 526 mg/dL.
It was not until the widespread uptake of serum cholesterol measurement in the 1950s that the association of hypercholesterolemia with ASCVD and cutaneous or tendon xanthomas was gradually revealed4). Fredrickson et al 5) studied the classification of hereditary lipoprotein metabolic diseases; however, they did not advance from the phenotypic classification to genotypic elucidation.
In this review article, I describe the clinical phenotypic and genotypic diagnosis of FH and the progress in its management mainly in Japan.
Molecular Pathogenesis of FH
In 1974, Brown and Goldstein (Fig. 6)6) initiated the pathogenetic elucidation of FH in a case of homo-FH with extremely high plasma LDL levels. They were awarded Nobel Prize in 1985. In accordance with their methodology, we also studied the LDL metabolism in cultured skin fibroblasts of our patients with hetero- and homo-FH (Fig. 7)7). The suppression of negative feedback from normal cells by the cholesterol that was dissolved in LDL or ethanol in culture medium was observed; however, negative feedback was not observed in the cells of homo-FH patients. Thus, FH was found to be an LDL metabolic disorder because of the abnormality in the LDL receptor (LDL-R)8).
Fig. 6.

In 1974, Brown and Goldstein succeeded in the elucidation of FH, and they were awarded Nobel Prize in 1985. They visited Kanazawa for giving us memorial lectures in 1992. (Left; M.S. Brown, Right; J.L. Goldstein. They permitted me to use this picture in this article.)
Fig. 7.
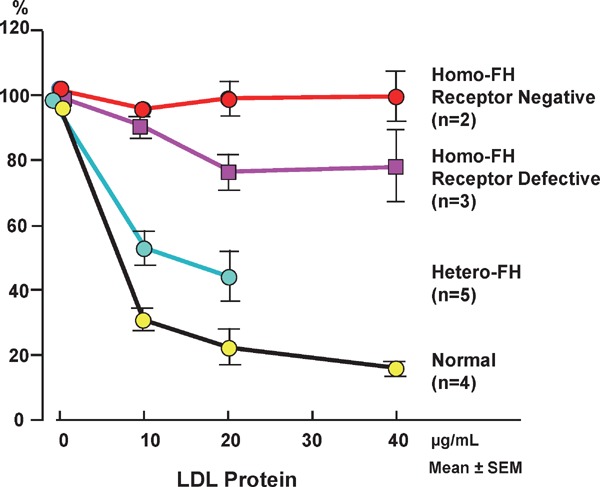
Effects of various concentrations of LDL on HMG-CoA reductase activity in cultured skin fibroblasts from unaffected subjects and hetero- and homo-FH patients.
Residual LDL-R activity was enough in normal subjects, and in hetero-FH patients residual LDL-R activity is about half of normal activity, and receptor activity in receptor-negative homo-FH patients is null, and the activity in the receptor-defective homo-FH is less than 20% of normal activity.
In association with Brown and Goldstein, Yamamoto9) successfully cloned the LDL-R gene, which is located on the short arm of chromosome 19 and encodes for 839 amino acids. Abnormalities in this gene are associated with FH; more than 1200 mutations in the LDL-R gene have been reported till date10). Recently, the gain-of-function mutation of proprotein convertase subtilisin/kexin type 9 (PCSK9) that resolves LDL-R was shown to reduce LDL-R; reduce the number and activities of LDL-R, which mimics the clinical picture of FH caused by LDL-R abnormalities (Fig. 8)11, 12).
Fig. 8.
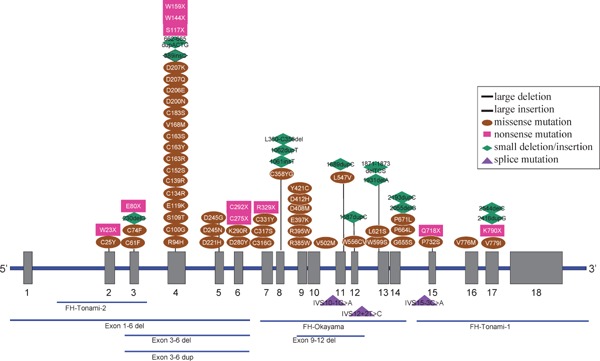
Location of point mutations and small deletions/insertions in LDL-R gene in Japanese FH patients in Hokuriku district.
FH caused by autosomal recessive hypercholesterolemia (ARH)13, 14) and signal-transducing adaptor family member 1 abnormality15) has also been reported. Similarly, the discovery of several other genes causing FH is anticipated. The frequencies of abnormalities in the FH-related genes identified till date in our laboratory are presented in Table 1 and Figs. 8 and 9.
Table 1. Incidence of FH gene mutant alleles in autosomal dominant FH.
| Genetic hypercholesterolemia | FH classification | FH gene mutation | Homozygote (n), (%) | Heterozygote (n), (%) | Total mutant alleles (n), (%) |
|---|---|---|---|---|---|
| Autosomal dominant hypercholesterolemia (ADH) | ADH1 | LDL-R | 69 (84.1%) | 757 (71.8%) | 826 (72.6%) |
| ADH2 | APO-B | 0 (0%) | 0 (0%) | 0 (0%) | |
| ADH3 | PCSK9 | 13 (15.9%) | 62 (5.9%) | 75 (6.6%) | |
| ADH4 | STAP1 * | 0 (0%) | 0 (0%) | 0 (0%) | |
| Unknown | 0 (0%) | 236 (22.4%) | 236 (20.8%) | ||
| Total | 82 (100%) | 1055 (100%) | 1137 (100%) | ||
FH genes: LDL-R gene, apolipoprotein B-100 (Apo-B) gene, PCSK9 gene and STAP1
STAP1*: not studied
Fig. 9.
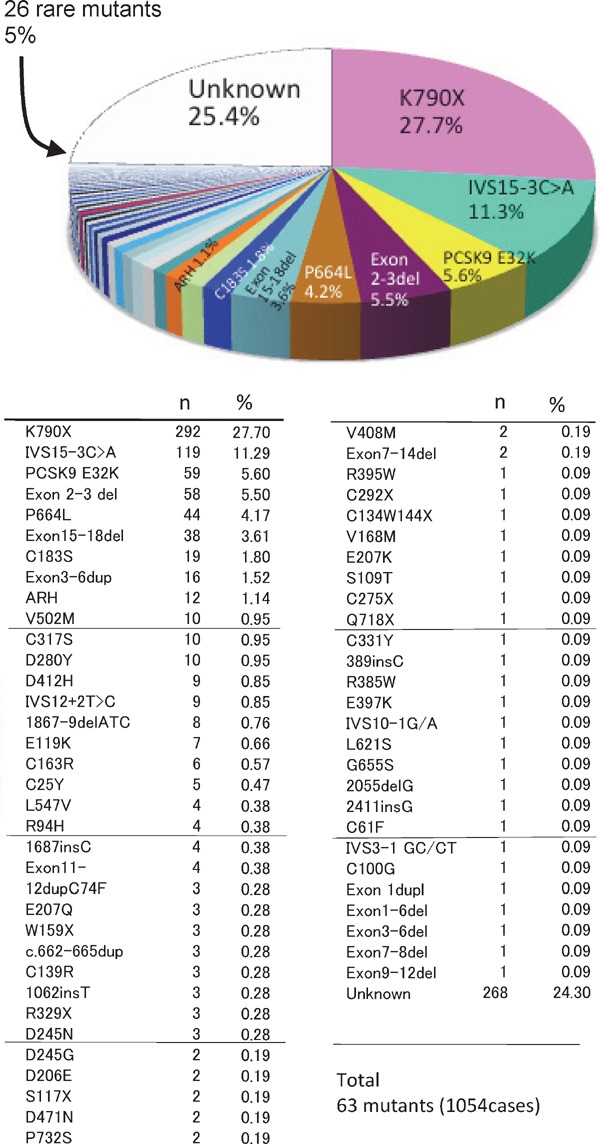
The frequency of FH gene mutations in LDL-receptor and PCSK9 in Japan.
Diagnostic Criteria of FH
Previously, the diagnosis of FH was based primarily on the presence of cutaneous and tendon xanthomas, and its association with hypercholesterolemia or ASCVD was not considered2). Although the presence of cutaneous or tendon xanthomas is a definitive sign of FH, its absence does not rule out FH. In other words, although xanthoma has a high specificity for the diagnosis of FH, its sensitivity is rather low. When we consulted a dermatologist because of xanthomas, most cases were simply diagnosed as homo-FH or seriously ill hetero-FH. When most FH cases were revealed only during dermatological consultations for xanthomas, most hetero-FH cases without xanthomas were never revealed and only homo-FH cases were sporadically reported15, 16).
The distribution of serum total cholesterol (TC) levels of unaffected subjects and hetero- and homo-FH patients are presented in Fig. 1017). Three peaks are observed in the serum cholesterol distribution among members of families affected by FH. These three peaks represent healthy individuals and hetero- and homo-FH patients. For the differential diagnosis of unaffected subjects and hetero-FH patients, establishing a threshold between these groups is essential. For such a threshold, we adapted the TC level for FH diagnosis and set the LDL-C reference value (Fig. 11). For the diagnosis of FH, the reference value of serum TC was 225 mg/dL and that of LDL-C was 162 mg/dL (Fig. 11)18, 19). Thus, the clinical diagnostic criteria of hetero-FH is defined as hyper-LDL cholesterolemia with tendon xanthomas (Table 2).
Fig. 10.
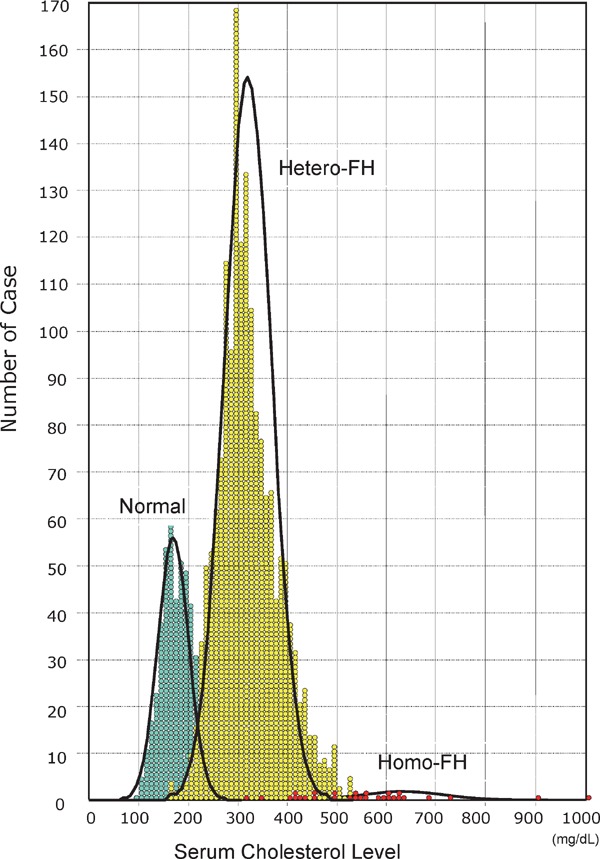
Plasma cholesterol distributions in homo-FH patients, hetero-FH patients and unaffected subjects. Clearly three peaks are observed in the serum cholesterol distribution among members of families affected by FH. These three peaks represent healthy individuals and hetero-FH and homo-FH patients.
Fig. 11.
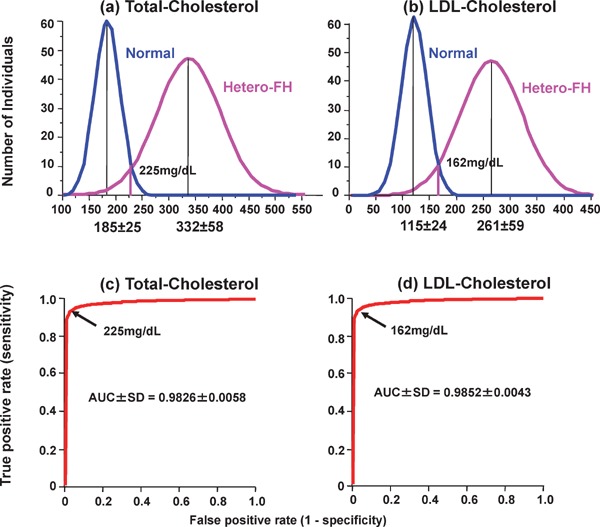
Distributions of (a) plasma total cholesterol and (b) LDL-cholesterol in FH and non-FH patients. Receiver operating characteristic (ROC) curves discriminating between FH and non-FH subjects by (c) total cholesterol and (d) LDL-cholesterol are shown. Areas under ROC curves (AUC) are shown as mean ± SD.
Table 2. Diagnostic criteria of familial hypercholesterolemia (FH).
| I. Hetero-FH | |
| a) Clinical diagnostic criteria | Hypercholesterolemia with tendon xanthomas, or hypercholesterolemia in the first- or second-degree relatives of FH patients. |
| b) Genetic diagnostic criteria | Heterozygous mutations of FH genes. |
| II. Homo-FH | |
| a) Clinical diagnostic criteria | Juvenile xanthomatosis with plasma cholesterol level about twice that of parents or other family members with hetero-FH |
| b) Genetic diagnostic criteria | True homozygotes, compound heterozygotes or double heterozygotes of FH genes |
Hypercholesterolemia: Hyper-LDL-cholesterolemia
FH genes: LDLR gene, apolipoprotein B-100 (ApoB) gene, PCSK9 gene and STAP1 gene
In previously obscure cases, the elucidation of the mechanism of LDL metabolism at the cellular level facilitated the diagnosis of FH by DNA analysis of FH-related genes. Consequently, the number of FH cases increased rapidly. The total number of cases in which we performed genotypic diagnosis is presented in Table 117, 18).
Clinical Severity of FH Diagnosed due to Abnormalities in the FH-related Genes
Hetero-FH cases diagnosed due to abnormalities in the FH-related genes are highly diverse, and it is clear that the serum cholesterol level varies depending on the particular mutation. The genotypic heterogeneity of LDL-R produces the clinical phenotypic variation (Fig. 12)12, 16). Similar variations with respect to other FH-related genes have also been identified. An apparent difference was found by the genetic investigation of homo-FH while comparing the plasma cholesterol levels in homo-FH patients in whom LDL-R mutations, PCSK9 mutations, and the classical gene mutations are unknown (Fig. 13). In terms of clinical severity, the order of FH is as follows: LDL-R mutation > Apo-B-100 mutation > PCSK9 mutation > ARH mutation. Furthermore, gene mutations should be identified and the associated clinical severity should be investigated. Even in cases with same gene mutation, the diagnosis may be hypolipidemic FH, normolipidemic FH, or hyperlipidemic FH. In this context, although the search for FH-related genes is a desirable endeavor, the precise genotypic diagnosis is unlikely to alter the treatment paradigm (achievement of target LDL-C level) or the choice of pharmacotherapeutic agents.
Fig. 12.
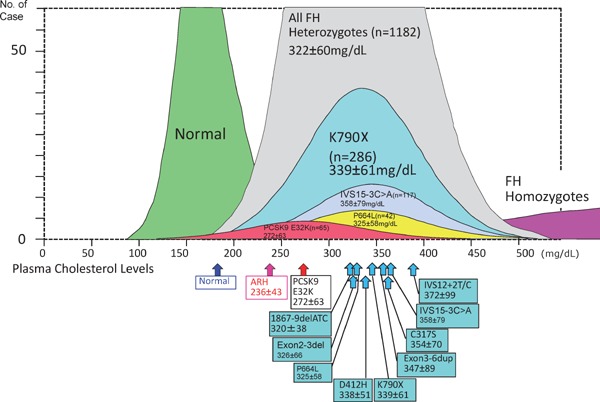
Plasma cholesterol distributions in each genotypic hetero-FH patient.
Genotypic variations show phenotypic heterogeneity.
Size of each boxes show mean ± SD.
Fig. 13.
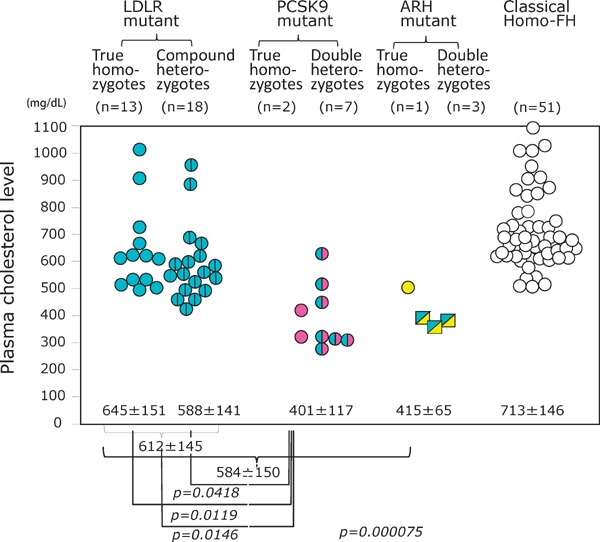
Plasma cholesterol levels in different genotypes of homo-FH patients.
Plasma cholesterol levels in the homo-FH due to LDL-R mutants are higher than those in the homo-FH due to PCSK9 E32K homo-FH or autosomal recessive homo-FH. Data were obtained from our previous studies by Mabuchi et. al.12) and Tada et. al.14)
Frequency of FH
A definitive diagnosis of FH is a necessity to accurately estimate the frequency of FH. In particular, the geographic region where a case is screened also remains an issue. On determining the total number of the homo-FH patients in a particular region, we can estimate the frequency of the hetero-FH patients using the Hardy-Weinberg equation. We studied FH in Hokuriku, Japan, which has a population of approximately 3,081,00012). The proportion of FH gene mutations in our patients was as follows: 73% in LDL-R and 6.6% in PCSK9, whereas no patient had Apo-B mutations (Table 1)17, 20). In 20.8% of subjects, no mutations were detected in the FH genes; thus, these patients may not have had FH or may have had FH due to unknown genetic mutations. Of the estimated prevalence of 1/500 hetero-FH patients in the general population, <1% of cases are diagnosed in most countries. Recently, direct screening in the general population in northern Europe revealed approximately 1/200 hetero-FH patients. Therefore, based on prevalence rates between 1/500 and 1/200, approximately 14 and 35 million individuals worldwide have FH21).
Serum cholesterol concentrations in all patients homo-FH (n = 31) due to LDL-R mutations were found to range widely from 362 to 1021 mg/dL, and the average cholesterol concentration was 612 ± 145 mg/dL (mean ± SD) (Fig. 13). Serum cholesterol concentrations in homo-FH due to PCSK9 E32K mutation ranged from 269 to 630 mg/dL, and the mean serum TC concentration (401 ± 117 mg/dL) in homozygotes caused by PCSK9 E32K was significantly lower than their counterparts with LDL-R mutation (p < 0.001). As a result, the frequency of LDL-R mutant heterozygotes is estimated to be 0.0034, which corresponds to 1/293 people in the general population12). The frequency of PCSK9 mutant heterozygotes is estimated to be 0.0016, which corresponds to 1/622 people in the general population12). Thus, it is estimated that heterozygous mutations in the LDL-R or the PCSK9 gene will occur at a combined frequency of 0.005, corresponding to 1/199 people in the general population of the Hokuriku district of Japan12). Of the investigated 149 myocardial infarction (MI) patients younger than 65 years, 18 patients (12%) were FH, and in the 43 MI patients younger than 40 years, 17 patients (41%) were FH22). Our assumption from this data states that coronary artery stenosis detectable by angiography occurs after 17 and 25 years of age in male and female heterozygotes, respectively, and the treatment of heterozygotes using lipid-lowering drugs can be delayed until late adolescence23).
Plasma cholesterol concentrations in patients with homo-FH (n = 30) due to PCSK9 mutation were found to range widely from 269 to 630 mg/dL. This variation was also observed in a pair of homo-FH brothers in a family with the same double mutations (PCSK9 E32K and LDL-R C183S) (Fig. 14)12). The genotype is a homo-FH of the same double heterozygote, whereas, as shown in the pictures by investigations of K.K. and M.K., the clinical phenotype is markedly different. Although determining the genotype is essential for the diagnosis of FH, the phenotypic characterization is the key for setting target levels for FH management. In this family the clinical phenotype is greatly different. Thus, a genotype is necessary for the diagnosis of FH; however, the phenotypic management target values are important for setting target values for FH management24).
Fig. 14.
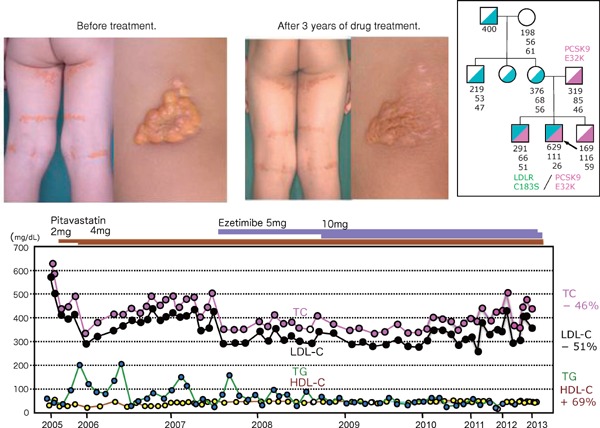
Two brothers with the same genotypic double heterozygotes due to LDL-R gene mutant C183S and PCSK9 E32K show quite different phenotypic characteristics.
The elder brother shows generalized planar xanthomatosis. His plasma TC was 629 mg/dL, LDL-C 581 mg/dL, and his plasma cholesterol responded very well to statin and ezetimibe, and his xanthomatosis became smaller and softer and almost disappeared without LDL-apheresis. But his younger brother has no xanthomas, and his TC was 291 mg/dL, and his LDL-C was 227 mg/dL and he had never been treated by statin or any other medication.
New Treatment of Homo-FH
The treatment of FH involves reducing plasma LDL-C in hetero-FH and homo-FH patients24). At present, it may be premature to introduce liver transplantation and LDL-R gene therapy for homo-FH patients, although a few patients have successfully undergone liver transplantation.
Despite the use of currently available lipid-lowering therapies, a significant proportion of patients with severe hypercholesterolemia do not achieve their treatment goals and consequently remain at an increased risk of ASCVD. Based on their clinical experience, these patients tend to have the most severe forms of FH and markedly elevated LDL-C levels but cannot tolerate statin therapy.
At present, an alternative therapy is the use of an antisense apoB-100 preparation (mipomersen)25) and microsomal triglyceride transfer protein (MTP) inhibitor (lomitapide)26, 27). MTP is indispensable for the formation of chylomicrons in the small intestine and very low-density lipoprotein in the liver. Because MTP controls triglycerides and the rearrangement of the apoB-100 synthesized in the liver and small intestine, an abetalipoproteinemic patient suffers from MTP deficiency26, 27) and lacks β-lipoprotein and plasma LDL-C. The serum cholesterol level in homo-FH patients is considered to moderately decrease upon MTP inhibition, as assumed from this case of MTP deficiency, and MTP inhibitor was developed and released recently. Plasma cholesterol decreases upon drug administration; however, fatty liver and diarrhea may occur due to its mechanism of action28). A male patient (S.Y.) with homo-FH due to true homozygote with LDL-R D280Y/D280Y has been treated by LDL-apheresis for more than 35 years. His severe xanthomatosis rapidly decreased in size and number, and his frequent anginal attacks almost disappeared after the long-term treatment with LDL-apheresis (Figs. 15 and 16)29).
Fig. 15.
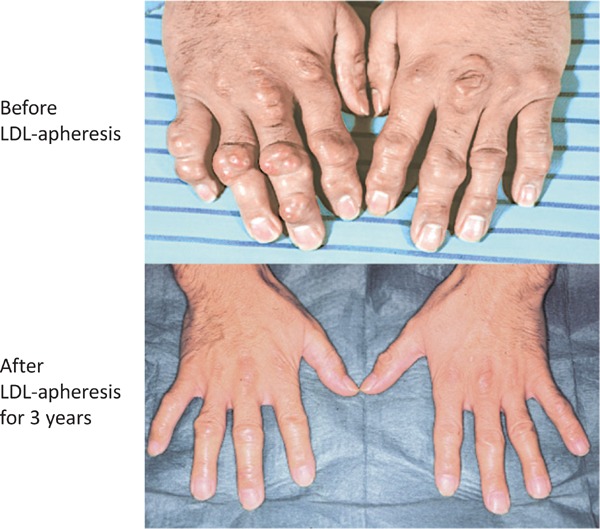
A homo-FH man had been treated by LDL-apheresis for more than 35 years.
After 3 years of LDL-apheresis, his xanthomatosis greatly decreased in size and hardness.
Fig. 16.
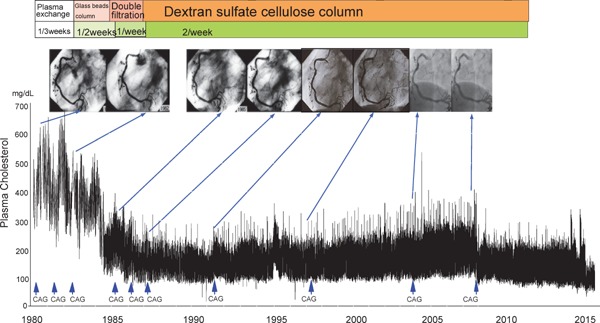
The male homo-FH patient (S.Y.) due to true homozygote with LDL-R D280Y/D280Y has been treated by LDL-apheresis for more than 35 years. His severe xanthomatosis rapidly decreased in size and number, and his frequent anginal attacks almost disappeared after the long-term treatment.
New Treatment of Hetero-FH
In 1971, Endo (Fig. 17) started the search for a cholesterol synthase (HMG-CoA reductase) inhibitor for the development of hypocholesterolemic agents. The metabolites from 6000 different kinds of mold were screened, and an HMG-CoA reductase inhibitor (compactin) was identified30). In this study, hetero-FH patients treated with compactin exhibited a decrease in LDL-C by 29%31), whereas no change in high-density lipoprotein cholesterol (HDL-C) was observed (Fig. 18). This finding was reported globally, including in the New York Times (Box 1) and Wall Street Journal, and even formed the opening scene of a play about the statin era (Boxes 2 and 3). Endo was awarded Lasker prize 2008 (Fig. 17).
Fig. 17.

Statin drugs discovered by Endo showed dramatic LDL-C reductions in hetero-FH and non-FH patients, and thus, compactin was called as “Penicillin for hypercholesterolemia”. (Left; A. Endo, Right; H. Mabuchi, A, Endo permitted me to use this picture.)
Fig. 18.
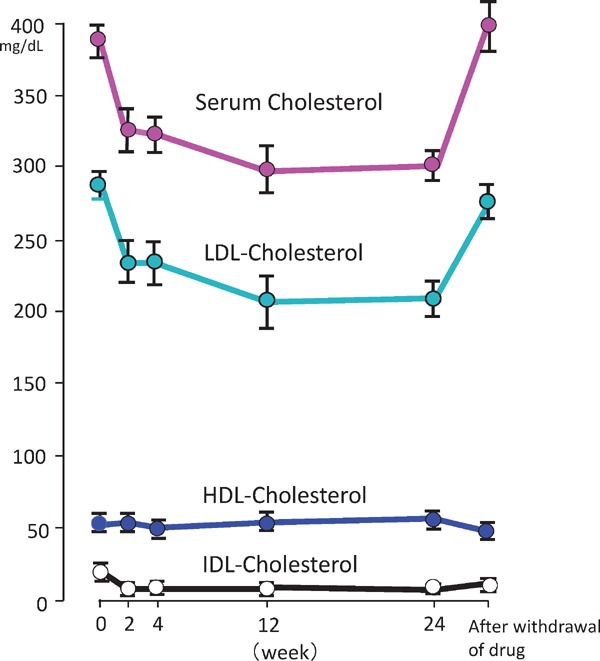
Effects of compactin on plasma total-, IDL-, LDL- and HDL-cholesterol levels in patients with hetero-FH.
LDL-C level was reduced by 29%, and HDL-C increased slightly.
Box 1.
New York Times. Aug. 26, 1981
New Drug May Prevent Hardening of Arteries
BOSTON, Aug. 26 (AP) — A new drug made from penicillin reduces levels of cholesterol in the blood and may prevent hardening of the arteries, an under-lying cause of the heart attacks and strokes that kill 800,000 Americans a year, researchers say.
A study shows that the experimental drug compactin is effective in people with an inherited defect that produces high cholesterol levels in the blood and leads to premature heart disease.
The new study was directed by Dr. Hiroshi Mabuchi at Kanazawa University School of Medicine in Japan and was published in the current issue of the New England Journal of Medicine.
Box 2.
Brown MS and Goldstein JL.
A tribute to Akira Endo, discoverer of a “Penicillin” for cholesterol.
Atherosclerosis Suppl. 5; 13–16, 2004
“Our confidence in statins was strongly reinforced when we saw Mabuchi's 1981 paper in The New England Journal of Medicine on the treatment of FH heterozygotes with compactin. Heterozygotes have one copy of a normal gene for LDL receptors, but this gene is partially suppressed by cholesterol. Compactin relieves this suppression, upregulating LDL receptors and lowering plasma LDL. We communicated our excitement in the editorial that we were invited to write to accompany Mabuchi's article, which we entitled “Lowering plasma cholesterol by raising LDL receptors”. Although we were enthusiastic, we cautioned that many hurdles had to be overcome before compactin would be a drug. “Many hurdles must be overcome before compactin or mevinolin can be accepted as a ‘penicillin’ for hypercholesterolemia. Among these was the ever-present concern about long-term toxicity, a concern that was not laid to rest until Merck conduced the landmark EXCEL study after the approval of lovastatin.”
Box 3.
Brown MS and Goldstein JL.
Editorial
Lowering plasma cholesterol by raising LDL receptors N Engl J Med. 305: 515–517, 1981
The goal of therapy in familial hypercholesterolemia is to reduce the concentration of LDL in plasma without disrupting cholesterol delivery to cells. The ideal approach is to stimulate the cells to produce more LDL receptors. When the number of receptors increases, the rate of LDL degradation will also initially increase. If the rate of LDL production does not change, the LDL level must decline.
In this context the recent discovery of compactin by Akira Endo, at the Sankyo Drug Company in Tokyo, assumes great importance. Isolated from a penicillin mold, compactin seems to have been designed by nature to be an ideal competitive inhibitor of HMG CoA reductase.
In this issue of the Journal, Mabuchi et al. reported a detailed study of compactin's effects on lipoprotein levels in human beings. They used extremely low doses of compactin, less than one tenth the amount used in the dog studies. Yet, they observed a dramatic 29 per cent reduction in plasma LDL levels in subjects with heterozygous familial hypercholesterolemia. Plasma HDL levels did not change.
Many hurdles must be overcome before compactin or mevinolin can be accepted as a “penicillin” for hypercholesterolemia. No long-term studies of toxicity have been reported in animals or patients. It is possible that these compounds will produce unexpected side effects and that new analogues will have to be developed. Yet the studies with the parent compounds compactin and mevinolin have established a general principle: interference with cholesterol synthesis can trigger an increase in LDL receptors, thereby reducing LDL levels in plasma without depleting vital body stores of cholesterol. This is indeed encouraging news.
Brown and Goldstein32) produced an editorial entitled “Lowering plasma cholesterol by raising LDL receptors” in association with this article and stated the following: “Mabuchi et al. examined the effect of compactin on the lipoprotein level in patients with FH as reported in the current issue of N Engl J Med 31) (Box 4) and announced that it did not reduce HDLC. We must overcome this barrier but comment that we can rightly estimate compactin as ‘penicillin for hypercholesterolemia'32).” Seven statin drugs have been used for treating hyper-LDL cholesterolemia, including that in FH patients. The rate of decline in LDL-C level due to statin is 20%–55%. This decline occurs via the release of a PCSK9 inhibitor, which increases LDL-R level33). Using an anion exchange resin, a dramatic decrease in LDL-C level was observed with the use of compactin, which increased LDL-R level, together with the anti-PCSK9 monoclonal antibody, which was recently released (Box 5). It is impossible to improve the abnormality of the LDL-R gene in hetero-FH patients, but statins can influence LDL-R activity by reinforcing other normal LDL-R gene remark34).
Box 4.
Kolata G.
Research News
Cholesterol–heart disease link illuminated.
Science 221: 1164–1166, 1983
New findings explain how blood cholesterol levels are controlled and how to lower them substantially in persons at high risk of heart disease
The relationship between cholesterol and heart disease has, until recently, eluded scientific explanation. It was known that extremely high concentrations of cholesterol in the blood can --- by themselves --- cause heart disease. It was known that cell surface receptors for a protein that carries cholesterol in the blood somehow are necessary for the control of blood cholesterol levels. But no one knew how blood cholesterol concentrations are controlled nor any logical way to substantially lower blood cholesterol in people at very high risk of heart disease. When given alone, either compactin or mevinolin will cause a decrease in cholesterol synthesis and a consequent increase in liver LDL receptors. But, says Brown, “The most dramatic effect is when one of these drugs is given with cholestyramine.” As first reported by Hiroshi Mabuchi and his associates at Kanazawa University in Japan, the two drug combination causes a 50 to 55 percent decrease in plasma LDL levels in heterozygotes for familial hypercholesterolemia --- which puts them in the normal range. Says Brown, “The important scientific point is that here is a genetic disease that is manifested in heterozygotes and we can stimulate the expression of normal gene.”
And by using these drugs, it may be possible to answer at last the question of whether lowering cholesterol levels lowers the risk of heart disease. For the first time, a treatment is available that lowers cholesterol enough that a difference in heart risk, if it occurs, should be readily apparent in a clinical trial.
Box 5.
Young SG and Fong LG.
Editorial
Lowering plasma cholesterol by raising LDL receptors–revisited
N Engl J Med 366; 1154–1155, 2012
Thirty-one years ago, Mabuchi and colleagues reported in the Journal that a statin called compactin reduced plasma low-density lipoprotein (LDL) cholesterol levels by 29% in patients with heterozygous familial hypercholesterolemia. This drug inhibits an early step in cholesterol synthesis catalyzed by the enzyme 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase. In an accompanying editorial entitled “Lowering Cholesterol by Raising LDL Receptors,” Brown and Goldstein noted that heterozygotes for familial hypercholesterolemia have a deficiency of LDL receptors on cells, resulting in reduced cholesterol uptake by cells and high levels of cholesterol in the plasma. They explained that cholesterol levels within cells are tightly controlled, in part by LDL receptors, and pointed out that the cells' machinery for regulating LDL receptors can be harnessed for therapeutic purposes. In the case of statins, blocking an early step in sterol synthesis deprives hepatocytes of cholesterol, leading to up-regulated LDL receptors, increased cholesterol uptake by cells, and lower plasma cholesterol levels. They predicted that statins, if proved to be safe, would delay the onset of atherosclerotic heart disease.
In the current issue of the Journal, Stein and co-workers report the results of phase 1 clinical trials of REGN727, a human PCSK9 monoclonal antibody manufactured by Regeneron Pharmaceuticals. REGN727 was administered either intravenously or subcutaneously to healthy control hypercholesterolemia, some of whom were receiving atorvastatin and some of whom were receiving a modified diet only. In a dose-dependent fashion, REGN727 lowered LDL cholesterol levels by up to 64%, and the percent lowering was similar regardless of whether patients were taking a statin. Cholesterol lowering was prompt and persisted for weeks, and the antibody had an acceptable side effect profile as compared with placebo. In their 1981 editorial, Brown and Goldstein concluded that the “important lesson” of the compactin studies was that “normal regulatory mechanisms can be exploited to lower plasma LDL.” The PCSK9 story reinforces this paradigm in an emphatic fashion. And although the discovery of PCSK9 is an exciting chapter in the cholesterol story, no one should assume it is the last.
As PCSK9 is a modulator of LDL-R, other mechanisms to control this modulation may exist. In a study by Stein, healthy subjects, non-FH patients, and FH patients were administered anti-PCSK9 monoclonal antibodies33). These drugs decreased LDL-C level by 41%–58%. An editorial associated with this article had the following title: “Lowering plasma cholesterol by raising LDL-Rs-Revisited,” which opened with the following remarks: “Thirty-one years ago, Mabuchi and colleagues reported in the journal that a statin called compactin reduced plasma LDL-C levels by 29% in patients with hetero-FH31, 32). This drug inhibits an early step in cholesterol synthesis catalyzed by HMG-CoA reductase. In an accompanying editorial titled “Lowering Cholesterol by Raising LDL Receptors,” Brown and Goldstein noted that “heterozygotes for FH have a deficiency of LDL receptors on cells, resulting in reduced cholesterol uptake by cells and high levels of cholesterol in the plasma. Here, repeated treatment of hetero-FH recovers 100% of residual LDL-R activity”(Fig. 7)34).
Causes of Death due to FH in the Pre-Statin and Statin Eras
A typical Japanese case of a 4-year-old boy with essential familial hypercholesterolemic xanthomatosis was reported in 196835). High serum cholesterol levels of 876 –1144 mg/dL and general xanthomatosis were suggestive of homo-FH. Profound narrowing of each coronary artery was also observed.
In the pre-statin era (before 1991), we studied the development of coronary artery disease (CAD) in 10 homozygous and 692 heterozygous FH patients. Of those affected by MI, 97 were male heterozygotes and 41 were female heterozygotes; this was first noted for men in their third decade of life and for women in their fourth decade of life (Fig. 19). Out of 107 deceased heterozygous patients 74 patients (69%) died of CAD (Table 3). The mean age at death was significantly lower in male heterozygotes (59 ± 13 years) than in their female counterparts (69 ± 9 years). Of those who underwent coronary angiographic evaluation, five were homo-FH patients and 105 were male and 56 were female hetero-FH patients. The regression equations between age (X) and coronary artery stenosis index (Y) were Y = 1.57X − 20.43 in the homozygotes, Y = 0.52X − 9.11 in the male heterozygotes, and Y = 0.47X − 12.57 in the female heterozygotes23). From these data, we can assume that coronary artery stenosis detectable by angiography will occur after 17 and 25 years of age in male and female heterozygotes, respectively, and the treatment of heterozygotes using lipid-lowering drugs can be delayed until late adolescence.
Fig. 19.
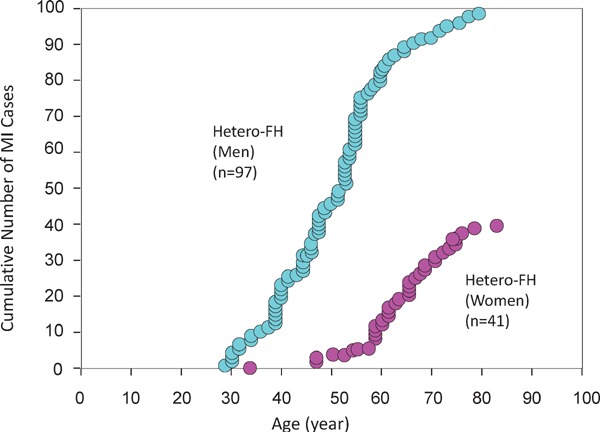
Cumulative number of myocardial infarction (MI) cases in male and female hetero-FH patients in prestatin era
MI was first noted in men in the 3 decade of life and in women in the 4 decade of life. The mean age of first MI attack is 50 years old in the male hetero-FH, and 65 years old in female hetero-FH patients. The MI patients younger than those ages could be called as “premature MI” cases
Table 3. Causes of death in hetero-FH patients in pre-statin era.
| Male hetero-FH |
Female hetero-FH |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | % | Age of Death (yrs) | TC | TG | n | % | Age of Death (yrs) | TC | TG | |
| ASCVD | 45 | 72.6 | 29 | 64.4 | ||||||
| Myocardial Infarction | 25 | 40.3 | 57 ± 12 | 358 ± 66 | 166 ± 66 | 20 | 44.4 | 70 ± 9 | 344 ± 72 | 158 ± 87 |
| Sudden Cardiac Death | 14 | 22.6 | 54 ± 15 | 361 ± 61 | 126 ± 75 | 6 | 13.3 | 69 ± 7 | 376 ± 54 | 152 ± 37 |
| Heart Failure | 2 | 3.2 | 76 ± 2 | 369 ± 40 | 111 | 3 | 6.7 | 68 ± 10 | 350 ± 26 | 140 ± 86 |
| Cardio-aortic Surgery | 4 | 6.5 | 62 ± 7 | 387 ± 119 | 151 ± 5 | 0 | 0 | --- | --- | --- |
| Stroke | 3 | 4.8 | 63 ± 5 | 379 ± 90 | 157 ± 78 | 7 | 15.6 | 71 ± 4 | 335 ± 116 | 136 ± 56 |
| Cancer | 9 | 14.5 | 60 ± 9 | 341 ± 68 | 191 ± 102 | 5 | 11.1 | 64 ± 13 | 373 ± 86 | 164 ± 35 |
| Others | 5 | 8.1 | 64 ± 20 | 334 ± 90 | 128 ± 60 | 4 | 8.9 | 73 ± 12 | 389 ± 90 | 84 ± 28 |
| Total | 62 | 100.0 | 59 ± 13 | 332 ± 104 | 148 ± 72 | 45 | 100.0 | 69 ± 9 | 355 ± 77 | 147 ± 69 |
ASCVD; Atherosclerotic Cardiovascular Disease
TC; total cholesterol (mg/dL), TG; triglyceride (mg/dL)
Data; mean ± SD
Statin was released in 1991 and contributed to the prevention and treatment of ASCVD in FH and non-FH patients worldwide. In this case, we investigated the causes of death in the FH patients in the pre-statin and statin eras. Regarding the causes of death in the hetero-FH patients in the pre-statin (Table 3)36), 72.6% of male hetero-FH patients died of ASCVD at a mean age of 59 ± 13 years, and 64.4% of female hetero-FH patients died of ASCVD at a mean age of 69 ± 9 years. Homo-FH patients died at a mean age of 28 ± 13 years in the pre-statin era, but this was prolonged by 30 years to 59 ± 20 years in the statin era (Table 4). These homo-FH patients had never been treated by LDL-apheresis. We also compared the age at death of hetero-FH patients between the pre-statin and statin eras (Table 5)33), which was increased by 12.6 years from 63.3 ± 12.3 years to 75.9 ± 12.4 years. Thus, the utility of statins for hetero-FH and homo-FH was demonstrated, even though double-blind study using placebo is ethically difficult to design.
Table 4. Causes of death in homo-FH patients in pre-statin and statin eras.
| <1991 (Pre-statin era) | ||||||||||
| No | Name | Sex | Entry year | Entry age | TC | TG | Death year | Death age | Cause of death | Genotype |
| 1 | M.I. | F | 1964 | 20 | 730 | 273 | 1967 | 23 | Cardiac death | K790X/K790X |
| 2 | Y.E. | M | 1966 | 11 | 908 | 300 | 1970 | 15 | Heart failure | IVS15-3C>A/IVS15-3C>A |
| 3 | K.Y. | F | 1977 | 27 | 609 | 126 | 1977 | 27 | Sudden cardiac death | D280Y/D280Y |
| 4 | K.M. | F | 1978 | 40 | 610 | 180 | 1979 | 41 | Sudden cardiac death | D280Y/D280Y |
| 5 | S.T. | M | 1984 | 17 | 529 | 177 | 1984 | 17 | Aortic valvular stenosis | ?/? |
| 6 | S.S. | F | 1987 | 26 | 1004 | 784 | 1987 | 26 | Sudden cardiac death | K90X/K790X |
| 7 | M.K. | F | 1980 | 40 | 550 | 143 | 1990 | 50 | Leukemia | Exon2-3del/Ex2-3del |
| Mean | 1977 | 26 | 706 | 283 | 1980 | 28 | ||||
| SD | 9 | 11 | 185 | 213 | 9 | 13 | ||||
| >1992 (Statin era) | ||||||||||
| No | Name | Sex | Entry year | Entry age | TC | TG | Death year | Death age | Cause of death | Genotype |
| 1 | N.Y. | F | 1992 | 36 | 590 | 93 | 1993 | 37 | Cardiac death | ?/? |
| 2 | T.T. | M | 1977 | 40 | 558 | 388 | 1995 | 58 | Sudden cardiac death | Exon2-3del/Exon2-3del |
| 3 | H.T. | M | 1985 | 59 | 493 | 128 | 1997 | 71 | Pancreas cancer | W159X/R94H |
| 4 | M.N. | F | 1994 | 73 | 599 | 107 | 1998 | 77 | Cardiac death | R395W/R395W |
| 5 | T.K. | M | 1986 | 35 | 633 | 312 | 2004 | 53 | After cardiac surgery | K790X/K790X |
| 6 | J.S. | M | 1979 | 5 | 853 | 123 | 2005 | 31 | After cardiac surgery | c2201-2202delCA/c2201-2202delCA |
| 7 | Y.I. | F | 1980 | 52 | 622 | 315 | 2012 | 84 | Interstitial pneumonia | Exon2-3del/Exon2-3del |
| Mean | 1985 | 43 | 621 | 209 | 2001 | 59 | ||||
| SD | 7 | 22 | 112 | 124 | 7 | 20 | ||||
TC; total cholesterol (mg/dL), TG; triglyceride (mg/dL)
Table 5. Clinical data and age of death in homo- and hetero-FH patients in pre-statin and statin eras.
| M/F | Entry year | Entry age (yr) | TC (mg/dL) at entry | Death year | Death age (yr) | |
|---|---|---|---|---|---|---|
| Homo-FH | ||||||
| Pre-statin (n = 7) | 2/5 | 1976.6 ± 5.4 | 25.9 ± 11.1 | 706 ± 185 | 1979.0 ± 9.0 | 28.4 ± 12.7 |
| Statin (n = 7) | 4/3 | 1985.0 ± 6.5 | 42.9 ± 11.8 | 621 ± 112 | 2001.0 ± 7.0 | 58.7 ± 20.0 |
| (Statin – Pre-statin) | +30.3 years P < 0.00001 |
|||||
| Hetero-FH | ||||||
| Pre-statin (n = 108) | 62/46 | 1980.2 ± 5.4 | 59.5 ± 12.6 | 357 ± 72 | 1984.1 ± 5.0 | 63.3 ± 12.3 |
| Statin (n = 93) | 51/42 | 1985.0 ± 7.4 | 57.4 ± 12.4 | 334 ± 59 | 2001.7 ± 6.9 | 75.9 ± 12.4 |
| (Statin – Pre-statin) | +12.6 years P < 0.000000054 |
|||||
TC; total cholesterol (mg/dL), TG; triglyceride (mg/dL)
Data; mean ± SD
Conclusions
FH is a common genetic disease and is highly associated with premature ASCVD. For the differential diagnosis of homo-FH and hetero-FH in patients, obtaining a pedigree (family tree) is simple and essential in planning cascade screening or family studies. For the diagnosis of hetero-FH, the detection of Achilles tendon xanthomas by palpation or on X-ray is an indispensable diagnostic skill in clinical lipidology. For the management of LDL-C level in homo- and hetero-FH patients, PCSK9 inhibitors, mipomersen, lomitapide, and LDL-apheresis in addition to statins should be used. It is important to sequentially unravel the unrecognized pathogenetic mechanisms of FH to reduce the under-recognition of FH and develop new management strategies for FH (Fig. 20).
Fig. 20.
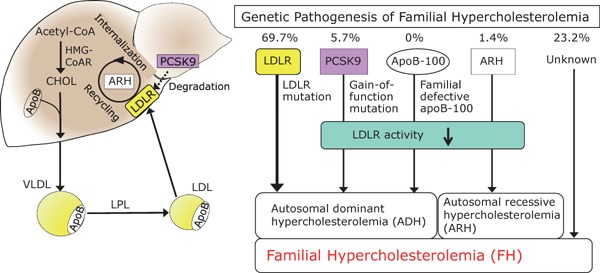
Molecular pathogenesis of FH
Genetic mechanism in the pathogenesis of FH is shown here (n = 1,067). Causes of FH are LDL-R mutations (69.7%), PCSK9 gain of function mutations (5.7%), ARH (1.4%) and other unknown mutation (23.2%).
Acknowledgements
My lifework about FH is a result of the Hokuriku FH Research Group members, and I express sincere gratitude for their long-term collaboration.
Conflict of Interest
H.M. has no conflict of interest concerning this publication.
References
- 1). Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In the Metabolic and molecular bases of inherited disease. ed. by Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, Vogelstein B. McGraw-Hill Professional; 8th ed. 2000 [Google Scholar]
- 2). Fagge CH. Xanthomatous disease of the skin. I. General xanthelasma or vitiligoides. Trans Pathol Soc Lond 24: 242-250, 1873 [Google Scholar]
- 3). Yamakawa S, Kashiwabara M. Relashionship between pathogenesis of xanthoma and disturbance of lipid metabolism. (in Japanese). Tohoku Igakuzasshi 6: 322-336, 1922 [Google Scholar]
- 4). Mabuchi H, Tatami R, Haba T, Ueda K, Ueda R, Kametani T, Itoh S, Koizumi J, Oota M, Miyamoto S, Takeda R, Takeshita H. Homozygous familial hypercholesterolemia in Japan. Am J Med. 65: 290-297, 1978 [DOI] [PubMed] [Google Scholar]
- 5). Fredrickson DS, Levy RI, Lees RS. Fat transport in lipoproteins - an integrated approach to mechanisms and disorders. N Engl J Med. 276: 34-42, 94,-103, 148,-156, 215,-225, 273-281, 1967 [DOI] [PubMed] [Google Scholar]
- 6). Brown MS, Goldstein JL. Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc Natl Acad Sci U S A. 71: 788-792, 1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7). Haba T, Mabuchi H, Yoshimura A, Watanabe A, Wakasugi T, Tatami R, Ueda K, Ueda R, Kametani T, Koizumi J, Miyamoto S, Takeda R, Takeshita H. Effects of ML-236b (compactin) on sterol synthesis and low density lipoprotein receptor activities in fibroblasts of patients with homozygous familial hypercholesterolemia. J Clin Invest. 67: 1532-1540, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 161: 161-172, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9). Yamamoto T, Davis CG, Brown MS, Schneider WJ, Casey ML, Goldstein JL, Russell DW. The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell. 39: 27-38, 1984 [DOI] [PubMed] [Google Scholar]
- 10). Usifo E, Leigh SE, Whittall RA, Lench N, Taylor A, Yeats C, Orengo CA, Martin AC, Celli J, Humphries SE. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Ann Hum Genet 76: 387-401, 2012 [DOI] [PubMed] [Google Scholar]
- 11). Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derré A, Villéger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 34: 154-156, 2003 [DOI] [PubMed] [Google Scholar]
- 12). Mabuchi H, Nohara A, Noguchi T, Kobayashi J, Kawashiri MA, Inoue T, Mori M, Tada H, Nakanishi C, Yagi K, Yamagishi M, Ueda K, Takegoshi T, Miyamoto S, Inazu A, Koizumi J, Hokuriku FH Study Group Genotypic and phenotypic features in homozygous familial hypercholesterolemia caused by proprotein convertase subtilisin/kexin type 9 (PCSK9) gain-of-function mutation. Atherosclerosis. 236: 54-61, 2014 [DOI] [PubMed] [Google Scholar]
- 13). Harada-Shiba M, Takagi A, Miyamoto Y, Tsushima M, Ikeda Y, Yokoyama S, Yamamoto A. Clinical features and genetic analysis of autosomal recessive hypercholesterolemia. J Clin Endocrinol Metab. 88: 2541-2547, 2003 [DOI] [PubMed] [Google Scholar]
- 14). Tada H, Kawashiri MA, Ohtani R, Noguchi T, Nakanishi C, Konno T, Hayashi K, Nohara A, Inazu A, Kobayashi J, Mabuchi H, Yamagishi M. A novel type of familial hypercholesterolemia: double heterozygous mutations in LDL receptor and LDL receptor adaptor protein 1 gene. Atherosclerosis. 219: 663-666, 2011 [DOI] [PubMed] [Google Scholar]
- 15). Fouchier SW, Dallinga-Thie GM, Meijers JC, Zelcer N, Kastelein JJ, Defesche JC, Hovingh GK. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ Res. 115: 552-555, 2014 [DOI] [PubMed] [Google Scholar]
- 16). Raal FJ, Sjouke B, Hovingh GK, Isaac BF. Phenotype diversity among patients with homozygous familial hypercholesterolemia: A cohort study Atherosclerosis. 248: 238-244, 2016 [DOI] [PubMed] [Google Scholar]
- 17). Yu W, Nohara A, Higashikata T, Lu H, Inazu A, Mabuchi H. Molecular genetic analysis of familial hypercholesterolemia: spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis. 165: 335-42, 2002. Erratum in: Atherosclerosis. 174: 399–400, 2004 [DOI] [PubMed] [Google Scholar]
- 18). Mabuchi H, Miyamoto S, Ueda K, Oota M, Takegoshi T, Wakasugi T, Takeda R. Causes of death in patients with familial hypercholesterolemia. Atherosclerosis. 61: 1-6, 1986 [DOI] [PubMed] [Google Scholar]
- 19). Mabuchi H, Higashikata T, Nohara A, Lu H, Yu WX, Nozue T, Noji Y, Katsuda S, Kawashiri MA, Inazu A, Kobayashi J, Koizumi J. Cutoff point separating affected and unaffected familial hypercholesterolemic patients validated by LDL-receptor gene mutants. J Atheroscler Thromb. 12: 35-40, 2005 [DOI] [PubMed] [Google Scholar]
- 20). Nohara A, Yagi K, Inazu A, Kajinami K, Koizumi J, Mabuchi H. Absence of familial defective apolipoprotein B-100 in Japanese patients with familial hypercholesterolaemia. Lancet. 345: 1438, 1995 [DOI] [PubMed] [Google Scholar]
- 21). Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjærg-Hansen A, European Atherosclerosis Society Consensus Panel Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 34: 3478-3490a, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22). Mabuchi H. The frequency of familial hypercholesterolemia (FH) in patients with coronary heart disease. In the Introductory textbook of dyslipidemia (In Japanese), Bunkoudou, Tokyo, p.42-43. 2005 [Google Scholar]
- 23). Mabuchi H, Koizumi J, Shimizu M, Takeda R. Development of coronary heart disease in familial hypercholesterolemia. Circulation. 79: 225-232, 1989 [DOI] [PubMed] [Google Scholar]
- 24). Harada-Shiba M, Arai H, Oikawa S, Ohta T, Okada T, Okamura T, Nohara A, Bujo H, Yokote K, Wakatsuki A, Ishibashi S, Yamashita S. Guidelines for the management of familial hypercholesterolemia. J Atheroscler Thromb. 19: 1043-1060, 2012 [DOI] [PubMed] [Google Scholar]
- 25). Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, Lachmann RH, Gaudet D, Tan JL, Chasan-Taber S, Tribble DL, Flaim JD, Crooke ST. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 375: 998-1006, 2010 [DOI] [PubMed] [Google Scholar]
- 26). Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 129: 1022-1032, 2014 [DOI] [PubMed] [Google Scholar]
- 27). Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, Averna MR, Sirtori CR, Shah PK, Gaudet D, Stefanutti C, Vigna GB, Du Plessis AM, Propert KJ, Sasiela WJ, Bloedon LT, Rader DJ, Phase 3 HoFH Lomitapide Study investigators Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 381: 40-46, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28). Mabuchi H, Nohara A. MTP inhibitor for treating severe LDL-cholesterolemia. Clin Lipidol 8: 387-389, 2013 [Google Scholar]
- 29). Thompson GR, Catapano A, Saheb S, Atassi-Dumont M, Barbir M, Eriksson M, Paulweber B, Sijbrands E, Stalenhoef AFh, Parhofer KG. Severe hypercholesterolaemia: therapeutic goals and eligibility criteria for LDL apheresis in Europe. Curr Opinion Lipidol 21: 492-498, 2010 [DOI] [PubMed] [Google Scholar]
- 30). Endo A. The discovery and development of HMG-CoA reductase inhibitors. J Lipid Res. 33: 1569-1582, 1992 [PubMed] [Google Scholar]
- 31). Mabuchi H, Haba T, Tatami R, Miyamoto S, Sakai Y, Wakasugi T, Watanabe A, Koizumi J, Takeda R. Effect of an inhibitor of 3-hydroxy-3-methyglutaryl coenzyme A reductase on serum lipoproteins and ubiquinone-10-levels in patients with familial hypercholesterolemia. N Engl J Med. 305: 478-482, 1981 [DOI] [PubMed] [Google Scholar]
- 32). Brown MS, Goldstein JL. Lowering plasma cholesterol by raising LDL receptors. N Engl J Med 305: 515-517, 1981 [DOI] [PubMed] [Google Scholar]
- 33). Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, Lisbon E, Gutierrez M, Webb C, Wu R, Du Y, Kranz T, Gasparino E, Swergold GD. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 366: 1108-1118, 2012 [DOI] [PubMed] [Google Scholar]
- 34). Young SG, Fong LG. Lowering plasma cholesterol by raising LDL receptors--revisited. N Engl J Med. 366: 1154-1155, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35). Watanabe T, Tanaka K, Yanai N. Essential familial hypercholesteremic xanthomatosis--an autopsy case with special reference to the pathogensis of cardiovascular lipidosis. Acta Pathol Jpn. 18: 319-331, 1968 [PubMed] [Google Scholar]
- 36). Mabuchi H, Nohara A, Kawashiri M-A, Mori M, Tada H, Nakanishi C, Yamagishi M, Yagi K, Inazu A, Kobayashi J, Hokuriku FH Research Group Causes of death in homo- and heterozygous familial hypercholesterolemia during pre-statin and statin eras. XV International Symposium on Atherosclerosis, Poster No.928, 2015, Amsterdam, Netherlands [Google Scholar]